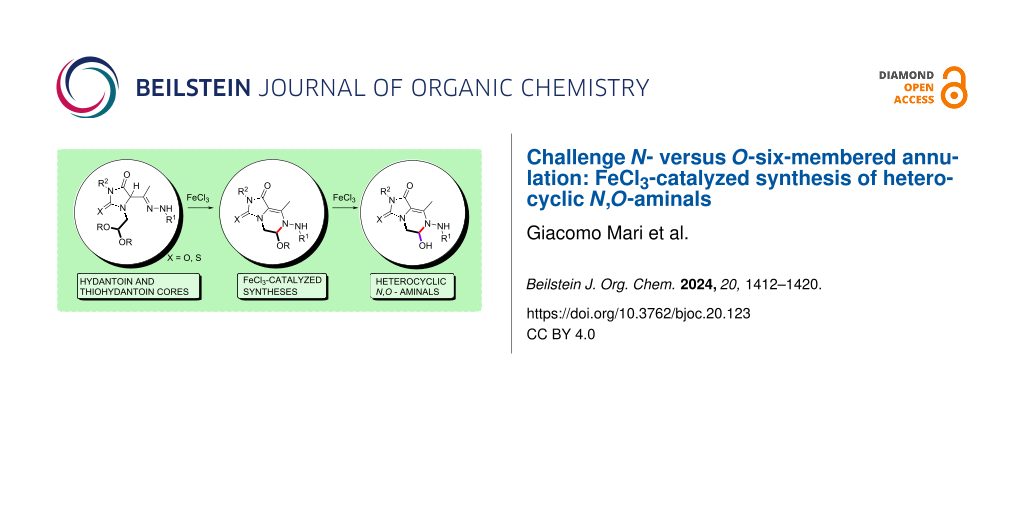
Abstract
A new class of heterocyclic N,O-aminal and hemiaminal scaffolds was successfully obtained by means of a three-component reaction (3-CR) of 1,2-diaza-1,3-dienes (DDs), α-aminoacetals and iso(thio)cyanates. These stable imine surrogates are generated from key-substituted (thio)hydantoin intermediates through selective FeCl3-catalyzed intramolecular N-annulation.

Graphical Abstract
Introduction
N-Fused heterocycles are ubiquitous within crucial molecules, including biologically active natural products, pharmaceuticals, and functional materials (Figure 1) [1-3].
![[1860-5397-20-123-1]](/bjoc/content/figures/1860-5397-20-123-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative examples of relevant N-fused heterocycles.
Figure 1: Representative examples of relevant N-fused heterocycles.
It has been assessed that almost one-third of the best-selling therapeutics contains fused heterocyclic structures [4]. Among the N-heterocycles, imidazopyrazine structures [5,6], derived from amalgamation of privileged imidazole and pyrazine pharmacophores, are well represented in the area of medicinal chemistry since they possess pharmacological properties as mammalian target of rapamycin (mTOR) inhibitors [7], adenosine triphosphate (ATP) competitive inhibitors of the insuline-like growth factor 1 (IGF-1) receptor related to Ewing sarcoma [8], IGF-1 receptor inhibitors [9] or act as ligands on corticotropin releasing hormone (CRH) [10], γ-aminobutyric acid (GABA) [11] and melanocortin receptors [12].
Given the established potencies of this class of N-ring-fused compounds, planned syntheses that simplify their preparation by using small building blocks and that lead, through appropriate transformations, to a product that becomes a substrate for another complexity-generating reaction, merit investigation [13-15].
Herein, we report a 3-CR-based synthesis of new properly decorated (thio)hydantoin framework able to afford, by a chemospecific Lewis acid-catalyzed ring-closure protocol, valuable heterocyclic N,O-aminals (Scheme 1).
![[1860-5397-20-123-i1]](/bjoc/content/inline/1860-5397-20-123-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Different acid-catalyzed six-membered ring cyclizations.
Scheme 1: Different acid-catalyzed six-membered ring cyclizations.
Results and Discussion
Since the direct functionalization of N-heterocycles offers an attractive entry to important molecular targets that might otherwise require lengthy synthetic procedures [16], our consolidated 3-CR strategy [17-19] implicates a careful selection of the starting components that ensures the installation of functionalities to be converged, by regioselective control, in different ring-closing processes [20,21].
With these considerations in mind and with the aim of diversity-oriented synthesis of N-heterocycles via sequential multicomponent approaches, we envisioned that α-aminoacetals could act as bifunctional building blocks along with 1,2-diaza-1,3-diene (DD) coupling partners [22,23], in obtaining functionalized N-aminohydrazones as key intermediates.
