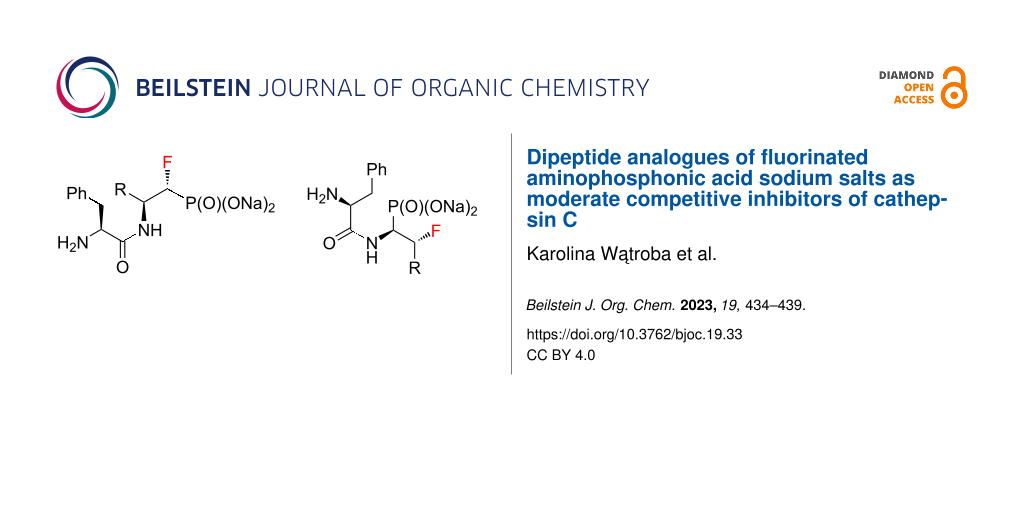
Abstract
In this paper, we present the solvolysis reaction of dipeptide analogues of fluorinated aminophosphonates with simultaneous quantitative deprotection of the amino group. To the best of our knowledge, this work is the first reported example of the application of fluorinated aminophosphonates in cathepsin C inhibition studies. The new molecules show moderate inhibition of the cathepsin C enzyme, which opens the door to consider them as potential therapeutic agents. Overall, our findings provide a new avenue for the development of fluorinated aminophosphonate-based inhibitors.

Graphical Abstract
Introduction
Cathepsin C, also known as dipeptidyl peptidase I (DPPI) belongs to the family of lysosomal cysteine proteases encompassing 11 human enzymes (cathepsins B, C, F, H, K, L, O, S, V, W, and X) [1]. Cathepsin C is considered a good target for designing new anticancer agents with broad substrate specificity [2].
Cathepsin C, which affects the processing of keratin, is of great importance in maintaining the structural organization of the epidermis, primarily the extremities, and the integrity of the teeth' tissues. Genetic studies have shown that a mutation in the gene encoding DPPI leads to early periodontitis, premature tooth loss, and keratosis of the palms and soles [3,4]. These conditions occur in Papillon–Lefevre syndrome and Haim–Munk syndrome [1]. Cathepsin C is an emerging pharmacological target due to its involvement in inflammatory and autoimmune diseases. Cathepsin C is upregulated in immune-related cells. DPPI also plays a role in the development of cancer – particularly in the liver and breast, hence the potential contribution of its inhibitors in chemotherapies to support traditional anticancer drugs. Moreover, there is a growing interest in the topic of cathepsin C inhibition, which directly affects serine protease activity [5,6]. Inhibitors of cathepsin C can be cystatins that show activity against a large group of cysteine proteases [7]. Other inhibitors are dipeptide derivatives showing substrate-like sequences. One of the most effective inhibitors is the dipeptide Gly-Phe-CHN2 (glycylphenylalanine-diazomethane), which, however, has not been used as a therapeutic substance due to the instability of the diazomethylketone group [8,9]. Based on its structure, many other inhibitors have been developed, such as vinyl sulfones, fluoromethyl ketones, and semicarbazides [8,9]. These inhibitors covalently bind to the nucleophilic thiol group of Cys234 in the active site of cathepsin C via a thioether bond.
Phosphonates have been identified as potential inhibitors of cathepsins. The phosphorus atom by default should mimic the tetrahedral intermediate, but this role may also be played by the hydroxy group present in hydroxyphosphonates, which mimic the carbonyl carbon in the peptide bond by forming a hydrogen bond with the amino group of the catalytic cysteine Cys234 [10]. Phosphonates, as well as their analogues phosphonic acids, can be modified in a number of ways, one of which is the introduction of a fluorine atom into their molecules by fluorination or alkylfluorination [11-14]. However, the reaction of β-aminoalcohols with nucleophilic deoxyfluorinating reagents often does not lead to the expected products with a fluorine atom in place of the –OH group. They usually undergo rearrangement, and intramolecular cyclization leading to products that are constitutional isomers [15].
