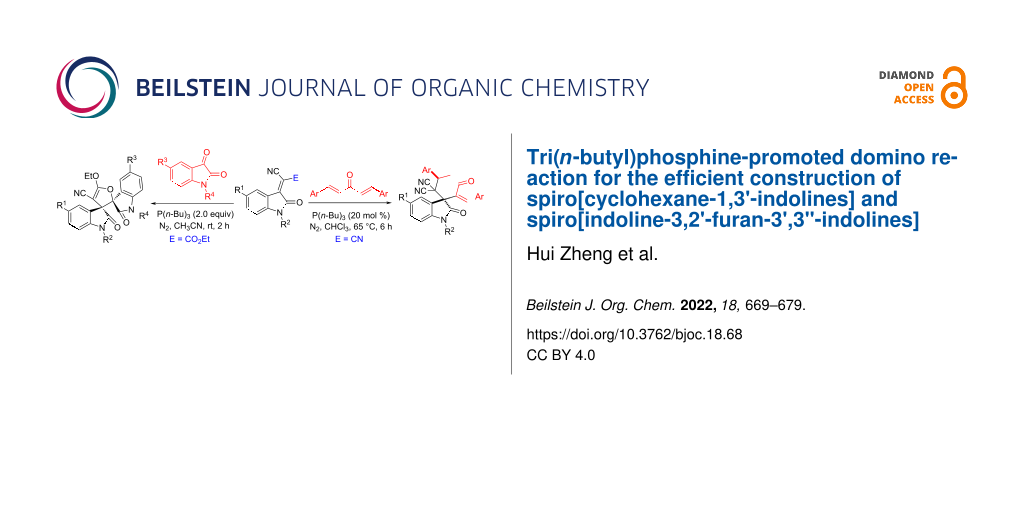
Abstract
The tri(n-butyl)phosphine-catalyzed reaction of isatylidene malononitriles and bis-chalcones in chloroform at 65 °C afforded functionalized spiro[cyclohexane-1,3'-indolines] in good yields and with good diastereoselectivity. On the other hand, the tri(n-butyl)phosphine-catalyzed reaction of 3-(ethoxycarbonylmethylene)oxindoles and bis-chalcones gave functionalized spiro[cyclohexane-1,3'-indolines] with different regioselectivity. Additionally, the tri(n-butyl)phosphine-promoted domino annulation reaction of isatins and ethyl isatylidene cyanoacetates produced spiro[indoline-3,2'-furan-3',3''-indolines] in satisfactory yields.

Graphical Abstract
Introduction
Spirooxindole is a privileged heterocyclic core existing in many natural products and medicinally relevant compounds. A variety of spirooxindole derivatives has been identified with a broad range of biological activities [1-5]. On the other hand, spirooxindoles can be constructed by introduction of various carbocyclic and heterocyclic units, which made them suitable for chemistry and many potential applications [6-9]. The development of unique spirooxindole systems and efficient synthetic methods for these compounds have attracted continual interests in many fields. A literature survey indicated that many convenient and atom-economic synthetic protocols have been developed for the synthesis of diverse spirooxindoles in recent years [10-16].
Nucleophilic tertiary phosphine-catalyzed reactions were successfully applied for the construction of diverse carbocyclic systems [17-24]. In these reactions, the tertiary phosphine firstly adds to electron-deficient alkenes, alkynes, and allenes to give active ionic intermediates. Then, the in situ-generated ionic intermediates further react with various reagents to give versatile cyclic compounds with recovery of the tertiary phosphine [25-34]. The superior catalytic ability of tertiary phosphines was primarily attributed to their excellent nucleophilicity as a nucleophile trigger and decent cleaving ability as a leaving group in the catalytic process [35-43]. The tertiary phosphine-catalyzed reactions have been widely applied to construct diverse spirooxindole systems by using readily available isatins and 3-methyleneoxindoles as key substrates [44-52]. In this respect, we have also developed several domino reactions by employing tertiary phosphine addition to electron-deficient alkynes as key protocol for the construction of diverse polycyclic spirooxindoles [53-59]. In continuation of our aim to explore elegant domino reactions for spiro compounds [60-66] and in order to demonstrate the potential synthetic value of the nucleophilic phosphine-catalyzed annulation reaction, herein we wish to report the tri(n-butyl)phosphine-catalyzed reaction of isatylidene malononitriles and bis-chalcones for the synthesis of functionalized various spiro[cyclohexane-1,3'-indolines] and related reactions.
Results and Discussion
Initially, the reaction conditions were optimized by using isatylidene malononitrile 1a and bis-chalcone 2a as standard reaction. Tertiary amines such as DMAP and DABCO did not catalyze this reaction (entries 1 and 2 in Table 1). Additionally, no reaction was observed when triphenylphosphine was used as catalyst (Table 1, entry 3). However, in the presence of tri(n-butyl)phosphine, the reaction in methylene dichloride, chloroform, and toluene at room temperature gave the expected functionalized spiro[cyclohexane-1,3'-indoline] 3a albeit with low yields (entries 4–6 in Table 1). The spiro[cyclohexane-1,3'-indoline] 3a was clearly produced by a tri(n-butyl)phosphine-catalyzed formal [4 + 2] cycloaddition reaction. This result showed that tri(n-butyl)phosphine has a higher nucleophilic catalytic ability than triphenylphosphine. It should be pointed out that the synthesis of polysubstituted cyclohexanones was reported by a tri(n-butyl)phosphine-catalyzed reaction of 1,4-dien-3-ones with 2-aryl-1,1-dicyanoalkenes [36]. At higher temperature (50 °C), the yield of the product 3a increased to 40% (in CH2Cl2), 65% (in toluene), and to 75% (in chloroform), respectively, in a Schlenk flask (entries 7–9 in Table 1). Therefore, chloroform was selected as the suitable solvent. The reaction in chloroform at 65 °C for six hours afforded the product 3a in 84% yield (entry 10, Table 1). By prolonging the reaction time, the yield of 3a did not increase further (entries 11 and 12, Table 1). At last, when 10% equiv tri(n-butyl)phosphine were used, the yield of product 3a decreased to 65% (entry 13, Table 1), whereas 50% equiv tri(n-butyl)phosphine did not improve the yield of 3a (82% yield, entry 14, Table 1). It should be pointed out that the total yield of the mixed two isomers was calculated in Table 1. Thus, the best results were obtained by carrying out the reaction in chloroform at 65 °C for six hours in the presence of 20% equiv tri(n-butyl)phosphine as catalyst.