Based on our previous findings [17-19], the initial nucleophilic addition of α-aminoacetals 2a,b as nitrogen source to the activated heterodiene system of 4-methoxycarbonyl-DDs 1a–f in dichloromethane (DCM) or ethanol (EtOH) at room temperature affords N-aminohydrazone derivatives I (Scheme 2), whose sequential acylation process by iso(thio)cyanates 3a–h gives rise to the asymmetric (thio)urea derivatives (intermediate II). The spontaneous nucleophilic attack of the (thio)amide nitrogen on the terminal methyl ester function at C-4 of the starting azo-ene system provides a regioselective heteroring closure, positioning appropriate functions both at N-3 and C-4 of the (thio)hydantoin frameworks 4a–r (30–81%) broadening their usable decorations (Scheme 2).
![[1860-5397-20-123-i2]](/bjoc/content/inline/1860-5397-20-123-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope for the assembly of suitably N-3-functionalized (thio)hydantoins 4a–r. aDCM was utilized as solvent with isothiocyanates 3a–f, while bEtOH was utilized with isocyanates 3g,h.
Scheme 2: Substrate scope for the assembly of suitably N-3-functionalized (thio)hydantoins 4a–r. aDCM was uti...
Recently, we reported that compounds 4a, 4f, and 4m undergo an intramolecular cyclization process through the involvement of the restored keto function of the hydrazone moiety and the open-chain hemiacetal or aldehyde hydrate in Brønsted acid medium to access 1H-imidazo[5,1-c][1,4]oxazine derivatives (Scheme 1) [21].
Considering that the hydrazone function at C-4 of 4a–r may exist in a tautomeric equilibrium with the corresponding ene-hydrazino form [17,24,25], we conceived the idea of reversing the reactivity of 4a–r in the six-membered cyclization process (N- vs O-annulation) through the generation of an electrophilic oxocarbenium [26,27] cation intermediate from the acetal residue at N-3 of the (thio)hydantoin core. To pursue our goal, different Lewis acids (10 mol %) such as Zn(OTf)2, CuCl2, and FeCl3 were screened at room temperature in different solvents, employing compound 4a as the model substrate (Table 1).
Table 1: Optimization conditions for the Lewis acid-catalyzed intramolecular cyclization of 4a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-123-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entrya | Lewis acid | Solvent | Time (h) | 5a (yield %)b | 6a (yield %)b |
| 1 | Zn(OTf)2 (10 mol %) | DCM | 42 | 38 | 22 |
| 2 | CuCl2 (10 mol %) | DCM | 96 | 28 | 15 |
| 3 | FeCl3 (10 mol %) | DCM | 86 | 44 | 31 |
| 4 | FeCl3 (20 mol %) | DCM | 38 | 63 | 18 |
| 5 | FeCl3 (30 mol %) | DCM | 2 | 73 | 8 |
| 6 | FeCl3 (30 mol %) | ACN | 2 | 68 | 13 |
| 7 | FeCl3 (30 mol %) | THF | 2 | 62 | 11 |
| 8 | FeCl3 (30 mol %) | EtOH | 48 | –c | –c |
aThe reactions were performed on a 0.5 mmol scale in 5 mL of solvent. bIsolated yields of products 5a and 6a based on starting 4a. cNot detected by TLC analysis.
From the set of data collected, both the formation of N,O-aminal 5a and corresponding hemiaminal 6a were observed (entries 1–7, Table 1). Similarly to what was observed by Yu and co-workers for the intramolecular cyclization of alkynyl aldehyde acetals [28,29], it was found that the use of FeCl3 provided the better result in terms of overall yield (entry 3, Table 1). Moreover, the choice of iron(III) seemed to have remarkable advantages such as an environmentally benign alternative to traditional transition-metal catalysis, a low cost, nontoxicity, good stability, and easy handling [30,31]. Upon increasing the amount of FeCl3 to 20 mol %, the time of the reaction was reduced from 86 to 38 hours, and the yield of 5a was incremented with respect to 6a (entry 4, Table 1). Rising the amount of FeCl3 to 30 mol %, the reaction was complete in 2 hours, enhancing the yield of 5a (73%) and minimizing the yield of 6a (8%) (entry 5, Table 1). In reactions carried out in acetonitrile (ACN) or tetrahydrofuran (THF) the yield of 5a decreased, while utilizing ethanol the reaction proceeded slowly and produced a complicated mixture in which both 5a and 6a were not detected (Table 1, entries 6–8).
With the optimized conditions in hand (Table 1, entry 5), a selection of N-3-functionalized (thio)hydantoins (4a–r, 1 mmol) were dissolved in DCM (10 mL), FeCl3 (30 mol %) added and magnetically stirred at room temperature. Within 2–30 h, the reactions went to completion (TLC monitoring), affording, at last, N,O-aminals 5a–r (42–82%) and the corresponding hemiaminals 6a–p (4–35%) after column chromatography (Scheme 3).
