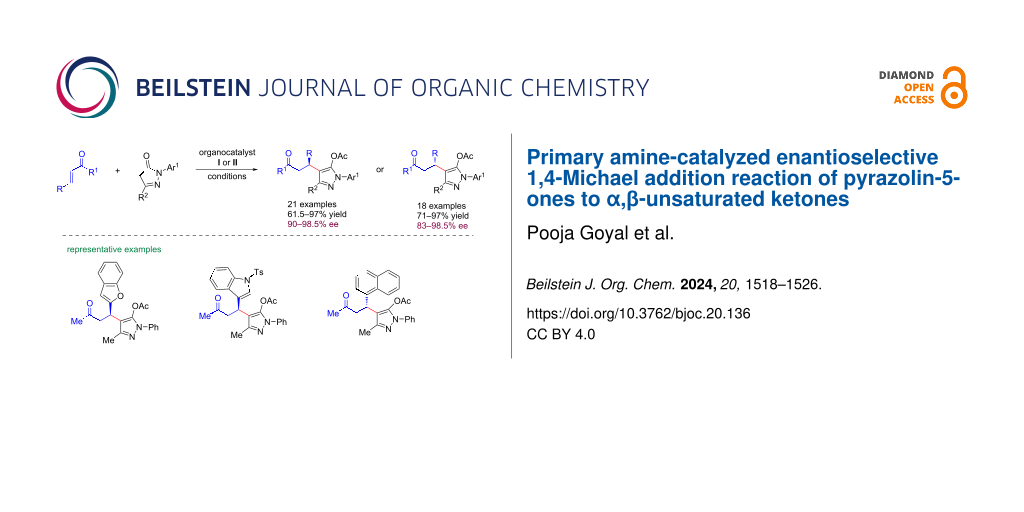
Abstract
The enantioselective 1,4-addition reaction of pyrazolin-5-ones to α,β-unsaturated ketones catalyzed by a cinchona alkaloid-derived primary amine–Brønsted acid composite is reported. Both enantiomers of the anticipated pyrazole derivatives were obtained in good to excellent yields (up to 97%) and high enantioselectivities (up to 98.5% ee) under mild reaction conditions. In addition, this protocol was further expanded to synthesize highly enantioenriched hybrid molecules bearing biologically relevant heterocycles.

Graphical Abstract
Introduction
N-Heterocycles are attractive molecules owing to their extensive applications in small-molecule drugs, natural products, and agrochemical products [1-3]. Among the N-heterocycles, pyrazole is an important structural scaffold, found in several marketed drugs and bioactive molecules (Figure 1) [4-7]. In addition, this moiety is an integral part of various agrochemical products and chelating agents [4-9]. Given the importance and widespread applications of pyrazoles, considerable efforts have been devoted to develop new protocols to access structurally diverse pyrazole derivatives [4-7,10-12].
![[1860-5397-20-136-1]](/bjoc/content/figures/1860-5397-20-136-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected examples of drugs and bioactive molecules bearing a pyrazole core.
Figure 1: Selected examples of drugs and bioactive molecules bearing a pyrazole core.
4-Unsubstituted pyrazolin-5-ones are well known precursors for the construction of optically active structurally diverse pyrazoles [10-12]. In this context, the organocatalyzed asymmetric Michael addition of 4-unsubstituted pyrazolin-5-ones to a variety of Michael acceptors has emerged as one of the most powerful strategies to access enantioenriched pyrazole derivatives [10-21]. In the majority of these cases, the reactivities of the pyrazolin-5-one derivatives were harnessed under non-covalent catalysis via bifunctional hydrogen-bonding organocatalysts. The C-4 nucleophilicity of pyrazolin-5-ones was also explored in enantioselective reactions with α,β-unsaturated carbonyl compounds through covalent catalysis with chiral amine-based catalysts; however, it has achieved limited success [10-21].