The solvolysis reaction of phosphonates to the corresponding phosphonic acids or their salts is often a necessary step to measure activity in enzyme inhibition bioassays [16-22]. Therefore, our goal was to determine the best conditions for carrying out the solvolysis reaction of synthesized dipeptide analogues of fluorinated aminophosphonates [23,24] with the simultaneous deprotection of the amino group. The free amines were subjected to kinetic studies to investigate their interaction with cathepsin C. The required steps should be simple and fast, and the conditions of the reactions should be as mild as possible. The reactions should proceed with high yields, and any byproducts should be easily removeable.
Results and Discussion
Dipeptide analogues of α- and β-fluorinated aminophosphonates 5 and 7 were obtained from the corresponding ʟ-amino acids [23,24] 1. In the key step of the synthesis fluorine was introduced to the corresponding α- and β-fluorinated aminophosphonates 4, 6 (Scheme 1) by regioselective deoxyfluorination reactions of α-hydroxy-β-aminophosphonates 2 [24-26].
![[1860-5397-19-33-i1]](/bjoc/content/inline/1860-5397-19-33-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic strategy towards 5 and 7.
Scheme 1: Synthetic strategy towards 5 and 7.
Next, the conditions for the solvolysis were carefully assembled (Scheme 2). The optimized reaction conditions included 8 equiv of trimethylsilyl bromide (TMSBr) and freshly distilled methylene chloride as a solvent. In each case the reactions were carried out at room temperature overnight under an argon atmosphere. The next day, the solvent, volatile byproducts, and TMSBr residues were thoroughly evaporated. Time of solvolysis reactions varies in the literature, ranging from 10 minutes to several hours [27-29]. In our case the alcoholysis was carried out for 30 minutes. During this process, disappearance of the brownish or yellowish color of the compounds was observed. According to the literature, addition of diethyl ether in the next step should make precipitation more efficient [10]. This was done for compound 8a, but no improvement was observed. Much better results were obtained with additional double wash of the precipitate with methanol combined with evaporation of the solvent under reduced pressure. As a result of the reactions carried out, the dipeptide analogues of α- and β-fluorinated aminophosphonic acids 8 and 10 were obtained. All the samples were solids, with very poor solubility in water and organic solvents such as DMSO and MeOH.
![[1860-5397-19-33-i2]](/bjoc/content/inline/1860-5397-19-33-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 9 and 11. (a) R = -CH3; (b) R = -CH(CH3)2; (c) R = -CH2CH(CH3)2; (d) R = -CH(CH3)CH2CH3, (e) R = -CH2Ph; i) (a) 5 or 7 (1 equiv), TMSBr (8 equiv), CH2Cl2, rt, 20 h, (b) MeOH, 30 min.; ii) 8 or 10 (1 equiv), 1 M NaOH (2 equiv), H2O, 15 min.
Scheme 2: Synthesis of 9 and 11. (a) R = -CH3; (b) R = -CH(CH3)2; (c) R = -CH2CH(CH3)2; (d) R = -CH(CH3)CH2CH3...
The final step of the synthesis was the reaction of the resulting phosphonic acids 8 and 10 with a 1 M aqueous NaOH solution. Based on the literature data, alternatively to this method [30-32], phosphonic acids can be passed through an ion exchange column [33]. The reactions of compounds 8 and 10 with 1 M NaOH were carried out at room temperature (Scheme 2). When a clear solutions were obtained, the reaction was carried out for another 15 minutes. The solutions were then concentrated under reduced pressure. The precipitated salts were washed with methanol [31] and the solvent evaporation procedure was repeated. Sublimation drying (lyophilization) was carried out, obtaining white powders with a yield of 99% in each case. The resulting sodium salts of phosphonic acids 9 and 11 were subjected to 1H, 13C, 19F, and 31P NMR spectroscopic analysis as well as mass spectrometry (MS) confirming their purity. The spectroscopic data of 9 and 11 are in agreement with the literature data of the starting esters 5 and 7 literature data [23,24]. A very good correlation of chemical shifts was also observed in the 13C NMR spectra for the key signals from the C1 and C2 atoms (Table 1). Each sample was pure; no byproducts were present.