![[1860-5397-20-123-i3]](/bjoc/content/inline/1860-5397-20-123-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of the iron(III)-catalyzed synthesis of functionalized heterocyclic N,O-aminals 5a–r and hemiaminals 6a–p.
Scheme 3: Substrate scope of the iron(III)-catalyzed synthesis of functionalized heterocyclic N,O-aminals 5a–r...
An increased yield of 6 was observed alongside a decreased yield of 5, in all those cases that required prolonged reaction times (24–30 h). This event led us to suppose the formation of carbinolamine 6 from N,O-aminal 5 owing to the nucleophilic attack of a water molecule, probably caused by the enriched moisture content of the reaction environment during the time.
Then, to explain the related formation of 5 and 6, we hypothesized a plausible reaction mechanism in which iron is involved in two concomitant catalytic cycles (Scheme 4). Initially, FeCl3 forms an acid–base complex with one of the alkoxy groups of 4 providing intermediate A. The latter, by loss of a trichloro(alkoxy)ferrate(III) anion, generates a strong electrophile such as the oxocarbenium cation intermediate B. The released trichloro(alkoxy)ferrate(III) splits into FeCl3, which enters the catalytic cycle, and a free alkoxide, which acts as a base, promoting, via hydrazone–enamine tautomerization [17,24,25], the nucleophilic addition which concludes with the construction of the heterocyclic N,O-aminal 5 through the intramolecular N–C bond formation. The FeCl3 can also interact with the newly formed N,O-aminals 5, giving rise to the second parallel catalytic cycle. Similar to what was previously observed, the elimination of the trichloro(alkoxy)ferrate(III) anion from intermediate C provides the iminium ion D, susceptible to nucleophilic attack by a water molecule present in the reaction medium, leading to the carbinolamines 6. This latter synthesis represents an interesting example of auto-tandem catalysis in which FeCl3 promotes two subsequent reactions.
![[1860-5397-20-123-i4]](/bjoc/content/inline/1860-5397-20-123-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism for the formation of N,O-aminals 5 and hemiaminals 6.
Scheme 4: Proposed mechanism for the formation of N,O-aminals 5 and hemiaminals 6.
For further confirmation to support our mechanistic hypothesis and in an attempt to switch the reaction toward the formation of hemiaminal 6a, we repeated the reaction of thiohydantoin 4j, (chosen as representative substrate), under the same previously optimized conditions, but extending the reaction time to 240 hours (experiment A in Scheme 5). In this case, the yield of hemiaminal 6a increased from 6% recorded after two hours at the complete conversion of 4j (Scheme 3) to 21% (experiment A, Scheme 5), in line with the values found for the slower reactions previously described (compounds 6b,d,g,i,n–p). By adding 500 μL of water to the medium the cyclization did not proceed, and the starting material 4j was recovered unchanged (experiment B, Scheme 5). This observation seems to suggest that the presence of a high water amount results in catalyst deactivation.
![[1860-5397-20-123-i5]](/bjoc/content/inline/1860-5397-20-123-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Control mechanistic experiments.
Scheme 5: Control mechanistic experiments.
Based on these results and what was observed in the optimization tests (Table 1, entry 6), we extended the reaction time but used ACN as solvent, which possesses a higher water content with respect to DCM (experiment C, Scheme 5). Gratifyingly, in this case, the formation of carbinolamine 6a becomes predominant (45%), despite a small quantity of N,O-aminal 5j (14%) is also produced, by virtue of the alcohol released from the starting acetal 4j. Probably, the higher water concentration in acetonitrile shifts the equilibrium in favour of 6a over the time (Scheme 4).
Within our proposed catalytic cycle, when compound 4j is utilized, methanol is released, due to the presence of a dimethyl acetal residue. Therefore, using molecular sieves (MS 4 Å) little alcohol molecules, such as MeOH, can be potentially trapped, allowing the insertion of a more encumbered alcohol, such as benzyl alcohol, which is not sequestered by MS 4 Å. As a matter of fact, the new benzylated N,O-aminal 7 was successfully obtained in 70% isolated yield as the sole product (experiment D, Scheme 5). In this latter case, the benzyl alcohol presumably reacts with the iminium ion D formed in the second catalytic cycle (Scheme 4), and its sole formation is ascribable to the capability of molecular sieves of sequestering MeOH eventually formed, shifting the equilibrium towards 7.
Noteworthy, in compounds 5a–r, 6a–p and 7, the newly created heterocyclic nucleus represents a new example of cyclic N,O-aminals and carbinolamine derivatives, an interesting class of organic compounds that are common structural motifs embedded within diverse biologically important natural products and pharmaceuticals [32-37]. On the other hand, the N,O-aminals are stable and very practical synthetic intermediates commonly employed for the in situ generation of highly electrophilic iminium ions [38-41].
