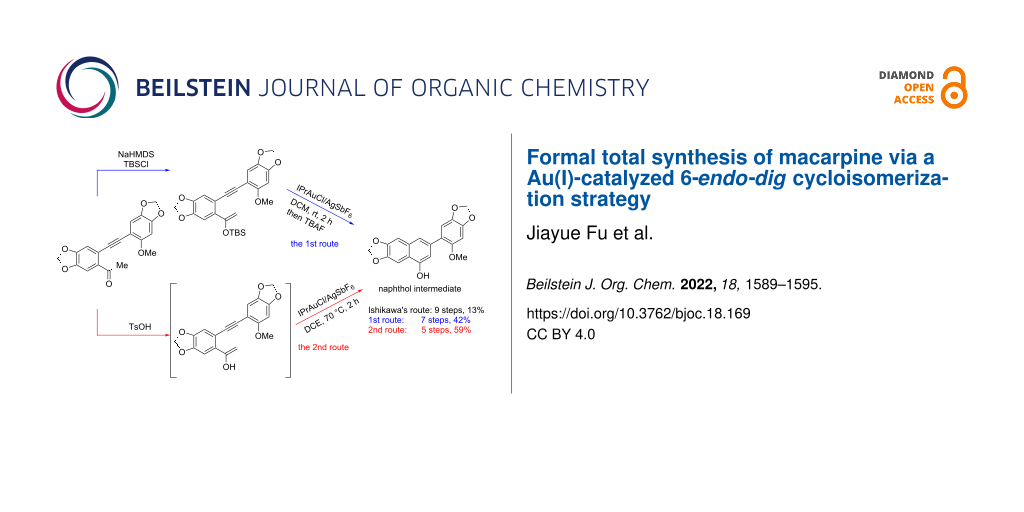
Abstract
The formal total synthesis of macarpine was accomplished by the construction of a naphthol intermediate in Ishikawa’s synthetic route with two different synthetic routes. The convergent synthetic strategies feature the utilization of Au(I)-catalyzed cycloisomerizations of a 1,5-enyne and alkynyl ketone substrates, which were prepared by Sonogashira coupling reactions.

Graphical Abstract
Introduction
Benzo[c]phenanthridine alkaloids are an ancient and influential category of isoquinoline alkaloids, mainly found in Papaveraceae and Rutaceae (Scheme 1) [1,2]. According to their oxidation states, benzo[c]phenanthridine alkaloids can be divided into two types: partially hydrogenated base and fully aromatized base, in which natural fully aromatic alkaloids can be further classified into three subclasses: O4-base, O5-base, and O6-base [3].
![[1860-5397-18-169-i1]](/bjoc/content/inline/1860-5397-18-169-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Classification of benzo[c]phenanthridine alkaloids.
Scheme 1: Classification of benzo[c]phenanthridine alkaloids.
Among these alkaloids macarpine is the most oxidized tetracyclic alkaloid with many bioactivities, including anesthesia, anticancer, anti-inflammatory [4-8], insecticidal, fungicidal, etc [9]. In addition to the above-mentioned activities, macarpine was also used as a DNA probe for flow cytometry and fluorescence microscopy due to its fluorescent properties [10]. Despite some research on the activities of macarpine had been performed, a more in-depth evaluation of the biological activities was still limited due to the need of its isolation from natural sources. Inspired by the requirement of further biological evaluation, the chemical syntheses of macarpine have been developed rapidly in the last three decades.
The benzo[c]phenanthridine skeleton consists of a phenanthridine (rings A, B, C) and a benzene (ring D), and most of the synthetic routes were completed in the last step by constructing ring B or ring C. Some representative examples and their key strategies are summarized in Scheme 2. In 1989, Hanaoka and co-workers developed the total synthesis of macarpine by Hofmann elimination from protoberberine by introducing rings B and C (Scheme 2a) [11]. In 1995, Ishikawa and co-workers accomplished the total synthesis via a Reformatsky reaction and aromatic nitrosation through the building of rings B and C (Scheme 2b) [12]. In 2010, Echavarren and co-worker completed the formal total synthesis via a Au(I)-catalyzed cyclization (Scheme 2c) [13]. In 2018, Pabbaraja and co-workers disclosed the synthesis of macarpine by constructing ring C through the domino Michael addition/SNAr reaction of nitromethane to an ynone precursor (Scheme 2d) [14].