Table 1: The 13C NMR chemical shifts of C1 and C2 carbon atoms.
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-33-i4.svg?max-width=637&scale=1.0)
|
||||
| R = | 5 C1 [ppm] | 9 C1 [ppm] | 5 C2 [ppm] | 9 C2 [ppm] |
| (a) -CH3 | 89.56 | 94.72 | 45.66 | 47.77 |
| (b) -CH(CH3)2 | 88.97 | 93.30 | 54.77 | 57.29 |
| (c) -CH2CH(CH3)2 | 91.33 | 95.36 | 48.62 | 50.22 |
| (d) -CH(CH3)CH2CH3 | 88.89 | 92.17 | 54.45 | 56.33 |
| (e) -CH2Ph | 88.05 | 94.79 | 51.39 | 52.93 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-33-i5.svg?max-width=637&scale=1.0)
|
||||
| R = | 7 C1 [ppm] | 11 C1 [ppm] | 7 C2 [ppm] | 11 C2 [ppm] |
| (a) -CH3 | 49.51 | 54.13 | 89.00 | 92.58 |
| (b) -CH(CH3)2 | 47.55 | 50.45 | 98.03 | 99.23 |
| (c) -CH2CH(CH3)2 | 50.00 | 54.09 | 92.21 | 94.76 |
| (d) -CH(CH3)CH2CH3 | 47.16 | 49.91 | 95.16 | 96.08 |
| (e) -CH2Ph | 48.69 | 53.55 | 93.24 | 96.21 |
Kinetic studies
Due to the homology and similar structural requirements, bovine cathepsin C is often used in research as a model for human cathepsin C as it was well documented by Poręba et al. [34] in the study of the substrate specificity of these two mammalian cathepsins. They showed the best fit of amino acids with larger side to the S1 pocket of the enzyme. In contrast, the S2 pocket preferably accommodates amino acids having short aliphatic side-chains, but also recognizes aromatic amino acids, preferably phenylalanine. To study the structural requirements of the S1 binding site of the enzyme, we synthesized a series of ten dipeptide analogues of fluorinated aminophosphonic acid sodium salts 9, 11 with phenylalanine at the N-terminus and evaluated their inhibitory activity against bovine cathepsin C. Inhibition kinetics were carried out at 37 °C for 10 minutes in acetate buffer at pH 5. Changes in product concentration versus time were monitored spectrophotometrically at λ = 405 nm. Seven of the tested compounds were moderate competitive inhibitors with millimolar inhibitory activity (Table 2). Three of them at higher concentrations precipitated from 0.1 M acetate buffer at pH 5.0. Dipeptide analogues of α-fluorinated aminophosphonic acid sodium salts 9 were more active against cathepsin C than β-fluorinated analogues 11.
Table 2: Inhibitory constants of the studied of α- and β-fluorinated aminophosphonic acid sodium salts towards bovine cathepsin C.
| Dipeptide |
![[Graphic 3]](/bjoc/content/inline/1860-5397-19-33-i6.svg?max-width=637&scale=1.0)
9 KI ± SD [mM] |
![[Graphic 4]](/bjoc/content/inline/1860-5397-19-33-i7.svg?max-width=637&scale=1.0)
11 KI ± SD [mM] |
|
Phe-Ala
(a) -CH3 |
0.603 ± 0.1 | 0.733 ± 0.087 |
|
Phe-Val
(b) -CH(CH3)2 |
0.0951 ± 0.05 | 1.869 ± 0.171 |
|
Phe-Leu
(c) -CH2CH(CH3)2 |
0.309 ± 0.066 | 0.847 ± 0.38 |
|
Phe-Ile
(d) -CH(CH3)CH2CH3 |
0.273 ± 0.15 |
up to a concentration of 0.37 mM, it does not inhibit activity;
at a higher concentration, it precipitates |
|
Phe-Phe
(e) -CH2Ph |
precipitates in a buffer | precipitates in buffer |
The dipeptide analogue of α-fluorinated aminophosphonic acid sodium salt bearing the valine residue as a second amino acid in the chain (9b) showed the greatest inhibitory power (Figure 1). The type of inhibition and the inhibition constant were determined from the Dixon-type linearization of Equation 1 [35]. For each of the simple data, Equation 1 determines the slope factor a, whereby a weighted fit was used. The statistical weight for each point in the above-mentioned transformation 1/V0,i = f(I) is equal to ![[Graphic 5]](/bjoc/content/inline/1860-5397-19-33-i8.svg?max-width=637&scale=1.18182) .
