Abstract
The monomer 2-methacrylamido-caprolactam (4) was synthesized from methacryloyl chloride (3) and racemic α-amino-ε-caprolactam (2). Copolymerization of 4 with N,N-dimethylacrylamide (5) was carried out by a free-radical mechanism using 2,2’-azobis(2-methylpropionitrile) (AIBN) as an initiator. The new copolymers show a lower critical solution temperature (LCST) in water and an upper critical solution temperature (UCST) in ethanol, 1-propanol, and 1-butanol. The solubility properties of the copolymers can be influenced significantly by the addition of randomly methylated β-cyclodextrin (CD). The complexation of the copolymers with CD, was confirmed by the use of ROESY-NMR-spectroscopy.

Graphical Abstract
Introduction
Recently, increasing interest has been spent on thermoresponsive polymer solutions, mainly because of their potential application in the field of drug delivery, gene delivery, or tissue engineering [1-3]. Especially polymers having a lower critical solution temperature (LCST) in water, like poly(N-isopropylacrylamide) or modified poly(N-vinylpyrrolidone) are extensively described in literature [4,5]. Below the critical temperature (TC) these polymers are soluble and they become insoluble by heating their solutions above the TC. In contrast, some polymers forming hydrogen bonds, like poly(acrylic acid) or polymers containing zwitter-ionic groups are soluble in water above a critical temperature (UCST) [6,7]. Only a few reports deal with polymers which have both, LCST- and UCST-behavior in water [8]. The TC of a thermoresponsive polymer solution can be altered easily by variation of the chemical structure and to a certain degree by variation of the chain length of the used polymer, also. Interestingly, the TC-values of suitable polymers in water can also be influenced significantly by supramolecular interactions with cyclodextrin [9]. Accordingly, we described in 2003 for the first time, that the LCST of a poly(N-adamantylacrylamide-co-N’-isopropylacrylamide) is influenced by CD [10]. Furthermore, the interaction of poly(pseudo-betaines) with CD leads to an UCST-behavior in water [11]. A precise control of TC via the addition of “effectors”, that induce polymer–polymer interactions has been described [12]. Recently, we published that CD can shift the UCST of copolymers obtained from N-vinylimidazole and adamantyl-modified N-vinylimidazole [13]. In the present study we evaluate the thermoresponsive behavior of solutions of polymers containing 2-methacrylamido-caprolactam as a comonomer. The used α-amino-ε-caprolactam is obtained from biomass through a combination of biological fermentation and chemical processes [14].
Results and Discussion
Racemic α-amino-ε-caprolactam (2) was obtained according to literature via cyclocondensation of L-lysine (1) [15]. Through amidation of the primary amine (2) with methacryloyl chloride (3) the polymerizable 2-methacrylamido-caprolactam (4) was obtained (Scheme 1) [14]. Free radical copolymerization of 4 in various comonomer compositions with N,N-dimethylacrylamide (5) leads to copolymers 6a–i (Scheme 1). 6a is soluble in water and methanol and insoluble in ethanol, 1-propanol, and 1-butanol. Copolymers 6b–e are soluble in water as well as in methanol, ethanol, 1-propanol, and 1-butanol (Figure 1 and Figure 3), respectively. According to 1H NMR spectroscopy, the monomer conversions were nearly quantitative. Therefore it is assumed, that the copolymer compositions are exactly identical with the monomer feed ratios.
![[1860-5397-10-203-i1]](/bjoc/content/inline/1860-5397-10-203-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of monomer 2-methacrylamido-caprolactam (4) and copolymers 6a–i based on 4 and N,N-dimethylacrylamide (5).
Scheme 1: Synthesis of monomer 2-methacrylamido-caprolactam (4) and copolymers 6a–i based on 4 and N,N-dimeth...