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-68-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst | Solvent | Temp [°C] | Time [h] | Yield [%]b |
| 1 | DABCO | CHCl3 | rt | 24 | – |
| 2 | DMAP | CHCl3 | rt | 24 | – |
| 3 | PPh3 | CHCl3 | rt | 24 | – |
| 4 | P(n-Bu)3 | CH2Cl2 | rt | 24 | 30 |
| 5 | P(n-Bu)3 | PhMe | rt | 24 | 35 |
| 6 | P(n-Bu)3 | CHCl3 | rt | 24 | 45 |
| 7 | P(n-Bu)3 | CH2Cl2 | 50 | 24 | 40 |
| 8 | P(n-Bu)3 | PhMe | 50 | 24 | 65 |
| 9 | P(n-Bu)3 | CHCl3 | 50 | 24 | 75 |
| 10 | P(n-Bu)3 | CHCl3 | 65 | 6 | 84 |
| 11 | P(n-Bu)3 | CHCl3 | 65 | 12 | 82 |
| 12 | P(n-Bu)3 | CHCl3 | 65 | 24 | 80 |
| 13 | P(n-Bu)3c | CHCl3 | 65 | 24 | 65 |
| 14 | P(n-Bu)3d | CHCl3 | 65 | 6 | 82 |
aReaction conditions: isatylidene malononitrile (0.5 mmol), bis-chalcone (0.6 mmol), catalyst (20% equiv), solvent (10 mL), N2 atmosphere in Schlenk tube; bisolated yields of the mixed diastereoisomers; cPBu3 (10% equiv) was used; dPBu3 (50% equiv) was used.
With the best reaction conditions in hand, the scope of the reaction was developed by using various substrates and the results are summarized in Table 2. It was found that all reactions proceeded smoothly to give the expected spiro[cyclohexane-1,3'-indolines] 3a–z in moderate to good yields. The isatylidene malononitriles with different substituents at the C5-position and C1-position can be successfully employed in the reaction. The reaction with bis-chalcones with electron-donating groups gave slightly higher yields of the products than that of bis-chalcones with electron-withdrawing groups. Because there are two chiral carbon atoms in the spiro[cyclohexane-1,3'-indolines] and the Z/E-configuration of the exocyclic C–C double bonds, several diastereoisomers might be formed in the obtained products. The 1H NMR spectra clearly indicated that the obtained products contain two isomers with ratios in the range of 3:1 to 6:1. Thus, it was disappointing to find that the diastereoselectivity of this reaction was not very good. Because the polarity of the two diastereoisomers were very similar, it was very difficult to isolate them as pure compounds by column chromatography. For convenience, only the pure major diastereoisomers of the spiro compounds 3a–w were successfully isolated and fully characterized, which also caused the isolated yields of the products slightly decreased. For comparison, the minor isomers 3f’ and 3o’ were also successfully separated and obtained as pure compounds and were fully characterized, respectively. Additionally, 1,5-di(thiophen-2-yl)penta-1,4-dien-3-one was also successfully employed in the reaction. The major isomers of the corresponding spiro compounds 3x and 3y were predominately produced in moderate yields. In order to determine the relative configuration of the spiro compounds, the single crystal structures of the major isomers 3l (Figure 1), 3s (Figure 2), and 3f’ (Figure 3) were determined by X-ray diffraction analysis. As can be seen from Figure 1 and Figure 2, the aryl group exists in the trans-position of the carbonyl group of the oxindole scaffold in the newly formed cyclohexyl ring. On the other hand, the aryl group exist on the cis-position of the carbonyl group on the exocyclic C=C bond (Z-configuration). Thus, it can be concluded that the major isomers 3a–z have this kind of the relative configuration. In Figure 3, the aryl group and the carbonyl group of the oxindole scaffold also exist on trans-position in the newly formed cyclohexyl ring, while an E-configuration was observed for the exocyclic C=C double bond. Therefore, the major and minor isomers were actually attributable to the Z/E-configuration of the C=C bond, not to the cis/trans-positions in the newly formed cyclohexyl ring.
Table 2: Reaction of isatylidene malononitriles and bis-chalcones.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-68-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Compound | R1 | R2 | Ar | Yield [%]b | Z/Ec |
| 1 | 3a | CH3 | Bn | p-CH3C6H4 | 67 | 4:1 |
| 2 | 3b | CH3 | Bn | p-t-BuC6H4 | 64 | 4:1 |
| 3 | 3c | CH3 | Bn | C6H5 | 64 | 4:1 |
| 4 | 3d | CH3 | Bn | p-ClC6H4 | 58 | 5:1 |
| 5 | 3e | CH3 | Bn | p-BrC6H4 | 62 | 5:1 |
| 6 | 3f (3f’)d | CH3 | CH3 | p-t-BuC6H4 | 42 (14) | 3:1 |
| 7 | 3g | CH3 | H | p-CH3C6H4 | 45 | 3:1 |
| 8 | 3h | CH3 | H | p-t-BuC6H4 | 49 | 3:1 |
| 9 | 3i | CH3 | n-Bu | p-BrC6H4 | 54 | 5:1 |
| 10 | 3j | CH3 | n-Bu | p-CH3C6H4 | 54 | 4:1 |
| 11 | 3k | CH3 | n-Bu | p-t-BuC6H4 | 58 | 5:1 |
| 12 | 3l | Cl | Bn | p-CH3C6H4 | 54 | 5:1 |
| 13 | 3m | Cl | Bn | p-t-BuC6H4 | 60 | 5:1 |
| 14 | 3n | Cl | Bn | p-iPrC6H4 | 56 | 4:1 |
| 15 | 3o (3o’)d | Cl | n-Bu | p-CH3C6H4 | 42 (8) | 5:1 |
| 16 | 3p | F | Bn | p-CH3C6H4 | 53 | 4:1 |
| 17 | 3q | F | Bn | p-iPrC6H4 | 58 | 4:1 |
| 18 | 3r | H | H | p-CH3C6H4 | 48 | 6:1 |
| 19 | 3s | H | Bn | p-CH3C6H4 | 58 | 4:1 |
| 20 | 3t | H | Bn | p-iPrC6H4 | 57 | 5:1 |
| 21 | 3u | H | Bn | C6H5 | 42 | 4:1 |
| 22 | 3v | H | Bn | p-ClC6H4 | 45 | 4:1 |
| 23 | 3w | H | Bn | p-BrC6H4 | 47 | 5:1 |
| 24 | 3x | CH3 | CH3 | 2-thiophenyl | 52 | trace |
| 25 | 3y | Cl | CH3 | 2-thiophenyl | 45 | trace |
| 26 | 3z | H | Bn | 2-thiophenyl | 53 | trace |
aReaction conditions: isatylidene malononitrile (0.5 mmol), bis-chalcone (0.6 mmol), P(n-Bu)3 (20% equiv), CHCl3 (10.0 mL), N2 atmosphere; bisolated yields; cthe Z/E ratio was determined by 1H NMR spectroscopy; dthe minor isomers 3f’ and 3o’ were isolated.
![[1860-5397-18-68-1]](/bjoc/content/figures/1860-5397-18-68-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Single crystal structure of compound 3l.
Figure 1: Single crystal structure of compound 3l.
![[1860-5397-18-68-2]](/bjoc/content/figures/1860-5397-18-68-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Single crystal structure of compound 3s.
Figure 2: Single crystal structure of compound 3s.
![[1860-5397-18-68-3]](/bjoc/content/figures/1860-5397-18-68-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Single crystal structure of compound 3f’.
Figure 3: Single crystal structure of compound 3f’.