Conclusion
In summary, we planned the synthesis of decorated imidazo skeletons accessible through a judicious choice of the starting components of a 3-CR process and developed a catalytic system-controlled selective intramolecular N-annulation process for ring-fused biheterocyclic N,O-aminal derivatives as stable imine equivalents and useful tools for new bond formation in view of further fused-heterocylization processes. Moreover, control experiments corroborate our mechanistic hypothesis related to the formation of both N,O-aminals and corresponding hemiaminals. In particular, the domino reaction that leads to the carbinolamines represents an interesting example of “auto-tandem catalysis” in which the FeCl3 catalyzes two different chemical transformations in a single reactor, reducing the number of steps and the amount of waste with consequent benefits of cost and environmental impact [42,43].
Supporting Information
| Supporting Information File 1: General experimental information, synthetic procedures, analytical data and NMR spectra for the reported compounds. | ||
| Format: PDF | Size: 8.8 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Taylor, R. D.; MacCoss, M.; Lawson, A. D. G. J. Med. Chem. 2014, 57, 5845–5859. doi:10.1021/jm4017625
Return to citation in text: [1] -
Bollini, M.; Casal, J. J.; Alvarez, D. E.; Boiani, L.; González, M.; Cerecetto, H.; Bruno, A. M. Bioorg. Med. Chem. 2009, 17, 1437–1444. doi:10.1016/j.bmc.2009.01.011
Return to citation in text: [1] -
Hao, Y.; Guo, J.; Wang, Z.; Liu, Y.; Li, Y.; Ma, D.; Wang, Q. J. Agric. Food Chem. 2020, 68, 5586–5595. doi:10.1021/acs.jafc.0c02101
Return to citation in text: [1] -
McGrath, N. A.; Brichacek, M.; Njardarson, J. T. J. Chem. Educ. 2010, 87, 1348–1349. doi:10.1021/ed1003806
Return to citation in text: [1] -
Goel, R.; Luxami, V.; Paul, K. Org. Biomol. Chem. 2015, 13, 3525–3555. doi:10.1039/c4ob01380h
Return to citation in text: [1] -
Mulvihill, M. J.; Ji, Q.-S.; Coate, H. R.; Cooke, A.; Dong, H.; Feng, L.; Foreman, K.; Rosenfeld-Franklin, M.; Honda, A.; Mak, G.; Mulvihill, K. M.; Nigro, A. I.; O’Connor, M.; Pirrit, C.; Steinig, A. G.; Siu, K.; Stolz, K. M.; Sun, Y.; Tavares, P. A. R.; Yao, Y.; Gibson, N. W. Bioorg. Med. Chem. 2008, 16, 1359–1375. doi:10.1016/j.bmc.2007.10.061
Return to citation in text: [1] -
Meng, Z.; Siddiqui, M. A.; Reddy, P. A. P. Imidazopyrazine Derivatives As Inhibitors of mTOR. International Pat. Appl. WO2013016160A1, Jan 31, 2013.
Return to citation in text: [1] -
Guaitoli, V.; Alvarez-Ginarte, Y. M.; Montero-Cabrera, L. A.; Bencomo-Martínez, A.; Pérez Badel, Y.; Giorgetti, A.; Suku, E. J. Mol. Model. 2020, 26, 222. doi:10.1007/s00894-020-04470-w
Return to citation in text: [1] -
Mulvihill, M. J.; Ji, Q.-S.; Werner, D.; Beck, P.; Cesario, C.; Cooke, A.; Cox, M.; Crew, A.; Dong, H.; Feng, L.; Foreman, K. W.; Mak, G.; Nigro, A.; O’Connor, M.; Saroglou, L.; Stolz, K. M.; Sujka, I.; Volk, B.; Weng, Q.; Wilkes, R. Bioorg. Med. Chem. Lett. 2007, 17, 1091–1097. doi:10.1016/j.bmcl.2006.11.016
Return to citation in text: [1] -
Hartz, R. A.; Gilligan, P. J.; Nanda, K. K.; Tebben, A. J.; Fitzgerald, L. W.; Miller, K. Bioorg. Med. Chem. Lett. 2002, 12, 291–294. doi:10.1016/s0960-894x(01)00741-7
Return to citation in text: [1] -
Bettati, M.; Chambers, M. S.; Goodacre, S. C.; Hallett, D. J.; Russel, M. G.; Street, L. J. Imidazo-pyrazine derivatives as ligands for GABA receptors. International Pat. Appl. WO03093272A1, May 2, 2002.
Return to citation in text: [1] -
Carpino, P. A.; Cole, B. M.; Morgan, B. P. Melanocortin receptor agonists. Eur. Pat. Appl. EP1294719A1, March 3, 2003.