Among the developed organocatalyzed enantioselective 1,4-addition reactions of pyrazolin-5-ones, the catalytic asymmetric reactions of pyrazolin-5-ones with α,β-unsaturated ketones are comparatively less studied. In 2009, Zhao’s group were the first who reported a chiral amine-catalysed aza-Michael addition reaction of pyrazolin-5-ones with α,β-unsaturated ketones to access β-(3-hydroxypyrazol-1-yl)ketones (Scheme 1a) [22]. The developed reaction was restricted to α,β-unsaturated ketones with aliphatic substituents (Scheme 1a) [22]. Ji and Wang disclosed organocatalyzed [5 + 1] double Michael additions between pyrazolones and dienones (Scheme 1b) [23]. Very recently, the Chimni group reported a cinchona-derived squaramide-catalyzed 1,4-Michael addition reaction of pyrazolin-5-ones with 2-enoylpyridines (Scheme 1c) [24]. Recently, we developed an organocatalyzed asymmetric Michael addition reaction of 4-monosubstituted pyrazol-5-ones to simple enones for the synthesis of pyrazolone derivatives [25]. Despite these progresses, arylidene/heteroarylideneacetones have remained untapped by 4-unsubstituted pyrazolin-5-ones under asymmetric organocatalytic or metal catalytic conditions. In continuation of our work in the field of organocatalysis [26-29], herein, we present the Michael addition reaction of 4-unsubstituted pyrazolin-5-ones with arylidene/heteroarylideneacetones using cinchona alkaloid-derived primary amine catalysts. The developed protocol delivered both enantiomers of the desired products in good to excellent yields and enantioselectivities.
![[1860-5397-20-136-i1]](/bjoc/content/inline/1860-5397-20-136-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Representative examples of asymmetric organocatalytic conjugate addition of pyrazolin-5-ones to α,β-unsaturated ketones and present study.
Scheme 1: Representative examples of asymmetric organocatalytic conjugate addition of pyrazolin-5-ones to α,β...
Results and Discussion
At the outset, the reaction between commercially available benzylideneacetone (1a) and 3-methyl-1-phenyl-2-pyrazolin-5-one (2a) was studied in the presence of a panel of primary amine catalysts (see Table S1 in Supporting Information File 1) in toluene at room temperature (30–32 °C). When the test reaction was conducted in the presence of 15 mol % of 9-amino-9-deoxy-epicinchonidine (I) as catalyst [30] for 12 h and treated with Ac2O followed by DABCO, the reaction gave the conjugate addition product 3aa in 58–62% yield with 74% ee (Table 1, entry 1). On the other hand, 9-amino-9-deoxyepicinchonine (II) [30] furnished the opposite enantiomer ent-3aa in 62% yield and 66% ee (Table 1, entry 2). Among the screened organocatalysts (see Table S1 in Supporting Information File 1), the catalyst I imparted the highest enantioselectivity (74% ee) of the Michael product 3aa (Table 1, entry 1). Different solvents (see details in Supporting Information File 1) were screened for the test reaction using 15 mol % of catalyst I. Among them, CHCl3 turned out to be the optimal solvent, as the product 3aa was isolated in reproducible yield (77%) and enantioselectivity 74% ee (Table 1, entry 3). Next, we explored a variety of achiral and/or chiral Brønsted acids A1–6 as additives in order to increase the yield and the enantioselectivity of the reaction (Table 1, entries 4–9). A marked increase in both the yield and enantioselectivity of the product 3aa were observed. Among the screened Brønsted acids A1–6, the combination of 15 mol % of the catalyst I and 30 mol % of (±)-mandelic acid (A5) was found to be superior in terms of enantioselectivity (92% ee) of the product 3aa (Table 1, entry 8). When the catalyst loading was lowered (10 mol % of I/20 mol % of A5), the desired product 3aa was obtained in 71% yield and 91% ee (Table 1, entry 10). Lowering the temperature (−20 °C) of the reaction improved the enantioselectivity of the product 3aa slightly but decreased its yield (Table 1, entry 11). Subsequently, the effect of concentration on the reaction outcome was also studied. In dilute conditions, both the yield and enantioselectivity of the product 3aa were improved to 80% and 94%, respectively, at room temperature (Table 1, entry 12).