.
![[1860-5397-19-33-1]](/bjoc/content/figures/1860-5397-19-33-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Dixon plot for the hydrolysis of Gly-Phe-pNA substrate catalyzed by bovine cathepsin C in the presence of 9b (T = 37 °C, pH 5.0).
Figure 1: Dixon plot for the hydrolysis of Gly-Phe-pNA substrate catalyzed by bovine cathepsin C in the prese...
![[1860-5397-19-33-i3]](/bjoc/content/inline/1860-5397-19-33-i3.svg?max-width=590&scale=1.18182)
where:
Vmax = maximum reaction velocity, KM = Michaelis constant, Kic = competitive inhibitory constant, Kiu = uncompetitive inhibitory constant, [S] = concentration of the substrate, [I] = concentration of the inhibitor.
Conclusion
In conclusion, we demonstrated the solvolysis reaction of dipeptide analogues of fluorinated aminophosphonates with the simultaneous deprotection of the amino group. The resulting acids were converted into the corresponding salts. All the reactions proceeded quantitatively. Obtained compounds were subjected to kinetic studies against cathepsin C, and the results indicated that they are moderate competitive inhibitors of this enzyme. This study presented the first kinetic investigation of fluorinated dipeptide derivatives of aminophosphonic acid salts against cathepsin C, thus contributing to the development of the novel cathepsin C inhibitors. We are currently working on the development of more effective fluorinated inhibitors of cathepsin C in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental procedures, biological protocol, NMR data. | ||
| Format: PDF | Size: 2.6 MB | Download |
References
-
Turk, D.; Janjić, V.; Štern, I.; Podobnik, M.; Lamba, D.; Dahl, S. W.; Lauritzen, C.; Pedersen, J.; Turk, V.; Turk, B. EMBO J. 2001, 20, 6570–6582. doi:10.1093/emboj/20.23.6570
Return to citation in text: [1] [2] -
Korkmaz, B.; Caughey, G. H.; Chapple, I.; Gauthier, F.; Hirschfeld, J.; Jenne, D. E.; Kettritz, R.; Lalmanach, G.; Lamort, A.-S.; Lauritzen, C.; Łȩgowska, M.; Lesner, A.; Marchand-Adam, S.; McKaig, S. J.; Moss, C.; Pedersen, J.; Roberts, H.; Schreiber, A.; Seren, S.; Thakker, N. S. Pharmacol. Ther. 2018, 190, 202–236. doi:10.1016/j.pharmthera.2018.05.011
Return to citation in text: [1] -
Rai, R.; Thiagarajan, S.; Mohandas, S.; Natarajan, K.; Shanmuga Sekar, C.; Ramalingam, S. Int. J. Dermatol. 2010, 49, 541–543. doi:10.1111/j.1365-4632.2010.04300.x
Return to citation in text: [1] -
Pham, C. T. N.; Ivanovich, J. L.; Raptis, S. Z.; Zehnbauer, B.; Ley, T. J. J. Immunol. 2004, 173, 7277–7281. doi:10.4049/jimmunol.173.12.7277
Return to citation in text: [1] -
Shen, X. B.; Chen, X.; Zhang, Z. Y.; Wu, F. F.; Liu, X. H. Eur. J. Med. Chem. 2021, 225, 113818. doi:10.1016/j.ejmech.2021.113818
Return to citation in text: [1] -
Xiao, Y.; Cong, M.; Li, J.; He, D.; Wu, Q.; Tian, P.; Wang, Y.; Yang, S.; Liang, C.; Liang, Y.; Wen, J.; Liu, Y.; Luo, W.; Lv, X.; He, Y.; Cheng, D.-d.; Zhou, T.; Zhao, W.; Zhang, P.; Zhang, X.; Xiao, Y.; Qian, Y.; Wang, H.; Gao, Q.; Yang, Q.-c.; Yang, Q.; Hu, G. Cancer Cell 2021, 39, 423–437.e7. doi:10.1016/j.ccell.2020.12.012