Return to citation in text: [1] -
Gopalsamy, A.; Shi, M. Org. Lett. 2003, 5, 3907–3909. doi:10.1021/ol035455i
Return to citation in text: [1] -
Trček, T.; Verček, B. Synthesis 2006, 3437–3442. doi:10.1055/s-2006-950211
Return to citation in text: [1] -
Shaabani, A.; Maleki, A.; Mofakham, H. Mol. Diversity 2009, 13, 63–67. doi:10.1007/s11030-008-9099-3
Return to citation in text: [1] -
Campos, K. R. Chem. Soc. Rev. 2007, 36, 1069–1084. doi:10.1039/b607547a
See for an excellent review on the direct functionalization of sp3 C–H bonds adjacent to nitrogen in heterocycles.
Return to citation in text: [1] -
Attanasi, O. A.; De Crescentini, L.; Favi, G.; Nicolini, S.; Perrulli, F. R.; Santeusanio, S. Org. Lett. 2011, 13, 353–355. doi:10.1021/ol102664n
Return to citation in text: [1] [2] [3] [4] -
Attanasi, O. A.; Bartoccini, S.; Favi, G.; Giorgi, G.; Perrulli, F. R.; Santeusanio, S. J. Org. Chem. 2012, 77, 1161–1167. doi:10.1021/jo2021949
Return to citation in text: [1] [2] -
Attanasi, O. A.; De Crescentini, L.; Filippone, P.; Giorgi, G.; Nicolini, S.; Perrulli, F. R.; Santeusanio, S. Tetrahedron 2014, 70, 7336–7343. doi:10.1016/j.tet.2014.07.038
Return to citation in text: [1] [2] -
Attanasi, O. A.; Favi, G.; Giorgi, G.; Majer, R.; Perrulli, F. R.; Santeusanio, S. Org. Biomol. Chem. 2014, 12, 4610–4619. doi:10.1039/c4ob00676c
Return to citation in text: [1] -
Mari, G.; De Crescentini, L.; Favi, G.; Mantellini, F.; Santeusanio, S. Eur. J. Org. Chem. 2022, e202201053. doi:10.1002/ejoc.202201053
Return to citation in text: [1] [2] -
Lopes, S. M. M.; Cardoso, A. L.; Lemos, A.; Pinho e Melo, T. M. V. D. Chem. Rev. 2018, 118, 11324–11352. doi:10.1021/acs.chemrev.8b00375
See for review on DDs.
Return to citation in text: [1] -
Attanasi, O. A.; De Crescentini, L.; Favi, G.; Filippone, P.; Mantellini, F.; Perrulli, F. R.; Santeusanio, S. Eur. J. Org. Chem. 2009, 3109–3127. doi:10.1002/ejoc.200900243
Return to citation in text: [1] -
Corrieri, M.; De Crescentini, L.; Mantellini, F.; Mari, G.; Santeusanio, S.; Favi, G. J. Org. Chem. 2021, 86, 17918–17929. doi:10.1021/acs.joc.1c02217
Return to citation in text: [1] [2] -
Mei, G.-J.; Tang, X.; Tasdan, Y.; Lu, Y. Angew. Chem., Int. Ed. 2020, 59, 648–652. doi:10.1002/anie.201911686
Return to citation in text: [1] [2] -
Liu, D.-Y.; Zhu, S.-M.; Li, R.-Q.; Tian, J.-S.; Loh, T.-P. Org. Lett. 2019, 21, 6357–6360. doi:10.1021/acs.orglett.9b02240
Return to citation in text: [1] -
Purohit, P.; Pandey, A. K.; Kumar, B.; Chauhan, P. M. S. RSC Adv. 2016, 6, 21165–21186. doi:10.1039/c5ra27090a
Return to citation in text: [1] -
Xu, T.; Yu, Z.; Wang, L. Org. Lett. 2009, 11, 2113–2116. doi:10.1021/ol9005689
Return to citation in text: [1] -
Xu, T.; Yang, Q.; Ye, W.; Jiang, Q.; Xu, Z.; Chen, J.; Yu, Z. Chem. – Eur. J. 2011, 17, 10547–10551. doi:10.1002/chem.201101667
Return to citation in text: [1] -
Bauer, I.; Knölker, H.-J. Chem. Rev. 2015, 115, 3170–3387. doi:10.1021/cr500425u
Return to citation in text: [1] -
Mari, G.; Corrieri, M.; De Crescentini, L.; Favi, G.; Santeusanio, S.; Mantellini, F. Eur. J. Org. Chem. 2021, 5202–5208. doi:10.1002/ejoc.202101046
Return to citation in text: [1] -
Ishiuchi, K.; Kubota, T.; Hoshino, T.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2006, 14, 5995–6000. doi:10.1016/j.bmc.2006.05.028
Return to citation in text: [1] -
Jiang, X.; Williams, N.; De Brabander, J. K. Org. Lett. 2007, 9, 227–230. doi:10.1021/ol062656o
Return to citation in text: [1] -
Rech, J. C.; Floreancig, P. E. Org. Lett. 2005, 7, 5175–5178. doi:10.1021/ol0520267
Return to citation in text: [1] -
He, F.; Bo, Y.; Altom, J. D.; Corey, E. J. J. Am. Chem. Soc. 1999, 121, 6771–6772. doi:10.1021/ja9915201
Return to citation in text: [1] -
Xu, Z.; Wang, Q.; Zhu, J. J. Am. Chem. Soc. 2015, 137, 6712–6724. doi:10.1021/jacs.5b03619
Return to citation in text: [1] -
Zhou, F.; Zeng, X.-P.; Wang, C.; Zhao, X.-L.; Zhou, J. Chem. Commun. 2013, 49, 2022–2024. doi:10.1039/c3cc38819k
Return to citation in text: [1] -
Huang, Y.-Y.; Cai, C.; Yang, X.; Lv, Z.-C.; Schneider, U. ACS Catal. 2016, 6, 5747–5763. doi:10.1021/acscatal.6b01725
And references cited therein.