![[1860-5397-18-169-i2]](/bjoc/content/inline/1860-5397-18-169-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Representative synthetic strategies for macarpine (1).
Scheme 2: Representative synthetic strategies for macarpine (1).
Results and Discussion
The efforts on developing efficient synthetic strategies to access macarpine never ceased during the last decades, and we have joined this meaningful research. Herein, a strategy involving the synthesis of an intermediate reported by Ishikawa in 1995 in the total synthesis of macarpine [12] is proposed via a Au(I)-catalyzed cycloisomerization reaction.
Retrosynthetically, the target molecule macarpine (1) could be disconnected into naphthol 12 (Scheme 3), a key intermediate reported by Ishikawa in the total synthesis of macarpine. This intermediate could be synthesized from silyl enol ether compound 10 via the Au(I)-catalyzed cycloisomerization reaction developed by our group [15]. The compound 10 could be constructed by the Sonogashira coupling reaction from readily prepared iodoarene 8 [12,16] and ketone 5, which could be synthesized by using cheap 6-bromopiperonal (2) as the starting material.
![[1860-5397-18-169-i3]](/bjoc/content/inline/1860-5397-18-169-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Retrosynthetic analysis of marcarpine precursor 12 for a partial synthesis.
Scheme 3: Retrosynthetic analysis of marcarpine precursor 12 for a partial synthesis.
To attempt the proposed synthetic strategy, ketone 5 and iodoarene 8 were prepared by following the synthetic route outlined in Scheme 4. Ketone 5 was prepared in a four-step procedure. Firstly, a Sonogashira coupling between 6-bromopiperonal (2) and trimethylsilylacetylene was performed to furnish aldehyde 3 [17,18] in 89% yield. A following nucleophilic addition reaction of aldehyde 3 by methylmagnesium bromide (MeMgBr) gave alcohol 4 in 99% yield, which was oxidized by pyridinium chlorochromate (PCC) leading to the formation of ketone compound and the deprotection of the silyl group was accomplished in the presence of potassium carbonate (K2CO3) and methanol to provide the terminal alkyne 5 in 96% yield in two steps. The iodoarene 8 [12,16] was facilely synthesized from sesamol (6) via methylation and iodination in an overall yield of 67%.
![[1860-5397-18-169-i4]](/bjoc/content/inline/1860-5397-18-169-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Syntheses of precursors 5 and 8.
Scheme 4: Syntheses of precursors 5 and 8.
With the building blocks 5 and 8 in hand, ketone 9 was prepared via a palladium-catalyzed Sonogashira coupling reaction in a yield of 95%. The precursor 10 for the gold(I)-catalyzed [19-24] cycloisomerization was then synthesized by treating ketone 9 with sodium bis(trimethylsilyl)amide (NaHMDS) and tert-butyldimethylsilyl chloride (TBSCl) (Scheme 5).
![[1860-5397-18-169-i5]](/bjoc/content/inline/1860-5397-18-169-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of enol silyl ether 10.
Scheme 5: Synthesis of enol silyl ether 10.