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-136-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | Cat. | Additive | Yield (%)b | ee (%)c |
| 1 | I | – | 58d | 74 |
| 2 | II | – | 62 | −66 |
| 3 | I | – | 77 | 74 |
| 4 | I | A1 | 80 | 90 |
| 5 | I | A2 | 76 | 88.5 |
| 6 | I | A3 | 86 | 83 |
| 7 | I | A4 | 83 | 87 |
| 8 | I | A5 | 77 | 92 |
| 9 | I | A6 | 77 | 90 |
| 10e | I | A5 | 71 | 91 |
| 11f | I | A5 | 58 | 94 |
| 12g | I | A5 | 80 | 94 |
| 13g | II | A5 | 76 | −87.5 |
aReaction conditions: 1a (0.3 mmol), 2a (0.2 mmol), 15 mol % of catalyst I or II in 0.5 mL toluene (entries 1 and 2) or catalyst I (15 mol %) and 30 mol % A in 0.5 mL CHCl3 (entries 3–9) for 12–14 h. Next, Ac2O (0.52 mmol, 50 µL) was added followed by DABCO (0.1 mmol, 11 mg) and the reaction mixture was further stirred for 2 h at 30–32 °C. bIsolated yield of 3aa or ent-3aa after column chromatography. cEnantiomeric excess (ee) was measured by HPLC analysis using a chiralcel OD-H column. dThe yield of the reaction product varied from 58–62%. eThe reaction was performed in the presence of 10 mol % I and 20 mol % A5. fThe reaction was performed at −20 °C using 15 mol % of catalyst I in combination with 30 mol % A5 in 0.5 mL CHCl3 for 24 h. gThe reaction was carried out using 15 mol % of catalyst I or II in combination with 30 mol % A5 in 1.0 mL CHCl3 for 14 h at 30–32 °C. Next, Ac2O (0.52 mmol, 50 µL) was added followed by DABCO (0.1 mmol, 11 mg) and the reaction mixture was further stirred for 2 h at 30–32 °C.
Taking into account the results of the optimization studies mentioned above, the catalytic system I (15 mol %)/A5 (30 mol %) in CHCl3 (1 mL) at room temperature (30–32 °C) was selected as the optimum reaction conditions (Table 1, entry 12). Under identical optimized reaction conditions, the catalytic system II (15 mol %)/A5 (30 mol %) furnished ent-3aa in 76% yield and 87.5% ee (Table 1, entry 13).
With the optimal reaction conditions at hands, the 1,4-conjugate addition reaction of a series of α,β-unsaturated ketones 1 with pyrazolin-5-one (2a) were studied next (Scheme 2). Aryl α,β-unsaturated ketones bearing a halogen, electron-withdrawing, or electron-donating group at the para-position of the benzene ring were compatible and led to the corresponding products 3ba–fa in good to excellent yields (72–97%) and enantioselectivities (90–95% ee). The α,β-unsaturated ketone 1f with a strong electron-withdrawing group (cyano) in the para-position of the benzene ring, was found to be more reactive as the reaction was completed within 4 h and the desired Michael adduct 3fa was isolated in 89% yield and 92% ee. Notably, the α,β-unsaturated ketone with a substituent in the meta-position of the benzene ring was also tolerated and the desired product 3ga was isolated in good yield (82%) and excellent enantioselectivity (95% ee). To our delight, the α,β-unsaturated ketone with a substituent in the ortho-position of the benzene ring, led to the product 3ha in good yield (76.5%) and highest enantioselectivity (98.5% ee). Moreover, 1-naphthyl-substituted and 2-thienyl-substituted α,β-unsaturated ketones also took part in the reaction and the desired products (3ia and 3ja) were isolated in good yields (77.5% and 80%, respectively) and enantioselectivities (92% ee and 90% ee, respectively). Interestingly, the reaction also worked well with (E)-1-phenylpent-1-en-3-one (1k) as α,β-unsaturated ketone. The corresponding product, 3ka was obtained in 91% yield and 95% ee. Moreover, ethyl (E)-5-oxohex-2-enoate (1l) also showed good reactivity and the expected product (−)-3la was isolated in 68% yield and 95% ee.
![[1860-5397-20-136-i2]](/bjoc/content/inline/1860-5397-20-136-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of substrates. Reaction conditions: 1 (0.3 mmol), 2 (0.2 mmol), 15 mol % of catalyst I, 30 mol % A5 (for 3) or 15 mol % catalyst II, 30 mol % A5 (for ent-3) in 1.0 mL CHCl3 for 4–14 h. Next, Ac2O (0.52 mmol, 50 µL) was added followed by DABCO (0.1 mmol, 11 mg) and the reaction mixture was further stirred for 2 h at 30–32 °C. aIsolated yield of 3 or ent-3 after column chromatography. bEnantiomeric excess (ee) was measured by HPLC analysis using a stationary phase chiral column. cValues in parentheses represent % ee after single recrystallization. dReaction time for the first step was 4 h.