In order to expand the scope of this reaction, 3-(ethoxycarbonylmethylene)oxindoles 4 were also employed in the reaction with bis-chalcones 2. We were pleased to find that the reaction proceeded smoothly in the presence of an excess amount of tri(n-butyl)phosphine under similar reaction conditions and the results are summarized in Table 3. Because there are three chiral carbon atoms in the molecule, several diastereoisomers can be formed in the reaction. The spiro[cyclohexane-1,3'-indolines] 5a–e were obtained in moderate to good yields. The single crystal structure of compound 5a was determined by X-ray diffraction method (Figure 4). It can be found that the ethoxycarbonyl group and the phenyl group of the oxindole moiety remained in the trans-position as in the starting 3-(ethoxycarbonylmethylene)oxindole. The aryl group and the carbonyl group exist on trans-position in the newly formed cyclohexyl ring. It should be pointed out that the exocyclic benzylidene group exists on the C3-position in the newly formed cyclohexyl ring, while it exists on the C6-position in the above obtained spiro compounds 3a–z.
Table 3: Reaction of 3-(ethoxycarbonylmethylene)oxindoles and bis-chalcones.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-68-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Compound | R1 | R2 | Ar | Yield (%)b |
| 1 | 5a | Cl | Et | p-CH3C6H4 | 60 |
| 2 | 5b | Cl | Et | p-CH3OC6H4 | 57 |
| 3 | 5c | Cl | Me | p-CH3C6H4 | 42 |
| 4 | 5d | Cl | Me | p-iPrC6H4 | 58 |
| 5 | 5e | H | Et | p-CH3C6H4 | 56 |
aReaction conditions: 3-(ethoxycarbonylmethylene)oxindole (0.5 mmol), bis-chalcone (0.6 mmol), P(n-Bu)3 (1.0 mmol), CHCl3 (10.0 mL), N2 atmosphere; bisolated yields.
![[1860-5397-18-68-4]](/bjoc/content/figures/1860-5397-18-68-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Single crystal structure of compound 5a.
Figure 4: Single crystal structure of compound 5a.
This result clearly indicated that these two reactions have the opposite regioselectivity. Another kind of readily available 1,3-dipolarophile, 3-phenacylidenoxindole, was also tested in the reaction. However, it was found that the C=C bond in 3-phenacylidenoxindole was directly reduced to give the corresponding saturated 3-(2-oxo-2-phenylethyl)indolin-2-one. A similar reduction reaction of 3-phenacylidenoxindoles by trialkylphosphine has been previously reported in the literature [65].
For explaining the formation of the two kinds of spiro[cyclohexane-1,3'-indolines] 3 and 5, a plausible reaction mechanism has been proposed (Scheme 1) on the basis of previously reported works and the obtained results from this work. Firstly, the nucleophilic addition of tributylphosphine to the bis-chalcone gives the active zwitterionic species (A). Secondly, the Michael addition of the zwitterionic species (A) to isatylidene malononitrile at the C3-position of the oxindole scaffold results in adduct (B). Thirdly, the intramolecular addition of the carbanion to the enone affords the cyclic intermediate (C), which in turn converts into the intermediate (D) by transfer of a negative charge. Finally, the spiro compound 3 is formed by elimination of tributylphosphine. When 3-(ethoxycarbonylmethylene)oxindole 4 is employed in the reaction, the nucleophilic addition of the zwitterion (A) to this compound takes place at the exocyclic position giving the adduct (E), which in turn proceeds with the intermediates (F) and (G) according to the above mentioned similar processes to give the spiro compound 5. The different addition direction of the zwitterion A to the isatylidene malononitrile 2 and 3-(ethoxycarbonylmethylene)oxindole 4 results in the different regioselectivity in the formation of the spiro compounds 3 and 5.
![[1860-5397-18-68-i1]](/bjoc/content/inline/1860-5397-18-68-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed reaction mechanism for the compounds 3 and 5.
Scheme 1: Proposed reaction mechanism for the compounds 3 and 5.
For further demonstrate the synthetic value of this procedure, ethyl isatylidene cyanoacetates 6 were also employed in the reaction. However, the reaction did not proceed to give the expected spiro[cyclohexane-1,3'-indoline], while a new spiro[indoline-3,2'-furan-3',3''-indoline] was obtained, which was clearly constructed from the annulation reaction of isatin with ethyl isatylidene cyanoacetate.
Therefore, we turned our attention to the examination of this unprecedented reaction of isatins with ethyl isatylidene cyanoacetates. At last, we successfully found that tri(n-butyl)phosphine promoted the reaction of ethyl isatylidene cyanoacetate 6a and isatin 7a always resulted in spiro[indoline-3,2'-furan-3',3''-indoline] 8a as the major product. The loading of tri(n-butyl)phosphine played an important role for the formation of the products. When 2.0 equiv of P(n-Bu)3 were used, the reaction gave the spiro compound 8a in 71% yield. Thus, this reaction is not a simple catalytic reaction and tri(n-butyl)phosphine acted not only as a catalyst. It was also noticed that when triphenylphosphine was used instead of tri(n-butyl)phosphine, the expected product was not obtained. Under the optimized reaction conditions, we next investigated the scope of the reaction by using various substituted ethyl isatylidene cyanoacetates and isatins and the results are summarized in Table 4. All reactions proceeded smoothly to give the expected spiro[indoline-3,2'-furan-3',3''-indolines] 8a–m in moderate to good yields. The substituents on the isatylidene cyanoacetates had only a marginal effect on the yields, while the presence of electron-donating methyl groups in the isatins gave higher yields than electron-withdrawing groups such as chloro and fluoro substituents. The structures of the spiro compounds 8a–m were fully characterized by their IR, HRMS, 1H and 13C NMR spectra. In addition, the single crystal structure of compound 8a (Figure 5) was determined by X-ray diffraction analysis. From Figure 5, it can be seen that the two oxindole moieties exist on the trans-configuration. Therefore, this reaction showed very high diastereoselectivity.
Table 4: Reaction of ethyl isatylidene cyanoacetates and isatins.a
![[Graphic 4]](/bjoc/content/inline/1860-5397-18-68-i6.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Compound | R1 | R2 | R3 | R4 | Yield (%)b |
| 1 | 8a | Cl | Bn | CH3 | Bn | 71 |
| 2 | 8b | Cl | Bn | CH3 | H | 70 |
| 3 | 8c | Cl | Bn | Cl | Bn | 57 |
| 4 | 8d | Cl | Bn | F | Bn | 56 |
| 5 | 8e | Cl | Bn | H | Bn | 54 |
| 6 | 8f | Cl | n-Bu | CH3 | Bn | 76 |
| 7 | 8g | Cl | n-Bu | CH3 | H | 73 |
| 8 | 8h | F | Bn | CH3 | Bn | 69 |
| 9 | 8i | F | Bn | Cl | Bn | 47 |
| 10 | 8j | F | Bn | F | Bn | 35 |
| 11 | 8k | CH3 | CH3 | CH3 | Bn | 77 |
| 12 | 8l | H | Bn | CH3 | Bn | 83 |
| 13 | 8m | H | Bn | CH3 | CH3 | 62 |
aReaction conditions: isatin (0.3 mmol), isatylidene cyanoacetate (0.3 mmol), P(n-Bu)3 (0.6 mmol), MeCN (5.0 mL), N2 atmosphere; bisolated yields.