Return to citation in text: [1] -
Wang, W.; Huang, H. Chem. Commun. 2019, 55, 3947–3950. doi:10.1039/c9cc01374a
Return to citation in text: [1] -
Chen, A.; Yu, H.; Yan, J.; Huang, H. Org. Lett. 2020, 22, 755–759. doi:10.1021/acs.orglett.9b04630
Return to citation in text: [1] -
Hou, J.; Xue, L.; Yu, T.; Li, J.; Yu, S.; Yao, C.; Li, Y.-M. Chem. Commun. 2023, 59, 7096–7099. doi:10.1039/d3cc01527k
Return to citation in text: [1] -
Shindoh, N.; Takemoto, Y.; Takasu, K. Chem. – Eur. J. 2009, 15, 12168–12179. doi:10.1002/chem.200901486
And references cited therein.
Return to citation in text: [1] -
Long, J.; Yu, R.; Gao, J.; Fang, X. Angew. Chem., Int. Ed. 2020, 59, 6785–6789. doi:10.1002/anie.202000704
Return to citation in text: [1]
| 28. | Xu, T.; Yu, Z.; Wang, L. Org. Lett. 2009, 11, 2113–2116. doi:10.1021/ol9005689 |
| 29. | Xu, T.; Yang, Q.; Ye, W.; Jiang, Q.; Xu, Z.; Chen, J.; Yu, Z. Chem. – Eur. J. 2011, 17, 10547–10551. doi:10.1002/chem.201101667 |
| 17. | Attanasi, O. A.; De Crescentini, L.; Favi, G.; Nicolini, S.; Perrulli, F. R.; Santeusanio, S. Org. Lett. 2011, 13, 353–355. doi:10.1021/ol102664n |
| 24. | Corrieri, M.; De Crescentini, L.; Mantellini, F.; Mari, G.; Santeusanio, S.; Favi, G. J. Org. Chem. 2021, 86, 17918–17929. doi:10.1021/acs.joc.1c02217 |
| 25. | Mei, G.-J.; Tang, X.; Tasdan, Y.; Lu, Y. Angew. Chem., Int. Ed. 2020, 59, 648–652. doi:10.1002/anie.201911686 |
| 26. | Liu, D.-Y.; Zhu, S.-M.; Li, R.-Q.; Tian, J.-S.; Loh, T.-P. Org. Lett. 2019, 21, 6357–6360. doi:10.1021/acs.orglett.9b02240 |
| 27. | Purohit, P.; Pandey, A. K.; Kumar, B.; Chauhan, P. M. S. RSC Adv. 2016, 6, 21165–21186. doi:10.1039/c5ra27090a |
| 1. | Taylor, R. D.; MacCoss, M.; Lawson, A. D. G. J. Med. Chem. 2014, 57, 5845–5859. doi:10.1021/jm4017625 |
| 2. | Bollini, M.; Casal, J. J.; Alvarez, D. E.; Boiani, L.; González, M.; Cerecetto, H.; Bruno, A. M. Bioorg. Med. Chem. 2009, 17, 1437–1444. doi:10.1016/j.bmc.2009.01.011 |
| 3. | Hao, Y.; Guo, J.; Wang, Z.; Liu, Y.; Li, Y.; Ma, D.; Wang, Q. J. Agric. Food Chem. 2020, 68, 5586–5595. doi:10.1021/acs.jafc.0c02101 |
| 8. | Guaitoli, V.; Alvarez-Ginarte, Y. M.; Montero-Cabrera, L. A.; Bencomo-Martínez, A.; Pérez Badel, Y.; Giorgetti, A.; Suku, E. J. Mol. Model. 2020, 26, 222. doi:10.1007/s00894-020-04470-w |
| 17. | Attanasi, O. A.; De Crescentini, L.; Favi, G.; Nicolini, S.; Perrulli, F. R.; Santeusanio, S. Org. Lett. 2011, 13, 353–355. doi:10.1021/ol102664n |
| 18. | Attanasi, O. A.; Bartoccini, S.; Favi, G.; Giorgi, G.; Perrulli, F. R.; Santeusanio, S. J. Org. Chem. 2012, 77, 1161–1167. doi:10.1021/jo2021949 |
| 19. | Attanasi, O. A.; De Crescentini, L.; Filippone, P.; Giorgi, G.; Nicolini, S.; Perrulli, F. R.; Santeusanio, S. Tetrahedron 2014, 70, 7336–7343. doi:10.1016/j.tet.2014.07.038 |