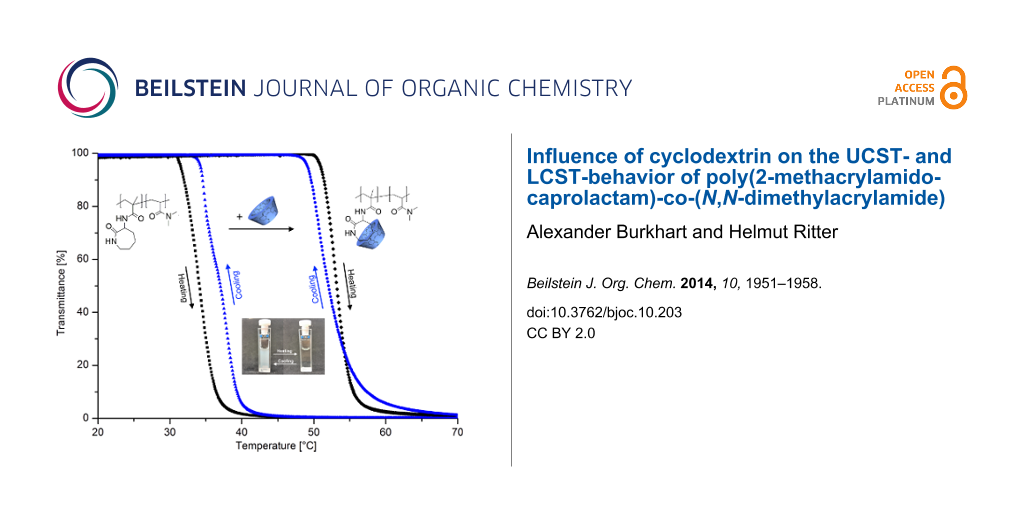
To investigate the solubility behavior of polymers 6 in water, turbidity measurements were performed. As shown in Figure 1, copolymer 6a has a typical LCST-behavior in water. The cloud point upon heating is 34 °C and upon cooling 37 °C, respectively. The hysteresis effect is a result of insoluble to soluble transition which depends on the shape and size distribution of dispersed polymer particles [16,17]. The more hydrophilic copolymer 6b has a higher cloud point at 67 °C whereas copolymer 6c is completely soluble in water up to >95 °C. Complexation of copolymer 6a with a 1.5 molar excess of CD, based on monomer 4, was carried out to yield the complexed copolymer 6aCD. Due to that complexation, the cloud point in the heating curves increases from 34 °C (6a) up to 47 °C (6aCD) with a sharp phase transition. Obviously, the complexation of the attached caprolactam ring with CD leads to a higher hydrophilicity of the copolymer and therefore to a better solubility in water.
![[1860-5397-10-203-1]](/bjoc/content/figures/1860-5397-10-203-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Turbidity curves upon heating and corresponding curves upon cooling of 10 mg ml−1 solution of polymer 6a and 6aCD at a heating/cooling rate of 1 °C min−1.
Figure 1: Turbidity curves upon heating and corresponding curves upon cooling of 10 mg ml−1 solution of polym...
The postulated complexation of the polymer attached caprolactam moiety in copolymer 6a with CD was principally demonstrated via ROESY-NMR-spectroscopy in solution using the monomer 4 (Figure 2a). The magnetic interaction of the protons of the cyclodextrin with protons from the lactam ring between 1.1 ppm and 2.0 ppm confirms the complexation. Furthermore, a Job plot has been carried out to identify the stoichiometry of the complex between the caprolactam and the CD. As shown in Figure 2b, a 1:1 complex was found.
![[1860-5397-10-203-2]](/bjoc/content/figures/1860-5397-10-203-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 2D NMR ROESY (300 MHz, D2O) spectrum of monomer 4CD (a), Job plot of 2 with RAMEB-CD (b).
Figure 2: 2D NMR ROESY (300 MHz, D2O) spectrum of monomer 4CD (a), Job plot of 2 with RAMEB-CD (b).
To prove that the increase of the TC (Figure 1) is a result of complexation with added CD and not a simple solvent effect, α-D(+)-glucose was added to copolymer 6a in an equimolar amount to the caprolactam groups. As expected, turbidity measurements indicate, that there is no significant increase of the TC.
Furthermore, it was found that the copolymer 6a is completely soluble in methanol. However, in less polar ethanol, 1-propanol, and 1-butanol heating is required to obtain solutions (Figure 3).
![[1860-5397-10-203-3]](/bjoc/content/figures/1860-5397-10-203-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Turbidity curves upon cooling of 10 mg ml−1 solution of copolymer 6a in various n-alcohols at a cooling rate of 1 °C min−1.
Figure 3: Turbidity curves upon cooling of 10 mg ml−1 solution of copolymer 6a in various n-alcohols at a coo...
The observed cloud points of copolymer 6a in alcohol solution increase from ethanol (35 °C), via 1-propanol (40 °C) up to less polar 1-butanol (54 °C). Since the content of hydrogen-bond forming amido-caprolactam units in copolymers 6b and 6c is reduced, the copolymers obviously become completely soluble in the used alcohols in the whole temperature range. The potential influence on the UCST-behavior by complexation with CD of polymer 6a in ethanol was evaluated by turbidity measurements (Figure 4).