Scheme 2: Scope of substrates. Reaction conditions: 1 (0.3 mmol), 2 (0.2 mmol), 15 mol % of catalyst I, 30 mo...
Next, we explored the scope of pyrazolin-5-ones 2 (Scheme 2, lower part) with diverse substituents (Br, Cl, F, Me or CN) in the para-position of the N-aryl group. These substrates reacted smoothly with α,β-unsaturated ketone 1c and the corresponding products 3cb–cf were obtained in good yields (61–85.5%) and good to excellent enantioselectivities (84.5–95% ee). In addition, pyrazolone 2g with a substituent in the meta-position of the N-aryl group also participated in the reaction and the desired product 3cg was isolated in 87% yield and 95% ee. Notably, a phenyl substituent at the C3 position of pyrazolone 2h was found to be compatible, and the desired product 3ch was obtained in 87% yield and 90% ee.
In general, enantiomers of a bioactive molecule have different biological activities. Therefore, there is a huge demand to develop methods to access both enantiomers of a chiral compound. We turned our attention to the synthesis of enantiomeric products ent-3. Under identical optimized reaction conditions (Table 1, entry 12), a panel of aryl/heteroaryl α,β-unsaturated ketones 1 and pyrazolin-5-ones 2 were studied (Scheme 2) using the catalytic system II (15 mol %)/A5 (30 mol %). To our delight, the enantiomeric products ent-3aa–ent-3cg (Scheme 2) were obtained in good to excellent yields (71–97%) and enantioselectivities (83.5–98% ee).
Molecules containing two or more biologically relevant heterocycle motifs are receiving attention in drug discovery research [31-33]. The enantioselective synthesis of such hybrid molecules is fascinating but at the same time challenging. Pyrazoles [4-7], benzofurans [34], and indoles [35,36] are popular scaffolds as they are prevalent in many bioactive molecules. Compounds bearing both pyrazole and indole moieties or pyrazole and benzofuran moieties (Figure 1) are highly attractive since such compounds might be endowed with potent biological activities.
Under the disclosed optimized reaction conditions, the reaction between pyrazolin-5-one (2a) and indole-derived α,β-unsaturated ketone 1m was performed. The resulting hybrid molecule 3ma was isolated in 96% yield and 90% ee (Scheme 3). On the other hand, the reaction of pyrazolin-5-one (2a) with benzofuran-derived α,β-unsaturated ketone 1n delivered the product 3na in 85.5% yield and 95.5% ee (Scheme 3). Moreover, by employing the catalytic composite II (15 mol %) and A5 (30 mol %) under otherwise identical optimized reaction conditions, the corresponding enantiomeric products (ent-3ma and ent-3na) were obtained (Scheme 3) in good yields (91.5% and 85.5%, respectively) and enantioselectivities (84% ee and 91.5% ee, respectively).
![[1860-5397-20-136-i3]](/bjoc/content/inline/1860-5397-20-136-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of pyrazole-benzofuran and pyrazole–indole hybrid molecules. Reaction conditions: 1m or 1n (0.3 mmol), 2a (0.2 mmol), 15 mol % of catalyst I, 30 mol % A5 (for 3) or 15 mol % catalyst II, 30 mol % A5 (for ent-3) in 1.0 mL CHCl3 for 14 h. Next, Ac2O (0.52 mmol, 50 µL) was added followed by DABCO (0.1 mmol, 11 mg) and the reaction mixture was further stirred for 2 h at 30–32°C. aIsolated yield of 3 or ent-3 after column chromatography. bEnantiomeric excess (ee) was measured by HPLC analysis using a stationary phase chiral column.
Scheme 3: Synthesis of pyrazole-benzofuran and pyrazole–indole hybrid molecules. Reaction conditions: 1m or 1n...
The practical utility of the developed method was demonstrated by carrying out the synthesis of 3aa on a 1 mmol scale under the optimized reaction conditions (Scheme 4). The product 3aa was isolated in slightly lower yield and similar enantioselectivity compared to the 0.2 mmol scale reaction.