![[1860-5397-18-68-5]](/bjoc/content/figures/1860-5397-18-68-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Single crystal struture of compound 8a.
Figure 5: Single crystal struture of compound 8a.
To explain the formation of the dispiro compounds 8, a plausible reaction mechanism has been proposed and is shown in Scheme 2. At first, the nucelophilic addition of tributylphosphine to isatylidene cyanoacetate gives a zwitterionic salt (H). Secondly, the addition of the carbanion to the carbonyl group of the isatin affords the adduct (I). Then, the intramolecular attack of the alkoxide to the carbonyl group in the ester moiety produces the cyclic intermediate (J). Finally, the dispiro compound 8 is formed by elimination of tri(n-butyl)phosphine oxide.
![[1860-5397-18-68-i2]](/bjoc/content/inline/1860-5397-18-68-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the formation of dispiro compounds 8.
Scheme 2: Proposed mechanism for the formation of dispiro compounds 8.
Conclusion
In summary, we have investigated tri(n-butyl)phosphine-catalyzed annulation reactions of bis-chalcones with isatylidene malononitriles or 3-(ethoxycarbonylmethylene)oxindoles for the efficient construction of two kinds of spiro[cyclohexane-1,3'-indolines] in good yields and with good diastereoselectivity. Additionally, tri(n-butyl)phosphine promoted the domino reaction of isatins and ethyl isatylidene cyanoacetates to give selectively spiro[indoline-3,2'-furan-3',3''-indolines] in satisfactory yields. The relative configuration of the complex spirooxindoles was confirmed by determination of several single crystal structures. Also, plausible reaction mechanisms have been proposed. This reaction has the advantages of using readily available substrates, simple operation, good yields, and molecular diversity, which enable it to find potential applications in heterocyclic and medicinal chemistry.
Experimental
1. General procedure for the preparation of the spiro[cyclohexane-1,3'-indolines] 3a–w: In an atmosphere of nitrogen, isatylidene malononitrile (0.5 mmol) and bis-chalcone (0.6 mmol) were dissolved in chloroform (10.0 mL) in a Schlenk bottle. Then, tri(n-butyl)phosphine (20% equiv) was added by syringe and the solution was stirred at 65 °C for six hours. After removing the solvent at reduced pressure, the residue was subjected to column chromatography with petroleum ether/ethyl acetate 15:1 (v/v) as eluent to give the pure product 3a–m for analysis.
rel-(1R,3R)-1'-Benzyl-5'-methyl-6-((Z)-4-methylbenzylidene)-2',5-dioxo-3-(p-tolyl)spiro[cyclohexane-1,3'-indoline]-2,2-dicarbonitrile (3a): white solid, 84% yield; mp 213–215 °C; 1H NMR (600 MHz, CDCl3) δ 7.70 (s, 1H, ArH), 7.43 (s, 2H, ArH), 7.33 (s, 4H, ArH), 7.29 (s, 1H, ArH), 7.25–7.22 (m, 4H, ArH), 7.19–7.18 (m, 1H, ArH), 7.13–7.11 (m, 2H, ArH), 6.76–6.75 (m, 1H, ArH), 6.63 (s, 1H, CH), 5.07 (d, J = 15.6 Hz, 1H, CH2), 5.02 (d, J = 13.2 Hz, 1H, CH2), 4.86 (d, J = 15.0 Hz, 1H, CH2), 3.46 (t, J = 13.8 Hz, 1H, CH), 3.08 (d, J = 15.6 Hz, 1H, CH2), 2.40 (s, 3H, CH3), 2.38 (s, 3H, CH3), 2.34 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 196.9, 171.7, 140.7, 140.4, 139.8, 139.6, 134.4, 134.0, 131.9, 131.6, 130.8, 130.1, 129.8, 129.3, 129.1, 129.0, 128.9, 128.0, 127.1, 127.0, 123.0, 112.3, 110.9, 60.3, 49.0, 44.6, 44.4, 42.6, 21.4, 21.2; IR (KBr) ν: 3727, 3405, 3029, 2921, 2863, 2317, 1911, 1709, 1609, 1501, 1443, 1362, 1295, 1185, 1049, 1022, 959, 920, 820, 732 cm−1; HRMS–ESI (m/z): [M + Na]+ calcd for C38H31NaN3O2, 584.2314; found, 584.2306.
2. General procedure for the preparation of the spiro[cyclohexane-1,3'-indolines] 5a–e: In an atmosphere of nitrogen, 3-(ethoxycarbonylmethyl)oxindole (0.5 mmol) and bis-chalcone (0.6 mmol) were dissolved in chloroform (10.0 mL) in a Schlenk bottle. Then, tri(n-butyl)phosphine (1.0 mmol) was added by syringe and the solution was stirred at 65 °C for six hours. After removing the solvent at reduced pressure, the residue was subjected to column chromatography with petroleum ether/ethyl acetate 15:1 (v/v) as eluent to give the pure product 5a–e for analysis.
Ethyl rel-(1S,2S,6R)-1'-benzyl-5'-chloro-3-((Z)-4-methylbenzylidene)-2',5-dioxo-6-(p-tolyl)spiro[cyclohexane-1,3'-indoline]-2-carboxylate (5a): white solid, 60% yield; mp 221–223 °C; 1H NMR (400 MHz, CDCl3) δ 7.67 (s, 1H, ArH), 7.29 (d, J = 7.6 Hz, 2H, ArH), 7.21–7.14 (m, 4H, ArH), 7.07 (t, J = 7.6 Hz, 2H, ArH), 7.01 (d, J = 8.4 Hz, 1H, ArH), 6.91 (d, J = 8.0 Hz, 2H, ArH), 6.84 (d, J = 8.0 Hz, 2H, ArH), 6.77 (d, J = 7.6 Hz, 2H, ArH), 6.34 (d, J = 8.4 Hz, 1H, CH), 4.70 (d, J = 16.0 Hz, 1H, CH2), 4.57 (d, J = 15.6 Hz, 1H, CH2), 4.52–4.47 (m, 1H, CH2), 4.36–4.24 (m, 2H, OCH2), 3.94 (s, 1H, CH), 3.80–3.72 (m, 1H, CH), 3.00–2.94 (m, 1H, CH2), 2.37 (s, 3H, CH3), 2.20 (s, 3H, CH3), 1.30 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 200.0, 175.1, 170.6, 140.6, 139.0, 138.8, 136.8, 135.0, 134.8, 131.6, 131.2, 130.1, 129.8, 129.3, 129.0, 128.6, 128.5, 127.5, 127.4, 127.1, 124.5, 110.2, 61.8, 53.0, 50.7, 43.4, 42.5, 42.0, 21.4, 21.0, 14.1; IR (KBr) ν: 3723, 3412, 2933, 2871, 2324, 1925, 1817, 1703, 1604, 1474, 1442, 1339, 1172, 1091, 1010, 904, 824, 716 cm−1; HRMS–ESI (m/z): [M + Na]+ calcd for C38H34ClNaNO4, 626.2069; found, 626.2066.