To find the best cycloisomerization conditions, the 1,5-enyne substrate 10 was subjected to different reaction conditions as listed in Table 1. It was observed that [1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene]gold(I) chloride (IPrAuCl) itself failed to catalyze the cycloisomerization (Table 1, entry 1). Evaluation of a number of silver salts illustrated that silver hexafluoroantimonate (AgSbF6) was the optimal additive to activate the gold catalyst (Table 1, entries 2, 3, and 7). Screening of the other ligands of Au(I) catalysts, including triphenylphosphane (Ph3P), [1,1'-biphenyl]-2-yl-di-tert-butylphosphane (JohnPhos) dicyclohexyl(2',4'-diisopropyl-3,6-dimethoxy-[1,1'-biphenyl]-2-yl)phosphane (BrettPhos) (Table 1, entries 4–6) revealed that 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) was still the best one (Table 1, entry 7). Neither decreasing nor increasing the loading of the catalyst gave better yields (Table 1, entries 8 and 9). Examination of the reaction time showed that 2 h was the shortest reaction time and that extending the reaction time did not help to increase the yield (Table 1, entries 10 and 11). Lowering or raising the reaction temperature resulted in lower yields (Table 1, entries 12 and 13). The solvent had less effect on the reaction, and combining various factors, DCM was used for the reaction (Table 1, entries 14 and 15). When AgSbF6 was utilized as the sole catalyst, not any product was generated indicating cationic Au(I) was the true catalyst (Table 1, entry 16). A control experiment using 2,6-di-tert-butylpyridine as a proton scavenger in the IPrAuCl/AgSbF6 system provided the product in good yield, which excluded the influence of trace amounts of acids on the reaction (Table 1, entry 17).
Table 1: Optimization of the Au(I)-catalyzed cycloisomerization conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-169-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | catalyst | solvent | additive | T (°C) | yield (%) |
| 1 | IPrAuCl | DCM | – | 23 | 0 |
| 2 | IPrAuCl | DCM | AgOTf | 23 | 61 |
| 3 | IPrAuCl | DCM | AgCO2CF3 | 23 | 23 |
| 4 | Ph3PAuCl | DCM | AgSbF6 | 23 | 77 |
| 5 | JohnPhosMeCNAuSbF6 | DCM | – | 23 | 64 |
| 6 | BrettPhosAuCl | DCM | AgSbF6 | 23 | 45 |
| 7 | IPrAuCl | DCM | AgSbF6 | 23 | 82 |
| 8 | IPrAuCl | DCM | AgSbF6 | 23 | 68a |
| 9 | IPrAuCl | DCM | AgSbF6 | 23 | 82b |
| 10 | IPrAuCl | DCM | AgSbF6 | 23 | 57c |
| 11 | IPrAuCl | DCM | AgSbF6 | 23 | 81d |
| 12 | IPrAuCl | DCM | AgSbF6 | 0 | 63 |
| 13 | IPrAuCl | DCM | AgSbF6 | 40 | 72 |
| 14 | IPrAuCl | toluene | AgSbF6 | 23 | 82 |
| 15 | IPrAuCl | THF | AgSbF6 | 23 | 80 |
| 16 | – | DCM | AgSbF6 | 23 | 0 |
| 17 | IPrAuCl | THF | AgSbF6 | 23 | 80e |
a3 mol % IPrAuCl and 3 mol % AgSbF6 were used. b10 mol % IPrAuCl and 10 mol % AgSbF6 were used. cThe reaction was run for 1 h. dThe reaction was run for 3 h. e5 mol % 2,6-di-tert-butylpyridine was added. IPr = [1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene]. JohnPhos = [[1,1'-biphenyl]-2-yldi-tert-butylphosphane]. BrettPhos = [dicyclohexyl(2',4'-diisopropyl-3,6-dimethoxy-[1,1'-biphenyl]-2-yl)phosphane].
The Au(I)-catalyzed cycloisomerization reaction of substrate 10 occurred under the catalysis of 5 mol % [1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene]gold(I) chloride (IPrAuCl) and 5 mol % silver hexafluoroantimonate (AgSbF6) [25,26] in anhydrous DCM at room temperature for 2 h forming a benzene ring smoothly, leading to the exclusive formation of biaryl intermediate 11 in a yield of 82%. It is worth noting that the methoxy substitution in the substrate played a crucial role in controlling the selectivity of the cycloisomerization according to our previous study [15]. It was rationalized that the electron-donating phenyl ring enabled the coordination of the alkyne with the Au+ complex in the α-position, which promoted the silyl ether to attack the β-position of the alkyne to promote a 6-endo-dig cyclization. Next, compound 11 was subjected to a solution of tetrabutylammonium fluoride (TBAF) in tetrahydrofuran (THF), resulting in the formation of naphthol 12 [12,13], a key intermediate in the previous total synthesis of macarpine (1) reported by Ishikawa (Scheme 6).