Return to citation in text: [1] -
Breznik, B.; Mitrović, A.; Lah, T. T.; Kos, J. Biochimie 2019, 166, 233–250. doi:10.1016/j.biochi.2019.05.002
Return to citation in text: [1] -
Kam, C.-M.; Götz, M. G.; Koot, G.; McGuire, M.; Thiele, D.; Hudig, D.; Powers, J. C. Arch. Biochem. Biophys. 2004, 427, 123–134. doi:10.1016/j.abb.2004.04.011
Return to citation in text: [1] [2] -
Mølgaard, A.; Arnau, J.; Lauritzen, C.; Larsen, S.; Petersen, G.; Pedersen, J. Biochem. J. 2007, 401, 645–650. doi:10.1042/bj20061389
Return to citation in text: [1] [2] -
Drąg, M.; Wieczerzak, E.; Pawełczak, M.; Berlicki, Ł.; Grzonka, Z.; Kafarski, P. Biochimie 2013, 95, 1640–1649. doi:10.1016/j.biochi.2013.05.006
Return to citation in text: [1] [2] -
Turcheniuk, K. V.; Kukhar, V. P.; Röschenthaler, G.-V.; Aceña, J. L.; Soloshonok, V. A.; Sorochinsky, A. E. RSC Adv. 2013, 3, 6693–6716. doi:10.1039/c3ra22891f
Return to citation in text: [1] -
Cytlak, T.; Kaźmierczak, M.; Skibińska, M.; Koroniak, H. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 602–620. doi:10.1080/10426507.2017.1287706
Return to citation in text: [1] -
Naydenova, E. D.; Todorov, P. T.; Troev, K. D. Amino Acids 2010, 38, 23–30. doi:10.1007/s00726-009-0254-7
Return to citation in text: [1] -
Kafarski, P. RSC Adv. 2020, 10, 25898–25910. doi:10.1039/d0ra04655h
Return to citation in text: [1] -
Kaźmierczak, M.; Bilska‐Markowska, M. Eur. J. Org. Chem. 2021, 5585–5604. doi:10.1002/ejoc.202101027
Return to citation in text: [1] -
Lejczak, B.; Kafarski, P.; Zygmunt, J. Biochemistry 1989, 28, 3549–3555. doi:10.1021/bi00434a060
Return to citation in text: [1] -
Herczegh, P.; Buxton, T. B.; McPherson, J. C.; Kovács-Kulyassa, Á.; Brewer, P. D.; Sztaricskai, F.; Stroebel, G. G.; Plowman, K. M.; Farcasiu, D.; Hartmann, J. F. J. Med. Chem. 2002, 45, 2338–2341. doi:10.1021/jm0105326
Return to citation in text: [1] -
Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. J. Med. Chem. 2003, 46, 2641–2655. doi:10.1021/jm030795v
Return to citation in text: [1] -
Pfund, E.; Lequeux, T.; Masson, S.; Vazeux, M.; Cordi, A.; Pierre, A.; Serre, V.; Hervé, G. Bioorg. Med. Chem. 2005, 13, 4921–4928. doi:10.1016/j.bmc.2005.05.026
Return to citation in text: [1] -
Drąg, M.; Grembecka, J.; Pawełczak, M.; Kafarski, P. Eur. J. Med. Chem. 2005, 40, 764–771. doi:10.1016/j.ejmech.2005.02.011
Return to citation in text: [1] -
Amin, S. A.; Adhikari, N.; Jha, T. J. Med. Chem. 2018, 61, 6468–6490. doi:10.1021/acs.jmedchem.7b00782
Return to citation in text: [1] -
Wanat, W.; Talma, M.; Pawełczak, M.; Kafarski, P. Pharmaceuticals 2019, 12, 139. doi:10.3390/ph12030139
Return to citation in text: [1] -
Kaźmierczak, M.; Koroniak, H. Beilstein J. Org. Chem. 2020, 16, 756–762. doi:10.3762/bjoc.16.69
Return to citation in text: [1] [2] [3] -
Kaźmierczak, M.; Dutkiewicz, G.; Cytlak, T. Synthesis 2020, 52, 2364–2372. doi:10.1055/s-0040-1707813