3. General procedure for the preparation of the spiro[cyclohexane-1,3'-indolines] 8a–m: In an atmosphere of nitrogen, isatylidene cyanoacetate (0.3 mmol) and isatin (0.3 mmol) were dissolved in acetonitrile (10.0 mL). Then, tri(n-butyl)phosphine (0.6 mmol) was added by syringe and the solution was stirred at room temperature for two hours. After removing the solvent at reduced pressure, the residue was subjected to column chromatography with petroleum ether/ethyl acetate 15:1 (v/v) as eluent to give the pure products 8a–m for analysis.
rel-(3R,3'R)-1,1''-Dibenzyl-5''-chloro-5'-ethoxy-5-methyl-2,2''-dioxodispiro[indoline-3,2'-furan-3',3''-indoline]-4'-carbonitrile (8a): white solid, 71% yield; mp 175–177 °C; 1H NMR (400 MHz, CDCl3) δ 7.64 (s, 1H, ArH), 7.53 (s, 1H, ArH), 7.18–7.08 (m, 5H, ArH), 7.03 (t, J = 6.8 Hz, 3H, ArH), 6.71 (d, J = 7.6 Hz, 2H, ArH), 6.54 (d, J = 7.2 Hz, 2H, ArH), 6.39 (d, J = 8.0 Hz, 1H, ArH), 6.34 (d, J = 8.4 Hz, 1H, ArH), 5.17 (d, J = 16.4 Hz, 1H, CH2), 5.07 (d, J = 16.0 Hz, 1H, CH2), 4.65 (q, J = 7.2 Hz, 2H, CH2), 4.35–4.30 (m, 2H, CH2), 2.12 (s, 3H, CH3), 1.54 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 174.0, 173.1, 170.9, 142.4, 141.5, 134.2, 134.2, 133.6, 132.2, 130.4, 129.2, 128.8, 128.7, 128.6, 127.7, 127.6, 127.4, 126.4, 126.0, 124.0, 120.7, 114.2, 110.4, 109.6, 89.0, 69.1, 62.8, 60.1, 43.9, 20.9, 14.7; IR (KBr) ν: 3467, 3063, 3035, 2990, 2919, 2205, 1739, 1706, 1632, 1602, 1496, 1454, 1434, 1408, 1381, 1361, 1333, 1293, 1258, 1215, 1199, 1186, 1167, 1133, 1089, 1070, 1026, 997, 962, 933, 911, 895, 875, 836, 818, 776, 747 cm−1; HRMS–ESI (m/z): [M + Na]+ calcd for C36H28NaClN3O4, 624.1666; found, 624.1660.
Supporting Information
Characterization data and 1H NMR, 13C NMR, HRMS spectra of the compounds are available. The crystallographic data of the compounds 3l (CCDC 2166451), 3s (CCDC 2166452), 3f’ (CCDC 2173182), 5a (CCDC 2166453), 8a (CCDC 2166454) have been deposited at the Cambridge Crystallographic Database Center (http://www.ccdc.cam.ac.uk).
| Supporting Information File 1: Characterization data and copies of NMR and HRMS spectra. | ||
| Format: PDF | Size: 7.4 MB | Download |
References
-
Williams, R. M.; Cox, R. J. Acc. Chem. Res. 2003, 36, 127–139. doi:10.1021/ar020229e
Return to citation in text: [1] -
Dounay, A. B.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2964. doi:10.1021/cr020039h
Return to citation in text: [1] -
Santos, M. M. M. Tetrahedron 2014, 70, 9735–9757. doi:10.1016/j.tet.2014.08.005
Return to citation in text: [1] -
Yu, B.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 97, 673–698. doi:10.1016/j.ejmech.2014.06.056
Return to citation in text: [1] -
Ye, N.; Chen, H.; Wold, E. A.; Shi, P.-Y.; Zhou, J. ACS Infect. Dis. 2016, 2, 382–392. doi:10.1021/acsinfecdis.6b00041
Return to citation in text: [1] -
Kotha, S.; Deb, A. C.; Lahiri, K.; Manivannan, E. Synthesis 2009, 165–193. doi:10.1055/s-0028-1083300
Return to citation in text: [1] -
Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003–3025. doi:10.1055/s-0029-1216975
Return to citation in text: [1] -
Ball-Jones, N. R.; Badillo, J. J.; Franz, A. K. Org. Biomol. Chem. 2012, 10, 5165–5181. doi:10.1039/c2ob25184a
Return to citation in text: [1] -
Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y
Return to citation in text: [1] -
Liu, Y.; Wang, H.; Wan, J. Asian J. Org. Chem. 2013, 2, 374–386. doi:10.1002/ajoc.201200180
Return to citation in text: [1] -
Cao, Z.-Y.; Wang, Y.-H.; Zeng, X.-P.; Zhou, J. Tetrahedron Lett. 2014, 55, 2571–2584. doi:10.1016/j.tetlet.2014.01.084
Return to citation in text: [1] -
Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F., III. ACS Catal. 2014, 4, 743–762. doi:10.1021/cs401172r
Return to citation in text: [1] -
Pavlovska, T. L.; Redkin, R. G.; Lipson, V. V.; Atamanuk, D. V. Mol. Diversity 2016, 20, 299–344. doi:10.1007/s11030-015-9629-8
Return to citation in text: [1] -
Bencivenni, G.; Wu, L.-Y.; Mazzanti, A.; Giannichi, B.; Pesciaioli, F.; Song, M.-P.; Bartoli, G.; Melchiorre, P. Angew. Chem., Int. Ed. 2009, 48, 7200–7203. doi:10.1002/anie.200903192
Return to citation in text: [1] -
Chen, G.-S.; Fang, Y.-B.; Ren, Z.; Tian, X.; Liu, Y.-L. Org. Chem. Front. 2021, 8, 3820–3828. doi:10.1039/d1qo00575h
Return to citation in text: [1] -
Balha, M.; Mondal, B.; Pan, S. C. Org. Biomol. Chem. 2019, 17, 6557–6561. doi:10.1039/c9ob01199d
Return to citation in text: [1] -
Zhang, C.; Lu, X. J. Org. Chem. 1995, 60, 2906–2908. doi:10.1021/jo00114a048
Return to citation in text: [1] -
Lu, X.; Zhang, C.; Xu, Z. Acc. Chem. Res. 2001, 34, 535–544. doi:10.1021/ar000253x
Return to citation in text: [1] -
Methot, J. L.; Roush, W. R. Adv. Synth. Catal. 2004, 346, 1035–1050. doi:10.1002/adsc.200404087
Return to citation in text: [1] -