| 7. | Meng, Z.; Siddiqui, M. A.; Reddy, P. A. P. Imidazopyrazine Derivatives As Inhibitors of mTOR. International Pat. Appl. WO2013016160A1, Jan 31, 2013. |
| 21. | Mari, G.; De Crescentini, L.; Favi, G.; Mantellini, F.; Santeusanio, S. Eur. J. Org. Chem. 2022, e202201053. doi:10.1002/ejoc.202201053 |
| 5. | Goel, R.; Luxami, V.; Paul, K. Org. Biomol. Chem. 2015, 13, 3525–3555. doi:10.1039/c4ob01380h |
| 6. | Mulvihill, M. J.; Ji, Q.-S.; Coate, H. R.; Cooke, A.; Dong, H.; Feng, L.; Foreman, K.; Rosenfeld-Franklin, M.; Honda, A.; Mak, G.; Mulvihill, K. M.; Nigro, A. I.; O’Connor, M.; Pirrit, C.; Steinig, A. G.; Siu, K.; Stolz, K. M.; Sun, Y.; Tavares, P. A. R.; Yao, Y.; Gibson, N. W. Bioorg. Med. Chem. 2008, 16, 1359–1375. doi:10.1016/j.bmc.2007.10.061 |
| 20. | Attanasi, O. A.; Favi, G.; Giorgi, G.; Majer, R.; Perrulli, F. R.; Santeusanio, S. Org. Biomol. Chem. 2014, 12, 4610–4619. doi:10.1039/c4ob00676c |
| 21. | Mari, G.; De Crescentini, L.; Favi, G.; Mantellini, F.; Santeusanio, S. Eur. J. Org. Chem. 2022, e202201053. doi:10.1002/ejoc.202201053 |
| 42. |
Shindoh, N.; Takemoto, Y.; Takasu, K. Chem. – Eur. J. 2009, 15, 12168–12179. doi:10.1002/chem.200901486
And references cited therein. |
| 43. | Long, J.; Yu, R.; Gao, J.; Fang, X. Angew. Chem., Int. Ed. 2020, 59, 6785–6789. doi:10.1002/anie.202000704 |
| 4. | McGrath, N. A.; Brichacek, M.; Njardarson, J. T. J. Chem. Educ. 2010, 87, 1348–1349. doi:10.1021/ed1003806 |
| 22. |
Lopes, S. M. M.; Cardoso, A. L.; Lemos, A.; Pinho e Melo, T. M. V. D. Chem. Rev. 2018, 118, 11324–11352. doi:10.1021/acs.chemrev.8b00375
See for review on DDs. |
| 23. | Attanasi, O. A.; De Crescentini, L.; Favi, G.; Filippone, P.; Mantellini, F.; Perrulli, F. R.; Santeusanio, S. Eur. J. Org. Chem. 2009, 3109–3127. doi:10.1002/ejoc.200900243 |
| 12. | Carpino, P. A.; Cole, B. M.; Morgan, B. P. Melanocortin receptor agonists. Eur. Pat. Appl. EP1294719A1, March 3, 2003. |
| 16. |
Campos, K. R. Chem. Soc. Rev. 2007, 36, 1069–1084. doi:10.1039/b607547a
See for an excellent review on the direct functionalization of sp3 C–H bonds adjacent to nitrogen in heterocycles. |
| 32. | Ishiuchi, K.; Kubota, T.; Hoshino, T.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2006, 14, 5995–6000. doi:10.1016/j.bmc.2006.05.028 |
| 33. | Jiang, X.; Williams, N.; De Brabander, J. K. Org. Lett. 2007, 9, 227–230. doi:10.1021/ol062656o |
| 34. | Rech, J. C.; Floreancig, P. E. Org. Lett. 2005, 7, 5175–5178. doi:10.1021/ol0520267 |
| 35. | He, F.; Bo, Y.; Altom, J. D.; Corey, E. J. J. Am. Chem. Soc. 1999, 121, 6771–6772. doi:10.1021/ja9915201 |
| 36. | Xu, Z.; Wang, Q.; Zhu, J. J. Am. Chem. Soc. 2015, 137, 6712–6724. doi:10.1021/jacs.5b03619 |
| 37. | Zhou, F.; Zeng, X.-P.; Wang, C.; Zhao, X.-L.; Zhou, J. Chem. Commun. 2013, 49, 2022–2024. doi:10.1039/c3cc38819k |