![[1860-5397-10-203-4]](/bjoc/content/figures/1860-5397-10-203-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Turbidity curves upon cooling of 10 mg ml−1 solution of polymer 6a and 6aCD at a cooling rate of 1 °C min−1 in ethanol.
Figure 4: Turbidity curves upon cooling of 10 mg ml−1 solution of polymer 6a and 6aCD at a cooling rate of 1 ...
Actually, the cloud point of copolymer 6a shifts from 34 °C down to 24 °C in ethanol, because of 6aCD formation. Due to that complexation of the polymer attached caprolactam-rings by CD the number of intermolecular hydrogen bonds is reduced which leads to a better solubility in the used alcohols, as mentioned above. The amount of CD was varied from 50 mol % up to 150 mol % based on the amount of caprolactam units of copolymer 6a. Surprisingly this variation of CD amount did not significantly influence the cloud point (24 °C) of copolymer 6aCD. Obviously, the dense packing of the caprolactam moieties in the copolymer allows the approach of the bulky CD rings only partially up to a certain limit to form those complexes. Molecular weights of copolymer 6a could not yet be detected by the use of gel-permeation chromatography (GPC) even under various conditions, probably due to strong interactions of the amide groups of the copolymer. Besides that, the measured hydrodynamic coil size of about 100 nm via dynamic light scattering (DLS) in different solutions indicates the formation of agglomerates. We also spent some interest in the thermal behavior of copolymers 6 in the condensed state. The observed glass-transition temperatures (Tg) of copolymers 6a–i in condensed phase, are summarized in Figure 5.
![[1860-5397-10-203-5]](/bjoc/content/figures/1860-5397-10-203-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Glass-transition temperature as a function of the amount of N,N-dimethylacrylamide (5) in the copolymers 6a–i.
Figure 5: Glass-transition temperature as a function of the amount of N,N-dimethylacrylamide (5) in the copol...
Generally with the increasing amount of flexible N,N-dimethylacrylamide (5) in the copolymer chains, the glass transition temperatures are decreasing, as expected. Due to the stronger intermolecular hydrogen-bond interactions in the relatively stiff homopolymer 6e the glass-transition temperature is higher than in the copolymers with various amounts of N,N-dimethylacrylamide (5).
Conclusion
It can be concluded from the above described results, that the novel copolymers from lysine based 2-methacrylamido-caprolactam (4) and N,N-dimethylacrylamide (5) show LCST-behavior in water and UCST-behavior in certain alcohols also, which can be controlled by the monomer ratios. Surprisingly, the complexation of the caprolactam-bearing units in the copolymer with CD leads to an increase of LCST values in water and vice versa, to a decrease of UCST in ethanol. According to that, these novel copolymers are of potential interest for the development of new types of smart materials.
Experimental
Materials and methods
All reactants were commercially available and used without further purification. All solvents used were of analytical purity or freshly distilled. L(+)-lysine monohydrochloride (99+%) was purchased from Acros Organics and N,N-dimethylacrylamide (99%) was obtained from Sigma-Aldrich. Methacryloyl chloride ( ≥99%) and triethylamine pure (99%) were purchased from AppliChem. RAMEB-CD was obtained from Wacker Chemie GmbH, Burghausen, Germany and was used after being dried with a vacuum oil pump over P4O10. 2,2’-Azobis(2-methylpropionitrile) (99%) was obtained from Sigma-Aldrich. α-D(+)-glucose (99+%) was obtained from Acros Organics.
1H NMR and 13C NMR spectra were recorded on a Bruker Avance DRX 300 by using dimethyl sulfoxide-d6 or chloroform-d as solvents. The chemical shifts (δ) are given in parts per million (ppm) using the solvent peak as an internal standard. 2D NMR-spectroscopy was performed using a Bruker Avance III DRX 300 at 300 MHz in D2O as a solvent. The samples were measured at room temperature. FTIR spectra were recorded on a Nicolet 6700 FTIR spectrometer equipped with an ATR unit. Differential scanning calorimetry (DSC) was performed using a Mettler Toledo DSC 822 instrument equipped with a sample robot TSO801RO. For calibration, standard tin, indium, and zinc samples were used. For Tg determination, heating and cooling curves were recorded between −50 and 330 °C at a heating rate of 15 K min−1. The Tg value was calculated from the arithmetic average of the inflection points of the second, third, and fourth heating curve. Turbidity measurements were accomplished using a TP1 turbidity photometer in a temperature range of 5 to 70 °C. During continuous stirring, the transparency of the sample was determined by a voltage-controlled semiconductor or laser and a silicon photodiode at a wavelength of 590 nm and a heating or cooling rate of 1 °C min−1. The critical temperature was determined at 50% of relative transmittance. Electrospray ionization mass spectrometry (ESIMS) was conducted on a Bruker maXis 4G mass spectrometer. Melting points were obtained using a Büchi Melting Point B-545 apparatus at a heating rate of 1 °C/min.