Wei, Y.; Shi, M. Acc. Chem. Res. 2010, 43, 1005–1018. doi:10.1021/ar900271g
Return to citation in text: [1] -
Guo, H.; Fan, Y. C.; Sun, Z.; Wu, Y.; Kwon, O. Chem. Rev. 2018, 118, 10049–10293. doi:10.1021/acs.chemrev.8b00081
Return to citation in text: [1] -
Ye, L.-W.; Zhou, J.; Tang, Y. Chem. Soc. Rev. 2008, 37, 1140–1152. doi:10.1039/b717758e
Return to citation in text: [1] -
Cowen, B. J.; Miller, S. J. Chem. Soc. Rev. 2009, 38, 3102–3116. doi:10.1039/b816700c
Return to citation in text: [1] -
Zhou, R.; Liu, R.; Li, R.; He, Z. Chin. J. Org. Chem. 2014, 34, 2385–2405. doi:10.6023/cjoc201406049
Return to citation in text: [1] -
Xie, P.; Huang, Y. Org. Biomol. Chem. 2015, 13, 8578–8595. doi:10.1039/c5ob00865d
Return to citation in text: [1] -
Zhou, R.; He, Z. Eur. J. Org. Chem. 2016, 1937–1954. doi:10.1002/ejoc.201501585
Return to citation in text: [1] -
Wei, Y.; Shi, M. Org. Chem. Front. 2017, 4, 1876–1890. doi:10.1039/c7qo00285h
Return to citation in text: [1] -
Karanam, P.; Reddy, G. M.; Koppolu, S. R.; Lin, W. Tetrahedron Lett. 2018, 59, 59–76. doi:10.1016/j.tetlet.2017.11.051
Return to citation in text: [1] -
Tan, B.; Candeias, N. R.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 4672–4675. doi:10.1021/ja110147w
Return to citation in text: [1] -
Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094
Return to citation in text: [1] -
Zhang, X.-C.; Cao, S.-H.; Wei, Y.; Shi, M. Org. Lett. 2011, 13, 1142–1145. doi:10.1021/ol1031798
Return to citation in text: [1] -
Deng, H.-P.; Wei, Y.; Shi, M. Org. Lett. 2011, 13, 3348–3351. doi:10.1021/ol201094f
Return to citation in text: [1] -
Gu, J.; Xiao, B.-X.; Chen, Y.-R.; Li, Q.-Z.; Ouyang, Q.; Du, W.; Chen, Y.-C. Org. Lett. 2018, 20, 2088–2091. doi:10.1021/acs.orglett.8b00649
Return to citation in text: [1] -
Mei, L.-y.; Wei, Y.; Xu, Q.; Shi, M. Organometallics 2013, 32, 3544–3556. doi:10.1021/om400473p
Return to citation in text: [1] -
Cai, L.; Zhang, B.; Wu, G.; Song, H.; He, Z. Chem. Commun. 2011, 47, 1045–1047. doi:10.1039/c0cc02817g
Return to citation in text: [1] -
Zhou, R.; Wang, J.; Tian, J.; He, Z. Org. Biomol. Chem. 2012, 10, 773–781. doi:10.1039/c1ob06187a
Return to citation in text: [1] [2] -
Zhou, R.; Yang, C.; Liu, Y.; Li, R.; He, Z. J. Org. Chem. 2014, 79, 10709–10715. doi:10.1021/jo502106c
Return to citation in text: [1] -
Qin, Z.; Liu, W.; Wang, D.; He, Z. J. Org. Chem. 2016, 81, 4690–4700. doi:10.1021/acs.joc.6b00596
Return to citation in text: [1] -
Yang, C.; Chen, X.; Tang, T.; He, Z. Org. Lett. 2016, 18, 1486–1489. doi:10.1021/acs.orglett.6b00456
Return to citation in text: [1] -
Zhu, C.-Z.; Sun, Y.-L.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2017, 359, 1263–1270. doi:10.1002/adsc.201700031
Return to citation in text: [1] -
Ramachary, D. B.; Prabhakar Reddy, T.; Suresh Kumar, A. Org. Biomol. Chem. 2017, 15, 9785–9789. doi:10.1039/c7ob02424j
Return to citation in text: [1] -
Zhang, J.; Wu, H.-H.; Zhang, J. Org. Lett. 2017, 19, 6080–6083. doi:10.1021/acs.orglett.7b02895
Return to citation in text: [1] -
Wang, C.; Gao, Z.; Zhou, L.; Wang, Q.; Wu, Y.; Yuan, C.; Liao, J.; Xiao, Y.; Guo, H. Chem. Commun. 2018, 54, 279–282. doi:10.1039/c7cc08380g
Return to citation in text: [1] -
Zhang, X.-C.; Cao, S.-H.; Wei, Y.; Shi, M. Chem. Commun. 2011, 47, 1548–1550. doi:10.1039/c0cc04289g
Return to citation in text: [1] -
Wang, Y.; Liu, L.; Zhang, T.; Zhong, N.-J.; Wang, D.; Chen, Y.-J. J. Org. Chem. 2012, 77, 4143–4147. doi:10.1021/jo3002535
Return to citation in text: [1] -
Gomez, C.; Gicquel, M.; Carry, J.-C.; Schio, L.; Retailleau, P.; Voituriez, A.; Marinetti, A. J. Org. Chem. 2013, 78, 1488–1496. doi:10.1021/jo302460d
Return to citation in text: [1] -
Hu, C.; Zhang, Q.; Huang, Y. Chem. – Asian J. 2013, 8, 1981–1984. doi:10.1002/asia.201300650
Return to citation in text: [1] -
Zhang, J.; Cheng, C.; Wang, D.; Miao, Z. J. Org. Chem. 2017, 82, 10121–10128. doi:10.1021/acs.joc.7b01582
Return to citation in text: [1] -
Chen, L.-J.; Yu, J.-H.; He, B.; Xie, J.-W.; Liu, Y.-X.; Zhu, W.-D. J. Org. Chem. 2020, 85, 7793–7802. doi:10.1021/acs.joc.0c00418
Return to citation in text: [1] -
Hu, F.-L.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2014, 356, 736–742. doi:10.1002/adsc.201301070
Return to citation in text: [1] -
Xiao, B.-X.; Jiang, B.; Song, X.; Du, W.; Chen, Y.-C. Chem. Commun. 2019, 55, 3097–3100. doi:10.1039/c9cc00386j
Return to citation in text: [1] -
He, T.; Peng, L.; Li, S.; Hu, F.; Xie, C.; Huang, S.; Jia, S.; Qin, W.; Yan, H. Org. Lett. 2020, 22, 6966–6971. doi:10.1021/acs.orglett.0c02519
Return to citation in text: [1] -
Han, Y.; Sheng, Y.-J.; Yan, C.-G. Org. Lett. 2014, 16, 2654–2657. doi:10.1021/ol5008394
Return to citation in text: [1] -
Gong, H.; Sun, J.; Yan, C. G. Synthesis 2014, 46, 2327–2332. doi:10.1055/s-0033-1339120