Return to citation in text: [1] [2] [3] [4] -
Kaźmierczak, M.; Koroniak, H. J. Fluorine Chem. 2012, 139, 23–27. doi:10.1016/j.jfluchem.2012.03.016
Return to citation in text: [1] -
Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Eur. J. Org. Chem. 2018, 3844–3852. doi:10.1002/ejoc.201800631
Return to citation in text: [1] -
Goldeman, W.; Nasulewicz-Goldeman, A. Bioorg. Med. Chem. Lett. 2014, 24, 3475–3479. doi:10.1016/j.bmcl.2014.05.071
Return to citation in text: [1] -
Xu, Y.; Jiang, G.; Tsukahara, R.; Fujiwara, Y.; Tigyi, G.; Prestwich, G. D. J. Med. Chem. 2006, 49, 5309–5315. doi:10.1021/jm060351+
Return to citation in text: [1] -
Chiminazzo, A.; Borsato, G.; Favero, A.; Fabbro, C.; McKenna, C. E.; Dalle Carbonare, L. G.; Valenti, M. T.; Fabris, F.; Scarso, A. Chem. – Eur. J. 2019, 25, 3617–3626. doi:10.1002/chem.201805436
Return to citation in text: [1] -
Goetz, D. B.; Varney, M. L.; Wiemer, D. F.; Holstein, S. A. Bioorg. Med. Chem. 2020, 28, 115604. doi:10.1016/j.bmc.2020.115604
Return to citation in text: [1] -
Grison, C.; Coutrot, P.; Comoy, C.; Balas, L.; Joliez, S.; Lavecchia, G.; Oliger, P.; Penverne, B.; Serre, V.; Hervé, G. Eur. J. Med. Chem. 2004, 39, 333–344. doi:10.1016/j.ejmech.2004.01.006
Return to citation in text: [1] [2] -
Liu, Y.; Wu, Y.; Sun, l.; Gu, Y.; Hu, L. Eur. J. Med. Chem. 2020, 191, 112181. doi:10.1016/j.ejmech.2020.112181
Return to citation in text: [1] -
Voos, K.; Schönauer, E.; Alhayek, A.; Haupenthal, J.; Andreas, A.; Müller, R.; Hartmann, R. W.; Brandstetter, H.; Hirsch, A. K. H.; Ducho, C. ChemMedChem 2021, 16, 1257–1267. doi:10.1002/cmdc.202000994
Return to citation in text: [1] -
Poreba, M.; Mihelic, M.; Krai, P.; Rajkovic, J.; Krezel, A.; Pawelczak, M.; Klemba, M.; Turk, D.; Turk, B.; Latajka, R.; Drag, M. Amino Acids 2014, 46, 931–943. doi:10.1007/s00726-013-1654-2
Return to citation in text: [1] -
Cortés, A.; Cascante, M.; Cárdenas, M. L.; Cornish-Bowden, A. Biochem. J. 2001, 357, 263–268. doi:10.1042/bj3570263
Return to citation in text: [1]
| 33. | Voos, K.; Schönauer, E.; Alhayek, A.; Haupenthal, J.; Andreas, A.; Müller, R.; Hartmann, R. W.; Brandstetter, H.; Hirsch, A. K. H.; Ducho, C. ChemMedChem 2021, 16, 1257–1267. doi:10.1002/cmdc.202000994 |
| 10. | Drąg, M.; Wieczerzak, E.; Pawełczak, M.; Berlicki, Ł.; Grzonka, Z.; Kafarski, P. Biochimie 2013, 95, 1640–1649. doi:10.1016/j.biochi.2013.05.006 |
| 30. | Goetz, D. B.; Varney, M. L.; Wiemer, D. F.; Holstein, S. A. Bioorg. Med. Chem. 2020, 28, 115604. doi:10.1016/j.bmc.2020.115604 |
| 31. | Grison, C.; Coutrot, P.; Comoy, C.; Balas, L.; Joliez, S.; Lavecchia, G.; Oliger, P.; Penverne, B.; Serre, V.; Hervé, G. Eur. J. Med. Chem. 2004, 39, 333–344. doi:10.1016/j.ejmech.2004.01.006 |
| 32. | Liu, Y.; Wu, Y.; Sun, l.; Gu, Y.; Hu, L. Eur. J. Med. Chem. 2020, 191, 112181. doi:10.1016/j.ejmech.2020.112181 |
| 1. | Turk, D.; Janjić, V.; Štern, I.; Podobnik, M.; Lamba, D.; Dahl, S. W.; Lauritzen, C.; Pedersen, J.; Turk, V.; Turk, B. EMBO J. 2001, 20, 6570–6582. doi:10.1093/emboj/20.23.6570 |