![[1860-5397-20-136-i4]](/bjoc/content/inline/1860-5397-20-136-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 3aa on preparative scale.
Scheme 4: Synthesis of 3aa on preparative scale.
Subsequently, we turned our attention to determine the absolute configuration of the newly formed chiral center. Under the disclosed optimized conditions, the product ent-3ba was isolated as white solid with 85% ee and the enantiopurity of the product could be enriched to 98% ee by single recrystallization. The absolute stereochemistry was determined to be “R” on the basis of single-crystal X-ray crystallography data of ent-3ba (Figure 2) [37]. The stereochemistry of the products in this series was assigned by analogy.
![[1860-5397-20-136-2]](/bjoc/content/figures/1860-5397-20-136-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Single crystal X-ray structure of ent-3ba (CCDC 2234286).
Figure 2: Single crystal X-ray structure of ent-3ba (CCDC 2234286).
Based on the observed absolute configuration of product ent-3ba and preceding literature reports [38,40], a plausible mechanistic pathway is outlined in Scheme 5. Initially, in the presence of one equivalent Brønsted acid additive A5, the catalyst II generates the monoprotonated diamine II-A5. The condensation of the primary amine moiety in II-A5 with the carbonyl group of the α,β-unsaturated ketone 1b in presence of the Brønsted acid leads to the formation of the iminium ion assembly 4 (Scheme 5). It is known that Brønsted acids facilitate the iminium ion formation step [38,39] and the counteranion of the acid plays an important role in the stereocontrolling event [38,40]. On the other hand, the protonated quinuclidine nitrogen atom of the catalyst II (in the iminium ion assembly) activates the pyrazol-5-one 2a through hydrogen bonding and forms the corresponding enol. Simultaneously, the enol form of the pyrazol-5-one attacks the Re-face of the α,β-unsaturated ketone 1b to provide the intermediate 5 (Scheme 5), which after hydrolysis leads to product ent-3ba'. In situ acetylation of the ent-3ba' using acetic anhydride and DABCO, furnishes the desired product ent-3ba.
![[1860-5397-20-136-i5]](/bjoc/content/inline/1860-5397-20-136-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have realized the Michael addition reaction of 4-unsubstituted pyrazolin-5-ones to α,β-unsaturated ketones under organocatalytic conditions. The developed protocol was efficiently applied to diverse α,β-unsaturated ketones and a pool of pyrazolin-5-ones. The formed Michael adducts were isolated in good to excellent yields and enantioselectivities. The method also led to enantioenriched hybrid molecules bearing pyrazole–indole moieties and pyrazole–benzofuranone moieties. It is worth mentioning that the current protocol delivers both enantiomers of the Michael adducts.
Experimental
General procedure for the synthesis of 3 and ent-3.
In an oven-dried 4 mL glass vial fitted with a magnetic stirring bar, the mixture of catalyst I (15 mol %, ≈9.0 mg) and (±)-mandelic acid (30 mol %, 9.0 mg) or catalyst II (15 mol %, ≈9.0 mg) and (±)-mandelic acid (30 mol %) in CHCl3 (1.0 mL) was stirred at room temperature (30–32 °C) for 5 min. Next, α,β-unsaturated ketone (0.3 mmol, 1.5 equiv) was added in one portion and the reaction mixture was further stirred for 5 min. Then, the pyrazolin-5-one 2 (0.2 mmol, 1.0 equiv) was added to the mixture and stirred for 4–14 h. Once the pyrazolin-5-one 2 was consumed (monitored by TLC), Ac2O (50 µL, ≈0.52 mmol, 2.6 equiv) and DABCO (11 mg, 50 mol %) were sequentially added. The resulting reaction mixture was further stirred for 2 h at room temperature. The crude reaction mixture was purified by silica gel (230–400 mesh) column chromatography (petroleum ether/EtOAc as the eluent) to give the product 3 or ent-3.
Supporting Information
| Supporting Information File 1: Additional optimization studies, characterization data of compounds 3aa–na and ent-3aa-ent-3na, 1H, 13C NMR spectra of 3aa–na, 1H NMR of ent-3aa–ent-3na and their HPLC traces and single crystal data of ent-3ba. | ||
| Format: PDF | Size: 8.0 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b
Return to citation in text: [1] -
Rajput, A. P.; Kankhare, A. R. Int. J. Pharm. Sci. Invent. 2017, 6, 19.