Return to citation in text: [1] -
Han, Y.; Qi, W.-J.; Shen, Y.-J.; Yan, C.-G. Tetrahedron Lett. 2015, 56, 5196–5198. doi:10.1016/j.tetlet.2015.07.071
Return to citation in text: [1] -
Qi, W.-J.; Han, Y.; Yan, C.-G. Tetrahedron 2016, 72, 5057–5063. doi:10.1016/j.tet.2016.06.020
Return to citation in text: [1] -
Liu, C.-Z.; Han, Y.; Qi, W.-J.; Yan, C.-G. Heterocycl. Commun. 2016, 22, 301–306. doi:10.1515/hc-2016-0123
Return to citation in text: [1] -
Qi, W.-J.; Han, Y.; Liu, C.-Z.; Yan, C.-G. Chin. Chem. Lett. 2017, 28, 442–445. doi:10.1016/j.cclet.2016.09.014
Return to citation in text: [1] -
Han, Y.; Zheng, H.; Zhang, Y.-Y.; Yan, C.-G. Chin. Chem. Lett. 2020, 31, 1337–1341. doi:10.1016/j.cclet.2019.10.042
Return to citation in text: [1] -
Sun, J.; Zhang, Y.; Shi, R.-G.; Yan, C.-G. Org. Biomol. Chem. 2019, 17, 3978–3983. doi:10.1039/c9ob00166b
Return to citation in text: [1] -
Ye, R.; Yan, C.-G. Eur. J. Org. Chem. 2019, 5882–5886. doi:10.1002/ejoc.201900955
Return to citation in text: [1] -
Cao, J.; Yang, F.; Sun, J.; Huang, Y.; Yan, C.-G. J. Org. Chem. 2019, 84, 622–635. doi:10.1021/acs.joc.8b02457
Return to citation in text: [1] -
Pan, L.-N.; Sun, J.; Shi, R.-G.; Yan, C.-G. Org. Chem. Front. 2020, 7, 3202–3208. doi:10.1039/d0qo00845a
Return to citation in text: [1] -
Zhan, S.-C.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Biomol. Chem. 2020, 18, 163–168. doi:10.1039/c9ob02013f
Return to citation in text: [1] -
Liu, D.; Cao, J.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Lett. 2020, 22, 8931–8936. doi:10.1021/acs.orglett.0c03331
Return to citation in text: [1] [2] -
Cao, S.-H.; Zhang, X.-C.; Wei, Y.; Shi, M. Eur. J. Org. Chem. 2011, 2668–2672. doi:10.1002/ejoc.201100017
Return to citation in text: [1]
| 1. | Williams, R. M.; Cox, R. J. Acc. Chem. Res. 2003, 36, 127–139. doi:10.1021/ar020229e |
| 2. | Dounay, A. B.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2964. doi:10.1021/cr020039h |
| 3. | Santos, M. M. M. Tetrahedron 2014, 70, 9735–9757. doi:10.1016/j.tet.2014.08.005 |
| 4. | Yu, B.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 97, 673–698. doi:10.1016/j.ejmech.2014.06.056 |
| 5. | Ye, N.; Chen, H.; Wold, E. A.; Shi, P.-Y.; Zhou, J. ACS Infect. Dis. 2016, 2, 382–392. doi:10.1021/acsinfecdis.6b00041 |
| 25. | Xie, P.; Huang, Y. Org. Biomol. Chem. 2015, 13, 8578–8595. doi:10.1039/c5ob00865d |
| 26. | Zhou, R.; He, Z. Eur. J. Org. Chem. 2016, 1937–1954. doi:10.1002/ejoc.201501585 |
| 27. | Wei, Y.; Shi, M. Org. Chem. Front. 2017, 4, 1876–1890. doi:10.1039/c7qo00285h |
| 28. | Karanam, P.; Reddy, G. M.; Koppolu, S. R.; Lin, W. Tetrahedron Lett. 2018, 59, 59–76. doi:10.1016/j.tetlet.2017.11.051 |
| 29. | Tan, B.; Candeias, N. R.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 4672–4675. doi:10.1021/ja110147w |
| 30. | Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094 |
| 31. | Zhang, X.-C.; Cao, S.-H.; Wei, Y.; Shi, M. Org. Lett. 2011, 13, 1142–1145. doi:10.1021/ol1031798 |
| 32. | Deng, H.-P.; Wei, Y.; Shi, M. Org. Lett. 2011, 13, 3348–3351. doi:10.1021/ol201094f |
| 33. | Gu, J.; Xiao, B.-X.; Chen, Y.-R.; Li, Q.-Z.; Ouyang, Q.; Du, W.; Chen, Y.-C. Org. Lett. 2018, 20, 2088–2091. doi:10.1021/acs.orglett.8b00649 |
| 34. | Mei, L.-y.; Wei, Y.; Xu, Q.; Shi, M. Organometallics 2013, 32, 3544–3556. doi:10.1021/om400473p |
| 17. | Zhang, C.; Lu, X. J. Org. Chem. 1995, 60, 2906–2908. doi:10.1021/jo00114a048 |
| 18. | Lu, X.; Zhang, C.; Xu, Z. Acc. Chem. Res. 2001, 34, 535–544. doi:10.1021/ar000253x |
| 19. | Methot, J. L.; Roush, W. R. Adv. Synth. Catal. 2004, 346, 1035–1050. doi:10.1002/adsc.200404087 |
| 20. | Wei, Y.; Shi, M. Acc. Chem. Res. 2010, 43, 1005–1018. doi:10.1021/ar900271g |
| 21. | Guo, H.; Fan, Y. C.; Sun, Z.; Wu, Y.; Kwon, O. Chem. Rev. 2018, 118, 10049–10293. doi:10.1021/acs.chemrev.8b00081 |
| 22. | Ye, L.-W.; Zhou, J.; Tang, Y. Chem. Soc. Rev. 2008, 37, 1140–1152. doi:10.1039/b717758e |
| 23. | Cowen, B. J.; Miller, S. J. Chem. Soc. Rev. 2009, 38, 3102–3116. doi:10.1039/b816700c |
| 24. | Zhou, R.; Liu, R.; Li, R.; He, Z. Chin. J. Org. Chem. 2014, 34, 2385–2405. doi:10.6023/cjoc201406049 |
| 10. | Liu, Y.; Wang, H.; Wan, J. Asian J. Org. Chem. 2013, 2, 374–386. doi:10.1002/ajoc.201200180 |
| 11. | Cao, Z.-Y.; Wang, Y.-H.; Zeng, X.-P.; Zhou, J. Tetrahedron Lett. 2014, 55, 2571–2584. doi:10.1016/j.tetlet.2014.01.084 |
| 12. | Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F., III. ACS Catal. 2014, 4, 743–762. doi:10.1021/cs401172r |
| 13. | Pavlovska, T. L.; Redkin, R. G.; Lipson, V. V.; Atamanuk, D. V. Mol. Diversity 2016, 20, 299–344. doi:10.1007/s11030-015-9629-8 |
| 14. | Bencivenni, G.; Wu, L.-Y.; Mazzanti, A.; Giannichi, B.; Pesciaioli, F.; Song, M.-P.; Bartoli, G.; Melchiorre, P. Angew. Chem., Int. Ed. 2009, 48, 7200–7203. doi:10.1002/anie.200903192 |