Synthesis of α-amino-ε-caprolactam (2)
A mixture of 40 g (219 mmol) of L-lysine monohydrochloride (1), 8.76 g (219 mmol) sodium hydroxide, 120 g (1.178 mol) aluminium oxide and 450 ml of n-butanol is heated to reflux for 48 h in a reaction vessel equipped with a water trap. Subsequently, the mixture is filtrated and the obtained pale yellow solution is concentrated under reduced pressure. After precipitation in diethyl ether, filtration and drying in vacuum 15.6 g (121 mmol, 55.6%) of colorless/pale yellow crystals are received. Mp 63–67 °C; FTIR (diamond), ν (cm−1): 3355, 3282 (br. NH), 2928 (s, CH), 2909 (s, CH), 2848 (m, CH), 1648 (s, amide), 1567, 1474, 1431, 1316; 1H NMR (300 MHz, CDCl3) δ 6.52 (s, 1H, NH), 3.51 (dd, J = 10.8, 2.0 Hz, 1H, CH), 3.19 (m, 2H, RHN-CH2-R), 2.1–1.3 (m, 6H, (CH2)3); 13C NMR (75 MHz, CDCl3) δ 179.65 (1C, C(O)), 54.06 (1C, CH), 42.04 (1C, RHN-CH2-R), 33.97 (1C, RHC-CH2-R), 29.20 (1C, CH2), 28.57 (1C, CH2); ESIMS (H2O) m/z: 129.1 [M + H]+.
Synthesis of 2-methacrylamido-caprolactam (4)
In a 100 mL round bottom flask 5 g (39.01 mmol) of α-amino-ε-caprolactam (2) is dissolved in 50 mL of dry chloroform. 7.89 g (78.02 mmol) of triethylamine is added and the solution is cooled in an ice bath to 0 °C. To this solution 4.89 g (46.8 mmol) of methacryloyl chloride (3), dissolved in 20 ml anhydrous chloroform, is added within 10 min under nitrogen atmosphere. The mixture is stirred for further 2 h at 0 °C, before warming up to room temperature and stirring for additional 12 h. Then the reaction mixture is washed with brine and water. The organic phase is dried over magnesium sulfate, filtered and the solvent is evaporated under reduced pressure. A colorless solid is obtained. Mp = 125 °C; Yield: 4.7 g (23.9 mmol, 61%); FTIR (diamond), ν (cm−1): 3252 (br. NH), 2923 (s, CH), 2857 (m, CH), 1650 (s, amide), 1513, 1476, 1332, 1029; 1H NMR (300 MHz, CDCl3) δ 6.29 (s, 1H, NH), 5.79 (p, J = 1 Hz, 1H, CH2), 5.35 (p, J = 1.5 Hz, 1H, CH2), 3.40–3.15 (m, 3H, RHN-CH2-R, CH), 2.35–1.2 (m, 9H, (CH2)3, CH3); 13C NMR (75 MHz, CDCl3) δ 174.21 (1C, C(O)), 166.04 (1C, NH-C(O)-C), 139.57 (1C, R-C=CH2), 119.48 (1C, H2C=C-R), 51.54 (1C, CH), 40.63 (1C, RHN-CH2-R), 30.78 (1C, RHC-CH2-R), 28.83 (1C, CH2), 27.66 (1C, CH2), 18.38 (1C, CH3); ESIMS (ethanol) m/z: 197.1 [M + H]+, 219.1 [M + Na]+.
General procedure for the preparation of copolymers 6a–i
The monomer 2-methacrylamido-caprolactam (4), was mixed in different molar ratios (according to Table 1) with N,N-dimethylacrylamide (5) in dimethyl sulfoxide (total monomer concentration 1.8 mol L−1). 1 mol % of 2,2’-azobis(2-methylpropionitrile) with respect to monomers 4 and 5 were added to the solution under nitrogen. Under stirring, the solution was heated to 110 °C. After 12 h, the polymerization was stopped by cooling the solution to room temperature. The copolymers were precipitated in ethyl acetate, filtered off and dried in vacuo. The obtained products are colorless solids. Analytical data refer to monomer ratio of 1:1. FTIR (diamond), ν (cm−1): 3392 (br. NH), 2929 (CH), 1632 (s, amide), 1476, 1435, 1333, 1136; 1H NMR (300 MHz, DMSO-d6) δ 7.96 (s, 1H, NH), 7.21 (s, 1H, NH), 4.23 (s, 1H, CH), 3.3-2.6 (m, 11H, RHN-CH2-R, N-CH3, CH2 backbone, CH, backbone), 2.1–0.6 (m, 11H, CH2, C-CH3, CH2).