| 5. | Shen, X. B.; Chen, X.; Zhang, Z. Y.; Wu, F. F.; Liu, X. H. Eur. J. Med. Chem. 2021, 225, 113818. doi:10.1016/j.ejmech.2021.113818 |
| 6. | Xiao, Y.; Cong, M.; Li, J.; He, D.; Wu, Q.; Tian, P.; Wang, Y.; Yang, S.; Liang, C.; Liang, Y.; Wen, J.; Liu, Y.; Luo, W.; Lv, X.; He, Y.; Cheng, D.-d.; Zhou, T.; Zhao, W.; Zhang, P.; Zhang, X.; Xiao, Y.; Qian, Y.; Wang, H.; Gao, Q.; Yang, Q.-c.; Yang, Q.; Hu, G. Cancer Cell 2021, 39, 423–437.e7. doi:10.1016/j.ccell.2020.12.012 |
| 24. | Kaźmierczak, M.; Dutkiewicz, G.; Cytlak, T. Synthesis 2020, 52, 2364–2372. doi:10.1055/s-0040-1707813 |
| 25. | Kaźmierczak, M.; Koroniak, H. J. Fluorine Chem. 2012, 139, 23–27. doi:10.1016/j.jfluchem.2012.03.016 |
| 26. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Eur. J. Org. Chem. 2018, 3844–3852. doi:10.1002/ejoc.201800631 |
| 1. | Turk, D.; Janjić, V.; Štern, I.; Podobnik, M.; Lamba, D.; Dahl, S. W.; Lauritzen, C.; Pedersen, J.; Turk, V.; Turk, B. EMBO J. 2001, 20, 6570–6582. doi:10.1093/emboj/20.23.6570 |
| 27. | Goldeman, W.; Nasulewicz-Goldeman, A. Bioorg. Med. Chem. Lett. 2014, 24, 3475–3479. doi:10.1016/j.bmcl.2014.05.071 |
| 28. | Xu, Y.; Jiang, G.; Tsukahara, R.; Fujiwara, Y.; Tigyi, G.; Prestwich, G. D. J. Med. Chem. 2006, 49, 5309–5315. doi:10.1021/jm060351+ |
| 29. | Chiminazzo, A.; Borsato, G.; Favero, A.; Fabbro, C.; McKenna, C. E.; Dalle Carbonare, L. G.; Valenti, M. T.; Fabris, F.; Scarso, A. Chem. – Eur. J. 2019, 25, 3617–3626. doi:10.1002/chem.201805436 |
| 3. | Rai, R.; Thiagarajan, S.; Mohandas, S.; Natarajan, K.; Shanmuga Sekar, C.; Ramalingam, S. Int. J. Dermatol. 2010, 49, 541–543. doi:10.1111/j.1365-4632.2010.04300.x |
| 4. | Pham, C. T. N.; Ivanovich, J. L.; Raptis, S. Z.; Zehnbauer, B.; Ley, T. J. J. Immunol. 2004, 173, 7277–7281. doi:10.4049/jimmunol.173.12.7277 |
| 23. | Kaźmierczak, M.; Koroniak, H. Beilstein J. Org. Chem. 2020, 16, 756–762. doi:10.3762/bjoc.16.69 |
| 24. | Kaźmierczak, M.; Dutkiewicz, G.; Cytlak, T. Synthesis 2020, 52, 2364–2372. doi:10.1055/s-0040-1707813 |
| 2. | Korkmaz, B.; Caughey, G. H.; Chapple, I.; Gauthier, F.; Hirschfeld, J.; Jenne, D. E.; Kettritz, R.; Lalmanach, G.; Lamort, A.-S.; Lauritzen, C.; Łȩgowska, M.; Lesner, A.; Marchand-Adam, S.; McKaig, S. J.; Moss, C.; Pedersen, J.; Roberts, H.; Schreiber, A.; Seren, S.; Thakker, N. S. Pharmacol. Ther. 2018, 190, 202–236. doi:10.1016/j.pharmthera.2018.05.011 |
| 23. | Kaźmierczak, M.; Koroniak, H. Beilstein J. Org. Chem. 2020, 16, 756–762. doi:10.3762/bjoc.16.69 |
| 24. | Kaźmierczak, M.; Dutkiewicz, G.; Cytlak, T. Synthesis 2020, 52, 2364–2372. doi:10.1055/s-0040-1707813 |
| 10. | Drąg, M.; Wieczerzak, E.; Pawełczak, M.; Berlicki, Ł.; Grzonka, Z.; Kafarski, P. Biochimie 2013, 95, 1640–1649. doi:10.1016/j.biochi.2013.05.006 |
| 15. | Kaźmierczak, M.; Bilska‐Markowska, M. Eur. J. Org. Chem. 2021, 5585–5604. doi:10.1002/ejoc.202101027 |