| 15. | Chen, G.-S.; Fang, Y.-B.; Ren, Z.; Tian, X.; Liu, Y.-L. Org. Chem. Front. 2021, 8, 3820–3828. doi:10.1039/d1qo00575h |
| 16. | Balha, M.; Mondal, B.; Pan, S. C. Org. Biomol. Chem. 2019, 17, 6557–6561. doi:10.1039/c9ob01199d |
| 6. | Kotha, S.; Deb, A. C.; Lahiri, K.; Manivannan, E. Synthesis 2009, 165–193. doi:10.1055/s-0028-1083300 |
| 7. | Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003–3025. doi:10.1055/s-0029-1216975 |
| 8. | Ball-Jones, N. R.; Badillo, J. J.; Franz, A. K. Org. Biomol. Chem. 2012, 10, 5165–5181. doi:10.1039/c2ob25184a |
| 9. | Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y |
| 60. | Sun, J.; Zhang, Y.; Shi, R.-G.; Yan, C.-G. Org. Biomol. Chem. 2019, 17, 3978–3983. doi:10.1039/c9ob00166b |
| 61. | Ye, R.; Yan, C.-G. Eur. J. Org. Chem. 2019, 5882–5886. doi:10.1002/ejoc.201900955 |
| 62. | Cao, J.; Yang, F.; Sun, J.; Huang, Y.; Yan, C.-G. J. Org. Chem. 2019, 84, 622–635. doi:10.1021/acs.joc.8b02457 |
| 63. | Pan, L.-N.; Sun, J.; Shi, R.-G.; Yan, C.-G. Org. Chem. Front. 2020, 7, 3202–3208. doi:10.1039/d0qo00845a |
| 64. | Zhan, S.-C.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Biomol. Chem. 2020, 18, 163–168. doi:10.1039/c9ob02013f |
| 65. | Liu, D.; Cao, J.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Lett. 2020, 22, 8931–8936. doi:10.1021/acs.orglett.0c03331 |
| 66. | Cao, S.-H.; Zhang, X.-C.; Wei, Y.; Shi, M. Eur. J. Org. Chem. 2011, 2668–2672. doi:10.1002/ejoc.201100017 |
| 65. | Liu, D.; Cao, J.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Lett. 2020, 22, 8931–8936. doi:10.1021/acs.orglett.0c03331 |
| 53. | Han, Y.; Sheng, Y.-J.; Yan, C.-G. Org. Lett. 2014, 16, 2654–2657. doi:10.1021/ol5008394 |
| 54. | Gong, H.; Sun, J.; Yan, C. G. Synthesis 2014, 46, 2327–2332. doi:10.1055/s-0033-1339120 |
| 55. | Han, Y.; Qi, W.-J.; Shen, Y.-J.; Yan, C.-G. Tetrahedron Lett. 2015, 56, 5196–5198. doi:10.1016/j.tetlet.2015.07.071 |
| 56. | Qi, W.-J.; Han, Y.; Yan, C.-G. Tetrahedron 2016, 72, 5057–5063. doi:10.1016/j.tet.2016.06.020 |
| 57. | Liu, C.-Z.; Han, Y.; Qi, W.-J.; Yan, C.-G. Heterocycl. Commun. 2016, 22, 301–306. doi:10.1515/hc-2016-0123 |
| 58. | Qi, W.-J.; Han, Y.; Liu, C.-Z.; Yan, C.-G. Chin. Chem. Lett. 2017, 28, 442–445. doi:10.1016/j.cclet.2016.09.014 |
| 59. | Han, Y.; Zheng, H.; Zhang, Y.-Y.; Yan, C.-G. Chin. Chem. Lett. 2020, 31, 1337–1341. doi:10.1016/j.cclet.2019.10.042 |
| 44. | Zhang, X.-C.; Cao, S.-H.; Wei, Y.; Shi, M. Chem. Commun. 2011, 47, 1548–1550. doi:10.1039/c0cc04289g |
| 45. | Wang, Y.; Liu, L.; Zhang, T.; Zhong, N.-J.; Wang, D.; Chen, Y.-J. J. Org. Chem. 2012, 77, 4143–4147. doi:10.1021/jo3002535 |
| 46. | Gomez, C.; Gicquel, M.; Carry, J.-C.; Schio, L.; Retailleau, P.; Voituriez, A.; Marinetti, A. J. Org. Chem. 2013, 78, 1488–1496. doi:10.1021/jo302460d |
| 47. | Hu, C.; Zhang, Q.; Huang, Y. Chem. – Asian J. 2013, 8, 1981–1984. doi:10.1002/asia.201300650 |
| 48. | Zhang, J.; Cheng, C.; Wang, D.; Miao, Z. J. Org. Chem. 2017, 82, 10121–10128. doi:10.1021/acs.joc.7b01582 |
| 49. | Chen, L.-J.; Yu, J.-H.; He, B.; Xie, J.-W.; Liu, Y.-X.; Zhu, W.-D. J. Org. Chem. 2020, 85, 7793–7802. doi:10.1021/acs.joc.0c00418 |
| 50. | Hu, F.-L.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2014, 356, 736–742. doi:10.1002/adsc.201301070 |
| 51. | Xiao, B.-X.; Jiang, B.; Song, X.; Du, W.; Chen, Y.-C. Chem. Commun. 2019, 55, 3097–3100. doi:10.1039/c9cc00386j |
| 52. | He, T.; Peng, L.; Li, S.; Hu, F.; Xie, C.; Huang, S.; Jia, S.; Qin, W.; Yan, H. Org. Lett. 2020, 22, 6966–6971. doi:10.1021/acs.orglett.0c02519 |
| 35. | Cai, L.; Zhang, B.; Wu, G.; Song, H.; He, Z. Chem. Commun. 2011, 47, 1045–1047. doi:10.1039/c0cc02817g |
| 36. | Zhou, R.; Wang, J.; Tian, J.; He, Z. Org. Biomol. Chem. 2012, 10, 773–781. doi:10.1039/c1ob06187a |
| 37. | Zhou, R.; Yang, C.; Liu, Y.; Li, R.; He, Z. J. Org. Chem. 2014, 79, 10709–10715. doi:10.1021/jo502106c |
| 38. | Qin, Z.; Liu, W.; Wang, D.; He, Z. J. Org. Chem. 2016, 81, 4690–4700. doi:10.1021/acs.joc.6b00596 |
| 39. | Yang, C.; Chen, X.; Tang, T.; He, Z. Org. Lett. 2016, 18, 1486–1489. doi:10.1021/acs.orglett.6b00456 |
| 40. | Zhu, C.-Z.; Sun, Y.-L.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2017, 359, 1263–1270. doi:10.1002/adsc.201700031 |
| 41. | Ramachary, D. B.; Prabhakar Reddy, T.; Suresh Kumar, A. Org. Biomol. Chem. 2017, 15, 9785–9789. doi:10.1039/c7ob02424j |
| 42. | Zhang, J.; Wu, H.-H.; Zhang, J. Org. Lett. 2017, 19, 6080–6083. doi:10.1021/acs.orglett.7b02895 |
| 43. | Wang, C.; Gao, Z.; Zhou, L.; Wang, Q.; Wu, Y.; Yuan, C.; Liao, J.; Xiao, Y.; Guo, H. Chem. Commun. 2018, 54, 279–282. doi:10.1039/c7cc08380g |
| 36. | Zhou, R.; Wang, J.; Tian, J.; He, Z. Org. Biomol. Chem. 2012, 10, 773–781. doi:10.1039/c1ob06187a |
© 2022 Zheng et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.