Return to citation in text: [1] -
Vinogradov, M. G.; Turova, O. V.; Zlotin, S. G. Org. Biomol. Chem. 2019, 17, 3670–3708. doi:10.1039/c8ob03034k
Return to citation in text: [1] -
Schmidt, A.; Dreger, A. Curr. Org. Chem. 2011, 15, 1423–1463. doi:10.2174/138527211795378263
Return to citation in text: [1] [2] [3] [4] -
Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. Chem. Rev. 2011, 111, 6984–7034. doi:10.1021/cr2000459
Return to citation in text: [1] [2] [3] [4] -
Kumar, V.; Kaur, K.; Gupta, G. K.; Sharma, A. K. Eur. J. Med. Chem. 2013, 69, 735–753. doi:10.1016/j.ejmech.2013.08.053
Return to citation in text: [1] [2] [3] [4] -
Neto, J. S. S.; Zeni, G. Chem. – Eur. J. 2020, 26, 8175–8189. doi:10.1002/chem.201905276
Return to citation in text: [1] [2] [3] [4] -
Stricker, L.; Fritz, E.-C.; Peterlechner, M.; Doltsinis, N. L.; Ravoo, B. J. J. Am. Chem. Soc. 2016, 138, 4547–4554. doi:10.1021/jacs.6b00484
Return to citation in text: [1] -
Orrego-Hernández, J.; Cobo, J.; Portilla, J. ACS Omega 2019, 4, 16689–16700. doi:10.1021/acsomega.9b02796
Return to citation in text: [1] -
Chauhan, P.; Mahajan, S.; Enders, D. Chem. Commun. 2015, 51, 12890–12907. doi:10.1039/c5cc04930j
Return to citation in text: [1] [2] [3] [4] -
Liu, S.; Bao, X.; Wang, B. Chem. Commun. 2018, 54, 11515–11529. doi:10.1039/c8cc06196c
Return to citation in text: [1] [2] [3] [4] -
Bao, X.; Wang, X.; Tian, J.-M.; Ye, X.; Wang, B.; Wang, H. Org. Biomol. Chem. 2022, 20, 2370–2386. doi:10.1039/d1ob02426d
Return to citation in text: [1] [2] [3] [4] -
Rao, K. S.; Ramesh, P.; Trivedi, R.; Kantam, M. L. Tetrahedron Lett. 2016, 57, 1227–1231. doi:10.1016/j.tetlet.2016.02.008
Return to citation in text: [1] [2] -
Kim, Y. H.; Yoon, J. H.; Lee, M. Y.; Kim, D. Y. Bull. Korean Chem. Soc. 2017, 38, 1242–1245. doi:10.1002/bkcs.11241
Return to citation in text: [1] [2] -
Phelan, J. P.; Ellman, J. A. Adv. Synth. Catal. 2016, 358, 1713–1718. doi:10.1002/adsc.201600110
Return to citation in text: [1] [2] -
Sharma, A.; Sharma, V.; Chimni, S. S. Org. Biomol. Chem. 2019, 17, 9514–9523. doi:10.1039/c9ob01700c
Return to citation in text: [1] [2] -
Yang, M.; Zhang, M.; Wang, Z.; Tang, L.; Chen, W.; Ban, S.; Li, Q. Chirality 2018, 30, 1096–1104. doi:10.1002/chir.23003
Return to citation in text: [1] [2] -
Aydin, A. E.; Culha, S. Chirality 2021, 33, 106–114. doi:10.1002/chir.23295
Return to citation in text: [1] [2] -
Sharma, V.; Kaur, J.; Chimni, S. S. Eur. J. Org. Chem. 2018, 3489–3495. doi:10.1002/ejoc.201800589
Return to citation in text: [1] [2] -
Carceller-Ferrer, L.; Vila, C.; Blay, G.; Fernández, I.; Muñoz, M. C.; Pedro, J. R. Org. Biomol. Chem. 2019, 17, 9859–9863. doi:10.1039/c9ob02252j
Return to citation in text: [1] [2] -
Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. Asian J. Org. Chem. 2021, 10, 1173–1183. doi:10.1002/ajoc.202100120
Return to citation in text: [1] [2] -