Table 1: Monomer amount of 4 and 5 for the copolymerization of copolymers 6a–i.
|
Polymer
6 |
2-methacrylamido-
caprolactam (4) |
N,N-dimethylacryl-
amide (5) |
||
|---|---|---|---|---|
| [mmol] | [g] | [mmol] | [g] | |
| a | 2.5 | 0.5 | 1 | 0.1 |
| b | 2.5 | 0.5 | 2.5 | 0.25 |
| c | 2.5 | 0.5 | 5.8 | 0.57 |
| d | 0 | 0 | 2.5 | 0.25 |
| e | 2.5 | 0.5 | 0 | 0 |
| f | 2.5 | 0.5 | 0.3 | 0.03 |
| g | 2.5 | 0.5 | 0.6 | 0.06 |
| h | 2.5 | 0.5 | 1.7 | 0.17 |
| i | 2.5 | 0.5 | 3.8 | 0.38 |
General Procedure for the preparation of the standard solutions for the Job Plot analysis
0.16 g of α-amino-ε-caprolactam is dissolved in 10 ml water to achieve a 0.125 M standard solution (A). A second standard solution (B) is received by dissolving 1,64 g RAMEB cyclodextrin in 10 ml water, 0.125 M. Both solutions were stirred for 24 h before mixing them together in different amounts (Table 2). These solutions are stirred for additional 24 h before recording an 1H NMR.
Supporting Information
| Supporting Information File 1: Characterization data of intermediates and copolymers including 1H NMR and 13C NMR spectra, turbidity- and DLS-measurements. | ||
| Format: PDF | Size: 702.9 KB | Download |
References
-
Schmaljohann, D. Adv. Drug Delivery Rev. 2006, 58, 1655–1670. doi:10.1016/j.addr.2006.09.020
Return to citation in text: [1] -
Twaites, B. R.; de las Heras Alarcón, C.; Lavigne, M.; Saulnier, A.; Pennadam, S. S.; Cunliffe, D.; Górecki, D. C.; Alexander, C. J. Controlled Release 2005, 108, 472–483. doi:10.1016/j.jconrel.2005.08.009
Return to citation in text: [1] -
Klouda, L.; Mikos, A. G. Eur. J. Pharm. Biopharm. 2008, 68, 34–45. doi:10.1016/j.ejpb.2007.02.025
Return to citation in text: [1] -
Schild, H. G. Prog. Polym. Sci. 1992, 17, 163–249. doi:10.1016/0079-6700(92)90023-R
Return to citation in text: [1] -
Trellenkamp, T.; Ritter, H. Macromol. Rapid Commun. 2009, 30, 1736–1740. doi:10.1002/marc.200900250
Return to citation in text: [1] -
Buscall, R.; Corner, T. Eur. Polym. J. 1982, 18, 967–974. doi:10.1016/0014-3057(82)90084-2
Return to citation in text: [1] -
Dong, Z.; Mao, J.; Wang, D.; Yang, M.; Wang, W.; Bo, S.; Ji, X. Macromol. Chem. Phys. 2014, 215, 111–120. doi:10.1002/macp.201300552
Return to citation in text: [1] -
Lee, H.-N.; Rosen, B. M.; Fenyvesi, G.; Sunkara, H. B. J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 4311–4315. doi:10.1002/pola.26242
Return to citation in text: [1] -
Fischer, J.; Ritter, H. Beilstein J. Org. Chem. 2013, 9, 2803–2811. doi:10.3762/bjoc.9.315
Return to citation in text: [1] -
Ritter, H.; Sadowski, O.; Tepper, E. Angew. Chem., Int. Ed. 2003, 42, 3171–3173. doi:10.1002/anie.200250814
Return to citation in text: [1] -
Gonsior, N.; Ritter, H. Macromol. Chem. Phys. 2012, 213, 382–388. doi:10.1002/macp.201100336
Return to citation in text: [1] -
Amemori, S.; Kokado, K.; Sada, K. J. Am. Chem. Soc. 2012, 134, 8344–8347. doi:10.1021/ja301688u
Return to citation in text: [1] -
Meiswinkel, G.; Ritter, H. Macromol. Chem. Phys. 2013, 214, 2835–2840. doi:10.1002/macp.201300560
Return to citation in text: [1] -
Tarkin-Tas, E.; Tas, H.; Mathias, L. J. Macromol. Symp. 2012, 313–314, 79–89. doi:10.1002/masy.201250309
Return to citation in text: [1] [2] -
Frost, J. W. Synthesis Of Caprolactam From Lysine. WO Patent 2005/123669 A1, 2005.