![[1860-5397-18-169-i6]](/bjoc/content/inline/1860-5397-18-169-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Formal total synthesis of macarpine (1).
Scheme 6: Formal total synthesis of macarpine (1).
To simplify the synthetic procedure, a more straightforward strategy was proposed by using alkynyl ketone 9 [27-29] as the substrate for the gold-catalyzed cycloisomerization in the presence of protonic acid. It was supposed that alkynyl ketone 9 would undergo enolization under the acidic conditions, followed by a gold-catalyzed cycloisomerization to provide the naphthol 12.
To test the idea, alkynyl ketone 9 was subjected to different reaction conditions as listed in Table 2. It was observed that both the acids and the temperatures had a great influence on the cycloisomerization. An attempt was also made by using only p-toluenesulfonic acid (TsOH) in the cycloisomerization step, but no corresponding product was obtained. Finally, the optimal conditions for the Au(I)-catalyzed cycloisomerization of alkynyl ketone 9 were determined as to stir the substrate under the catalysis of 5 mol % IPrAuCl/AgSbF6 with 2 equiv of TsOH as the additive at 70 °C for 2 h (Table 2, entry 3). It is notable that our synthetic route to naphthol 9 is shorter and proceeds with higher yield (5 steps, 59% yield) than Ishikawa’s route (9 steps, 13% yield).
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-169-i8.svg?max-width=637&scale=1.0)
Conclusion
In summary, the formal total synthesis of the natural product macarpine was achieved through two synthetic routes by synthesizing Ishikawa’s naphthol intermediate via Au(I)-catalyzed cycloisomerizations. Compared to the route reported in the literature, these routes are more concise and easier to perform. This gold-catalyzed strategy provides a new approach to macarpine and related benzo[c]phenanthridine alkaloids and the application of this strategy to access benzo[c]phenanthridine derivatives and further assessments of their bioactivities are currently in progress in our laboratory.
Supporting Information
| Supporting Information File 1: Synthetic procedures and characterization data for compounds 3–5, 8–12, and their 1H NMR and 13C NMR spectra. | ||
| Format: PDF | Size: 1.3 MB | Download |
Funding
Y.L. acknowledges the financial support from the National Natural Science Foundation of China (No. 21977073), Natural Science Foundation of Liaoning, China (No. 2022-MS-245) and the Liaoning Provincial Foundation of Educational Department (No. LJKZ0908). The authors would like to thank the Program for the Innovative Research Team of the Ministry of Education and the Program for the Liaoning Innovative Research Team in University.
References
-
Slaninová, I.; Slunská, Z.; Šinkora, J.; Vlková, M.; Táborská, E. Pharm. Biol. (Abingdon, U. K.) 2007, 45, 131–139. doi:10.1080/13880200601113099
Return to citation in text: [1] -
Al-Snafi, A. E. Indo Am. J. Pharm. Sci. 2017, 4, 257–263.