Gogoi, S.; Zhao, C.-G.; Ding, D. Org. Lett. 2009, 11, 2249–2252. doi:10.1021/ol900538q
Return to citation in text: [1] [2] -
Wu, B.; Chen, J.; Li, M.-Q.; Zhang, J.-X.; Xu, X.-P.; Ji, S.-J.; Wang, X.-W. Eur. J. Org. Chem. 2012, 1318–1327. doi:10.1002/ejoc.201101529
Return to citation in text: [1] -
Sharma, V.; Kaur, A.; Sahoo, S. C.; Chimni, S. S. Org. Biomol. Chem. 2018, 16, 6470–6478. doi:10.1039/c8ob01588k
Return to citation in text: [1] -
Goyal, P.; Dubey, A. K.; Chowdhury, R. Eur. J. Org. Chem. 2024, 27, e202400002. doi:10.1002/ejoc.202400002
Return to citation in text: [1] -
Chowdhury, R.; Kumar, M.; Ghosh, S. K. Org. Biomol. Chem. 2016, 14, 11250–11260. doi:10.1039/c6ob02104b
Return to citation in text: [1] -
Vamisetti, G. B.; Chowdhury, R.; Ghosh, S. K. Org. Biomol. Chem. 2017, 15, 3869–3873. doi:10.1039/c7ob00796e
Return to citation in text: [1] -
Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. Eur. J. Org. Chem. 2020, 2962–2972. doi:10.1002/ejoc.202000306
Return to citation in text: [1] -
Dubey, A. K.; Chowdhury, R. Beilstein J. Org. Chem. 2021, 17, 2642–2649. doi:10.3762/bjoc.17.177
Return to citation in text: [1] -
Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s
Return to citation in text: [1] [2] -
Shaveta; Mishra, S.; Singh, P. Eur. J. Med. Chem. 2016, 124, 500–536. doi:10.1016/j.ejmech.2016.08.039
Return to citation in text: [1] -
Viegas-Junior, C.; Danuello, A.; da Silva Bolzani, V.; Barreiro, E. J.; Fraga, C. A. M. Curr. Med. Chem. 2007, 14, 1829–1852. doi:10.2174/092986707781058805
Return to citation in text: [1] -
Fu, R.-g.; Sun, Y.; Sheng, W.-b.; Liao, D.-f. Eur. J. Med. Chem. 2017, 136, 195–211. doi:10.1016/j.ejmech.2017.05.016
Return to citation in text: [1] -
Khanam, H.; Shamsuzzaman. Eur. J. Med. Chem. 2015, 97, 483–504. doi:10.1016/j.ejmech.2014.11.039
Return to citation in text: [1] -
Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Eur. J. Med. Chem. 2019, 183, 111691. doi:10.1016/j.ejmech.2019.111691
Return to citation in text: [1] -
Thanikachalam, P. V.; Maurya, R. K.; Garg, V.; Monga, V. Eur. J. Med. Chem. 2019, 180, 562–612. doi:10.1016/j.ejmech.2019.07.019
Return to citation in text: [1] -
The crystallographic data (CCDC 2234286) for ent-3ba, can be obtained free of charge from the Cambridge crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 9748–9770. doi:10.1002/anie.201109036
Return to citation in text: [1] [2] [3] -
Hine, J.; Via, F. A. J. Org. Chem. 1977, 42, 1972–1978. doi:10.1021/jo00431a031
Return to citation in text: [1] -
Moran, A.; Hamilton, A.; Bo, C.; Melchiorre, P. J. Am. Chem. Soc. 2013, 135, 9091–9098. doi:10.1021/ja404784t
Return to citation in text: [1] [2]
| 35. | Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Eur. J. Med. Chem. 2019, 183, 111691. doi:10.1016/j.ejmech.2019.111691 |
| 36. | Thanikachalam, P. V.; Maurya, R. K.; Garg, V.; Monga, V. Eur. J. Med. Chem. 2019, 180, 562–612. doi:10.1016/j.ejmech.2019.07.019 |