| 11. | Bettati, M.; Chambers, M. S.; Goodacre, S. C.; Hallett, D. J.; Russel, M. G.; Street, L. J. Imidazo-pyrazine derivatives as ligands for GABA receptors. International Pat. Appl. WO03093272A1, May 2, 2002. |
| 17. | Attanasi, O. A.; De Crescentini, L.; Favi, G.; Nicolini, S.; Perrulli, F. R.; Santeusanio, S. Org. Lett. 2011, 13, 353–355. doi:10.1021/ol102664n |
| 18. | Attanasi, O. A.; Bartoccini, S.; Favi, G.; Giorgi, G.; Perrulli, F. R.; Santeusanio, S. J. Org. Chem. 2012, 77, 1161–1167. doi:10.1021/jo2021949 |
| 19. | Attanasi, O. A.; De Crescentini, L.; Filippone, P.; Giorgi, G.; Nicolini, S.; Perrulli, F. R.; Santeusanio, S. Tetrahedron 2014, 70, 7336–7343. doi:10.1016/j.tet.2014.07.038 |
| 38. |
Huang, Y.-Y.; Cai, C.; Yang, X.; Lv, Z.-C.; Schneider, U. ACS Catal. 2016, 6, 5747–5763. doi:10.1021/acscatal.6b01725
And references cited therein. |
| 39. | Wang, W.; Huang, H. Chem. Commun. 2019, 55, 3947–3950. doi:10.1039/c9cc01374a |
| 40. | Chen, A.; Yu, H.; Yan, J.; Huang, H. Org. Lett. 2020, 22, 755–759. doi:10.1021/acs.orglett.9b04630 |
| 41. | Hou, J.; Xue, L.; Yu, T.; Li, J.; Yu, S.; Yao, C.; Li, Y.-M. Chem. Commun. 2023, 59, 7096–7099. doi:10.1039/d3cc01527k |
| 10. | Hartz, R. A.; Gilligan, P. J.; Nanda, K. K.; Tebben, A. J.; Fitzgerald, L. W.; Miller, K. Bioorg. Med. Chem. Lett. 2002, 12, 291–294. doi:10.1016/s0960-894x(01)00741-7 |
| 30. | Bauer, I.; Knölker, H.-J. Chem. Rev. 2015, 115, 3170–3387. doi:10.1021/cr500425u |
| 31. | Mari, G.; Corrieri, M.; De Crescentini, L.; Favi, G.; Santeusanio, S.; Mantellini, F. Eur. J. Org. Chem. 2021, 5202–5208. doi:10.1002/ejoc.202101046 |
| 9. | Mulvihill, M. J.; Ji, Q.-S.; Werner, D.; Beck, P.; Cesario, C.; Cooke, A.; Cox, M.; Crew, A.; Dong, H.; Feng, L.; Foreman, K. W.; Mak, G.; Nigro, A.; O’Connor, M.; Saroglou, L.; Stolz, K. M.; Sujka, I.; Volk, B.; Weng, Q.; Wilkes, R. Bioorg. Med. Chem. Lett. 2007, 17, 1091–1097. doi:10.1016/j.bmcl.2006.11.016 |
| 13. | Gopalsamy, A.; Shi, M. Org. Lett. 2003, 5, 3907–3909. doi:10.1021/ol035455i |
| 14. | Trček, T.; Verček, B. Synthesis 2006, 3437–3442. doi:10.1055/s-2006-950211 |
| 15. | Shaabani, A.; Maleki, A.; Mofakham, H. Mol. Diversity 2009, 13, 63–67. doi:10.1007/s11030-008-9099-3 |
| 17. | Attanasi, O. A.; De Crescentini, L.; Favi, G.; Nicolini, S.; Perrulli, F. R.; Santeusanio, S. Org. Lett. 2011, 13, 353–355. doi:10.1021/ol102664n |
| 24. | Corrieri, M.; De Crescentini, L.; Mantellini, F.; Mari, G.; Santeusanio, S.; Favi, G. J. Org. Chem. 2021, 86, 17918–17929. doi:10.1021/acs.joc.1c02217 |
| 25. | Mei, G.-J.; Tang, X.; Tasdan, Y.; Lu, Y. Angew. Chem., Int. Ed. 2020, 59, 648–652. doi:10.1002/anie.201911686 |
© 2024 Mari et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.