Return to citation in text: [1] -
Cheng, P.; Zeng, J. Chin. J. Org. Chem. 2012, 32, 1605–1619. doi:10.6023/cjoc201204008
Return to citation in text: [1] -
Orhan, I.; Özçelik, B.; Karaoğlu, T.; Şener, B. Z. Naturforsch., C: J. Biosci. 2007, 62c, 19–26. doi:10.1515/znc-2007-1-204
Return to citation in text: [1] -
Wang, Q.; Dai, P.; Bao, H.; Liang, P.; Wang, W.; Xing, A.; Sun, J. Exp. Ther. Med. 2017, 13, 263–268. doi:10.3892/etm.2016.3947
Return to citation in text: [1] -
Balažová, A.; Urdová, J.; Forman, V.; Mučaji, P. Molecules 2020, 25, 1261. doi:10.3390/molecules25061261
Return to citation in text: [1] -
Gaziano, R.; Moroni, G.; Buè, C.; Miele, M. T.; Sinibaldi-Vallebona, P.; Pica, F. World J. Gastrointest. Oncol. 2016, 8, 30–39. doi:10.4251/wjgo.v8.i1.30
Return to citation in text: [1] -
Pica, F.; Balestrieri, E.; Serafino, A.; Sorrentino, R.; Gaziano, R.; Moroni, G.; Moroni, N.; Palmieri, G.; Mattei, M.; Garaci, E.; Sinibaldi-Vallebona, P. Anti-Cancer Drugs 2012, 23, 32–42. doi:10.1097/cad.0b013e32834a0c8e
Return to citation in text: [1] -
Schmeller, T.; Latz-Brüning, B.; Wink, M. Phytochemistry 1997, 44, 257–266. doi:10.1016/s0031-9422(96)00545-6
Return to citation in text: [1] -
Slaninová, I.; López-Sánchez, N.; Šebrlová, K.; Vymazal, O.; Frade, J. M.; Táborská, E. Biol. Cell 2016, 108, 1–18. doi:10.1111/boc.201500047
Return to citation in text: [1] -
Hanaoka, M.; Cho, W. J.; Yoshida, S.; Fueki, T.; Mukai, C. Heterocycles 1989, 29, 857–860. doi:10.3987/com-89-4926
Return to citation in text: [1] -
Ishikawa, T.; Saito, T.; Ishii, H. Tetrahedron 1995, 51, 8447–8458. doi:10.1016/0040-4020(95)00460-p
Return to citation in text: [1] [2] [3] [4] [5] -
Solorio-Alvarado, C. R.; Echavarren, A. M. J. Am. Chem. Soc. 2010, 132, 11881–11883. doi:10.1021/ja104743k
Return to citation in text: [1] [2] -
Singh, S.; Samineni, R.; Pabbaraja, S.; Mehta, G. Angew. Chem., Int. Ed. 2018, 57, 16847–16851. doi:10.1002/anie.201810652
Return to citation in text: [1] -
Fu, J.; Li, B.; Wang, X.; Liang, Q.; Peng, X.; Yang, L.; Wan, T.; Wang, X.; Lin, B.; Cheng, M.; Liu, Y. Chin. J. Chem. 2022, 40, 46–52. doi:10.1002/cjoc.202100582
Return to citation in text: [1] [2] -
Huang, Q.; Wang, P.; Tian, Y.; Song, N.; Ren, S.; Tao, J.; Hang, K.; Li, M. Synlett 2015, 26, 1385–1390. doi:10.1055/s-0034-1380520
Return to citation in text: [1] [2] -
Xie, F.; Zhang, B.; Chen, Y.; Jia, H.; Sun, L.; Zhuang, K.; Yin, L.; Cheng, M.; Lin, B.; Liu, Y. Adv. Synth. Catal. 2020, 362, 3886–3897. doi:10.1002/adsc.202000755
Return to citation in text: [1] -
Liu, X.-J.; Zhou, S.-Y.; Xiao, Y.; Sun, Q.; Lu, X.; Li, Y.; Li, J.-H. Org. Lett. 2021, 23, 7839–7844. doi:10.1021/acs.orglett.1c02858
Return to citation in text: [1] -
Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k
Return to citation in text: [1] -
Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/c1cs15279c
Return to citation in text: [1] -
Pflästerer, D.; Hashmi, A. S. K. Chem. Soc. Rev. 2016, 45, 1331–1367. doi:10.1039/c5cs00721f