Return to citation in text: [1] -
Lu, Y.; Zhou, K.; Ding, Y.; Zhang, G.; Wu, C. Phys. Chem. Chem. Phys. 2010, 12, 3188–3194. doi:10.1039/b918969f
Return to citation in text: [1] -
Maeda, Y.; Nakamura, T.; Ikeda, I. Macromolecules 2001, 34, 1391–1399. doi:10.1021/ma001306t
Return to citation in text: [1]
| 1. | Schmaljohann, D. Adv. Drug Delivery Rev. 2006, 58, 1655–1670. doi:10.1016/j.addr.2006.09.020 |
| 2. | Twaites, B. R.; de las Heras Alarcón, C.; Lavigne, M.; Saulnier, A.; Pennadam, S. S.; Cunliffe, D.; Górecki, D. C.; Alexander, C. J. Controlled Release 2005, 108, 472–483. doi:10.1016/j.jconrel.2005.08.009 |
| 3. | Klouda, L.; Mikos, A. G. Eur. J. Pharm. Biopharm. 2008, 68, 34–45. doi:10.1016/j.ejpb.2007.02.025 |
| 9. | Fischer, J.; Ritter, H. Beilstein J. Org. Chem. 2013, 9, 2803–2811. doi:10.3762/bjoc.9.315 |
| 8. | Lee, H.-N.; Rosen, B. M.; Fenyvesi, G.; Sunkara, H. B. J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 4311–4315. doi:10.1002/pola.26242 |
| 6. | Buscall, R.; Corner, T. Eur. Polym. J. 1982, 18, 967–974. doi:10.1016/0014-3057(82)90084-2 |
| 7. | Dong, Z.; Mao, J.; Wang, D.; Yang, M.; Wang, W.; Bo, S.; Ji, X. Macromol. Chem. Phys. 2014, 215, 111–120. doi:10.1002/macp.201300552 |
| 16. | Lu, Y.; Zhou, K.; Ding, Y.; Zhang, G.; Wu, C. Phys. Chem. Chem. Phys. 2010, 12, 3188–3194. doi:10.1039/b918969f |
| 17. | Maeda, Y.; Nakamura, T.; Ikeda, I. Macromolecules 2001, 34, 1391–1399. doi:10.1021/ma001306t |
| 4. | Schild, H. G. Prog. Polym. Sci. 1992, 17, 163–249. doi:10.1016/0079-6700(92)90023-R |
| 5. | Trellenkamp, T.; Ritter, H. Macromol. Rapid Commun. 2009, 30, 1736–1740. doi:10.1002/marc.200900250 |
| 13. | Meiswinkel, G.; Ritter, H. Macromol. Chem. Phys. 2013, 214, 2835–2840. doi:10.1002/macp.201300560 |
| 15. | Frost, J. W. Synthesis Of Caprolactam From Lysine. WO Patent 2005/123669 A1, 2005. |
| 12. | Amemori, S.; Kokado, K.; Sada, K. J. Am. Chem. Soc. 2012, 134, 8344–8347. doi:10.1021/ja301688u |
| 14. | Tarkin-Tas, E.; Tas, H.; Mathias, L. J. Macromol. Symp. 2012, 313–314, 79–89. doi:10.1002/masy.201250309 |
| 11. | Gonsior, N.; Ritter, H. Macromol. Chem. Phys. 2012, 213, 382–388. doi:10.1002/macp.201100336 |
| 10. | Ritter, H.; Sadowski, O.; Tepper, E. Angew. Chem., Int. Ed. 2003, 42, 3171–3173. doi:10.1002/anie.200250814 |
| 14. | Tarkin-Tas, E.; Tas, H.; Mathias, L. J. Macromol. Symp. 2012, 313–314, 79–89. doi:10.1002/masy.201250309 |
© 2014 Burkhart and Ritter; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)