| 34. | Poreba, M.; Mihelic, M.; Krai, P.; Rajkovic, J.; Krezel, A.; Pawelczak, M.; Klemba, M.; Turk, D.; Turk, B.; Latajka, R.; Drag, M. Amino Acids 2014, 46, 931–943. doi:10.1007/s00726-013-1654-2 |
| 8. | Kam, C.-M.; Götz, M. G.; Koot, G.; McGuire, M.; Thiele, D.; Hudig, D.; Powers, J. C. Arch. Biochem. Biophys. 2004, 427, 123–134. doi:10.1016/j.abb.2004.04.011 |
| 9. | Mølgaard, A.; Arnau, J.; Lauritzen, C.; Larsen, S.; Petersen, G.; Pedersen, J. Biochem. J. 2007, 401, 645–650. doi:10.1042/bj20061389 |
| 16. | Lejczak, B.; Kafarski, P.; Zygmunt, J. Biochemistry 1989, 28, 3549–3555. doi:10.1021/bi00434a060 |
| 17. | Herczegh, P.; Buxton, T. B.; McPherson, J. C.; Kovács-Kulyassa, Á.; Brewer, P. D.; Sztaricskai, F.; Stroebel, G. G.; Plowman, K. M.; Farcasiu, D.; Hartmann, J. F. J. Med. Chem. 2002, 45, 2338–2341. doi:10.1021/jm0105326 |
| 18. | Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. J. Med. Chem. 2003, 46, 2641–2655. doi:10.1021/jm030795v |
| 19. | Pfund, E.; Lequeux, T.; Masson, S.; Vazeux, M.; Cordi, A.; Pierre, A.; Serre, V.; Hervé, G. Bioorg. Med. Chem. 2005, 13, 4921–4928. doi:10.1016/j.bmc.2005.05.026 |
| 20. | Drąg, M.; Grembecka, J.; Pawełczak, M.; Kafarski, P. Eur. J. Med. Chem. 2005, 40, 764–771. doi:10.1016/j.ejmech.2005.02.011 |
| 21. | Amin, S. A.; Adhikari, N.; Jha, T. J. Med. Chem. 2018, 61, 6468–6490. doi:10.1021/acs.jmedchem.7b00782 |
| 22. | Wanat, W.; Talma, M.; Pawełczak, M.; Kafarski, P. Pharmaceuticals 2019, 12, 139. doi:10.3390/ph12030139 |
| 35. | Cortés, A.; Cascante, M.; Cárdenas, M. L.; Cornish-Bowden, A. Biochem. J. 2001, 357, 263–268. doi:10.1042/bj3570263 |
| 8. | Kam, C.-M.; Götz, M. G.; Koot, G.; McGuire, M.; Thiele, D.; Hudig, D.; Powers, J. C. Arch. Biochem. Biophys. 2004, 427, 123–134. doi:10.1016/j.abb.2004.04.011 |
| 9. | Mølgaard, A.; Arnau, J.; Lauritzen, C.; Larsen, S.; Petersen, G.; Pedersen, J. Biochem. J. 2007, 401, 645–650. doi:10.1042/bj20061389 |
| 31. | Grison, C.; Coutrot, P.; Comoy, C.; Balas, L.; Joliez, S.; Lavecchia, G.; Oliger, P.; Penverne, B.; Serre, V.; Hervé, G. Eur. J. Med. Chem. 2004, 39, 333–344. doi:10.1016/j.ejmech.2004.01.006 |
| 7. | Breznik, B.; Mitrović, A.; Lah, T. T.; Kos, J. Biochimie 2019, 166, 233–250. doi:10.1016/j.biochi.2019.05.002 |
| 11. | Turcheniuk, K. V.; Kukhar, V. P.; Röschenthaler, G.-V.; Aceña, J. L.; Soloshonok, V. A.; Sorochinsky, A. E. RSC Adv. 2013, 3, 6693–6716. doi:10.1039/c3ra22891f |
| 12. | Cytlak, T.; Kaźmierczak, M.; Skibińska, M.; Koroniak, H. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 602–620. doi:10.1080/10426507.2017.1287706 |
| 13. | Naydenova, E. D.; Todorov, P. T.; Troev, K. D. Amino Acids 2010, 38, 23–30. doi:10.1007/s00726-009-0254-7 |
| 14. | Kafarski, P. RSC Adv. 2020, 10, 25898–25910. doi:10.1039/d0ra04655h |
| 23. | Kaźmierczak, M.; Koroniak, H. Beilstein J. Org. Chem. 2020, 16, 756–762. doi:10.3762/bjoc.16.69 |
| 24. | Kaźmierczak, M.; Dutkiewicz, G.; Cytlak, T. Synthesis 2020, 52, 2364–2372. doi:10.1055/s-0040-1707813 |
© 2023 Wątroba et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.