Return to citation in text: [1] -
Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553–11554. doi:10.1021/ja005570d
Return to citation in text: [1] -
Hashmi, A. S. K.; Rudolph, M.; Siehl, H.-U.; Tanaka, M.; Bats, J. W.; Frey, W. Chem. – Eur. J. 2008, 14, 3703–3708. doi:10.1002/chem.200701795
Return to citation in text: [1] -
Dankwardt, J. W. Tetrahedron Lett. 2001, 42, 5809–5812. doi:10.1016/s0040-4039(01)01146-7
Return to citation in text: [1] -
Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A. S. K. Adv. Synth. Catal. 2018, 360, 2493–2502. doi:10.1002/adsc.201800233
Return to citation in text: [1] -
Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A. S. K. Adv. Synth. Catal. 2018, 360, 3949–3959. doi:10.1002/adsc.201800629
Return to citation in text: [1] -
Song, L.; Tian, G.; Van der Eycken, E. V. Chem. – Eur. J. 2019, 25, 7645–7648. doi:10.1002/chem.201901860
Return to citation in text: [1] -
Guan, Y.; Lu, Z.; Yin, X.; Mohammadlou, A.; Staples, R. J.; Wulff, W. D. Synthesis 2020, 52, 2073–2091. doi:10.1055/s-0039-1690860
Return to citation in text: [1] -
Makra, F.; Rohloff, J. C.; Muehldorf, A. V.; Link, J. O. Tetrahedron Lett. 1995, 36, 6815–6818. doi:10.1016/00404-0399(50)14035-
Return to citation in text: [1]
| 27. | Song, L.; Tian, G.; Van der Eycken, E. V. Chem. – Eur. J. 2019, 25, 7645–7648. doi:10.1002/chem.201901860 |
| 28. | Guan, Y.; Lu, Z.; Yin, X.; Mohammadlou, A.; Staples, R. J.; Wulff, W. D. Synthesis 2020, 52, 2073–2091. doi:10.1055/s-0039-1690860 |
| 29. | Makra, F.; Rohloff, J. C.; Muehldorf, A. V.; Link, J. O. Tetrahedron Lett. 1995, 36, 6815–6818. doi:10.1016/00404-0399(50)14035- |
| 15. | Fu, J.; Li, B.; Wang, X.; Liang, Q.; Peng, X.; Yang, L.; Wan, T.; Wang, X.; Lin, B.; Cheng, M.; Liu, Y. Chin. J. Chem. 2022, 40, 46–52. doi:10.1002/cjoc.202100582 |
| 12. | Ishikawa, T.; Saito, T.; Ishii, H. Tetrahedron 1995, 51, 8447–8458. doi:10.1016/0040-4020(95)00460-p |
| 13. | Solorio-Alvarado, C. R.; Echavarren, A. M. J. Am. Chem. Soc. 2010, 132, 11881–11883. doi:10.1021/ja104743k |
| 1. | Slaninová, I.; Slunská, Z.; Šinkora, J.; Vlková, M.; Táborská, E. Pharm. Biol. (Abingdon, U. K.) 2007, 45, 131–139. doi:10.1080/13880200601113099 |
| 2. | Al-Snafi, A. E. Indo Am. J. Pharm. Sci. 2017, 4, 257–263. |
| 10. | Slaninová, I.; López-Sánchez, N.; Šebrlová, K.; Vymazal, O.; Frade, J. M.; Táborská, E. Biol. Cell 2016, 108, 1–18. doi:10.1111/boc.201500047 |
| 19. | Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k |
| 20. | Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/c1cs15279c |
| 21. | Pflästerer, D.; Hashmi, A. S. K. Chem. Soc. Rev. 2016, 45, 1331–1367. doi:10.1039/c5cs00721f |
| 22. | Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553–11554. doi:10.1021/ja005570d |
| 23. | Hashmi, A. S. K.; Rudolph, M.; Siehl, H.-U.; Tanaka, M.; Bats, J. W.; Frey, W. Chem. – Eur. J. 2008, 14, 3703–3708. doi:10.1002/chem.200701795 |
| 24. | Dankwardt, J. W. Tetrahedron Lett. 2001, 42, 5809–5812. doi:10.1016/s0040-4039(01)01146-7 |
| 9. | Schmeller, T.; Latz-Brüning, B.; Wink, M. Phytochemistry 1997, 44, 257–266. doi:10.1016/s0031-9422(96)00545-6 |
| 25. | Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A. S. K. Adv. Synth. Catal. 2018, 360, 2493–2502. doi:10.1002/adsc.201800233 |
| 26. | Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A. S. K. Adv. Synth. Catal. 2018, 360, 3949–3959. doi:10.1002/adsc.201800629 |
| 4. | Orhan, I.; Özçelik, B.; Karaoğlu, T.; Şener, B. Z. Naturforsch., C: J. Biosci. 2007, 62c, 19–26. doi:10.1515/znc-2007-1-204 |
| 5. | Wang, Q.; Dai, P.; Bao, H.; Liang, P.; Wang, W.; Xing, A.; Sun, J. Exp. Ther. Med. 2017, 13, 263–268. doi:10.3892/etm.2016.3947 |
| 6. | Balažová, A.; Urdová, J.; Forman, V.; Mučaji, P. Molecules 2020, 25, 1261. doi:10.3390/molecules25061261 |
| 7. | Gaziano, R.; Moroni, G.; Buè, C.; Miele, M. T.; Sinibaldi-Vallebona, P.; Pica, F. World J. Gastrointest. Oncol. 2016, 8, 30–39. doi:10.4251/wjgo.v8.i1.30 |
| 8. | Pica, F.; Balestrieri, E.; Serafino, A.; Sorrentino, R.; Gaziano, R.; Moroni, G.; Moroni, N.; Palmieri, G.; Mattei, M.; Garaci, E.; Sinibaldi-Vallebona, P. Anti-Cancer Drugs 2012, 23, 32–42. doi:10.1097/cad.0b013e32834a0c8e |
| 17. | Xie, F.; Zhang, B.; Chen, Y.; Jia, H.; Sun, L.; Zhuang, K.; Yin, L.; Cheng, M.; Lin, B.; Liu, Y. Adv. Synth. Catal. 2020, 362, 3886–3897. doi:10.1002/adsc.202000755 |
| 18. | Liu, X.-J.; Zhou, S.-Y.; Xiao, Y.; Sun, Q.; Lu, X.; Li, Y.; Li, J.-H. Org. Lett. 2021, 23, 7839–7844. doi:10.1021/acs.orglett.1c02858 |
| 3. | Cheng, P.; Zeng, J. Chin. J. Org. Chem. 2012, 32, 1605–1619. doi:10.6023/cjoc201204008 |
| 12. | Ishikawa, T.; Saito, T.; Ishii, H. Tetrahedron 1995, 51, 8447–8458. doi:10.1016/0040-4020(95)00460-p |
| 16. | Huang, Q.; Wang, P.; Tian, Y.; Song, N.; Ren, S.; Tao, J.; Hang, K.; Li, M. Synlett 2015, 26, 1385–1390. doi:10.1055/s-0034-1380520 |
| 14. | Singh, S.; Samineni, R.; Pabbaraja, S.; Mehta, G. Angew. Chem., Int. Ed. 2018, 57, 16847–16851. doi:10.1002/anie.201810652 |
| 15. | Fu, J.; Li, B.; Wang, X.; Liang, Q.; Peng, X.; Yang, L.; Wan, T.; Wang, X.; Lin, B.; Cheng, M.; Liu, Y. Chin. J. Chem. 2022, 40, 46–52. doi:10.1002/cjoc.202100582 |
| 13. | Solorio-Alvarado, C. R.; Echavarren, A. M. J. Am. Chem. Soc. 2010, 132, 11881–11883. doi:10.1021/ja104743k |
| 12. | Ishikawa, T.; Saito, T.; Ishii, H. Tetrahedron 1995, 51, 8447–8458. doi:10.1016/0040-4020(95)00460-p |
| 16. | Huang, Q.; Wang, P.; Tian, Y.; Song, N.; Ren, S.; Tao, J.; Hang, K.; Li, M. Synlett 2015, 26, 1385–1390. doi:10.1055/s-0034-1380520 |
| 12. | Ishikawa, T.; Saito, T.; Ishii, H. Tetrahedron 1995, 51, 8447–8458. doi:10.1016/0040-4020(95)00460-p |
| 11. | Hanaoka, M.; Cho, W. J.; Yoshida, S.; Fueki, T.; Mukai, C. Heterocycles 1989, 29, 857–860. doi:10.3987/com-89-4926 |
| 12. | Ishikawa, T.; Saito, T.; Ishii, H. Tetrahedron 1995, 51, 8447–8458. doi:10.1016/0040-4020(95)00460-p |
© 2022 Fu et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.