| 4. | Schmidt, A.; Dreger, A. Curr. Org. Chem. 2011, 15, 1423–1463. doi:10.2174/138527211795378263 |
| 5. | Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. Chem. Rev. 2011, 111, 6984–7034. doi:10.1021/cr2000459 |
| 6. | Kumar, V.; Kaur, K.; Gupta, G. K.; Sharma, A. K. Eur. J. Med. Chem. 2013, 69, 735–753. doi:10.1016/j.ejmech.2013.08.053 |
| 7. | Neto, J. S. S.; Zeni, G. Chem. – Eur. J. 2020, 26, 8175–8189. doi:10.1002/chem.201905276 |
| 34. | Khanam, H.; Shamsuzzaman. Eur. J. Med. Chem. 2015, 97, 483–504. doi:10.1016/j.ejmech.2014.11.039 |
| 1. | Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b |
| 2. | Rajput, A. P.; Kankhare, A. R. Int. J. Pharm. Sci. Invent. 2017, 6, 19. |
| 3. | Vinogradov, M. G.; Turova, O. V.; Zlotin, S. G. Org. Biomol. Chem. 2019, 17, 3670–3708. doi:10.1039/c8ob03034k |
| 10. | Chauhan, P.; Mahajan, S.; Enders, D. Chem. Commun. 2015, 51, 12890–12907. doi:10.1039/c5cc04930j |
| 11. | Liu, S.; Bao, X.; Wang, B. Chem. Commun. 2018, 54, 11515–11529. doi:10.1039/c8cc06196c |
| 12. | Bao, X.; Wang, X.; Tian, J.-M.; Ye, X.; Wang, B.; Wang, H. Org. Biomol. Chem. 2022, 20, 2370–2386. doi:10.1039/d1ob02426d |
| 30. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s |
| 4. | Schmidt, A.; Dreger, A. Curr. Org. Chem. 2011, 15, 1423–1463. doi:10.2174/138527211795378263 |
| 5. | Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. Chem. Rev. 2011, 111, 6984–7034. doi:10.1021/cr2000459 |
| 6. | Kumar, V.; Kaur, K.; Gupta, G. K.; Sharma, A. K. Eur. J. Med. Chem. 2013, 69, 735–753. doi:10.1016/j.ejmech.2013.08.053 |
| 7. | Neto, J. S. S.; Zeni, G. Chem. – Eur. J. 2020, 26, 8175–8189. doi:10.1002/chem.201905276 |
| 10. | Chauhan, P.; Mahajan, S.; Enders, D. Chem. Commun. 2015, 51, 12890–12907. doi:10.1039/c5cc04930j |
| 11. | Liu, S.; Bao, X.; Wang, B. Chem. Commun. 2018, 54, 11515–11529. doi:10.1039/c8cc06196c |
| 12. | Bao, X.; Wang, X.; Tian, J.-M.; Ye, X.; Wang, B.; Wang, H. Org. Biomol. Chem. 2022, 20, 2370–2386. doi:10.1039/d1ob02426d |
| 31. | Shaveta; Mishra, S.; Singh, P. Eur. J. Med. Chem. 2016, 124, 500–536. doi:10.1016/j.ejmech.2016.08.039 |
| 32. | Viegas-Junior, C.; Danuello, A.; da Silva Bolzani, V.; Barreiro, E. J.; Fraga, C. A. M. Curr. Med. Chem. 2007, 14, 1829–1852. doi:10.2174/092986707781058805 |
| 33. | Fu, R.-g.; Sun, Y.; Sheng, W.-b.; Liao, D.-f. Eur. J. Med. Chem. 2017, 136, 195–211. doi:10.1016/j.ejmech.2017.05.016 |
| 4. | Schmidt, A.; Dreger, A. Curr. Org. Chem. 2011, 15, 1423–1463. doi:10.2174/138527211795378263 |
| 5. | Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. Chem. Rev. 2011, 111, 6984–7034. doi:10.1021/cr2000459 |
| 6. | Kumar, V.; Kaur, K.; Gupta, G. K.; Sharma, A. K. Eur. J. Med. Chem. 2013, 69, 735–753. doi:10.1016/j.ejmech.2013.08.053 |
| 7. | Neto, J. S. S.; Zeni, G. Chem. – Eur. J. 2020, 26, 8175–8189. doi:10.1002/chem.201905276 |
| 8. | Stricker, L.; Fritz, E.-C.; Peterlechner, M.; Doltsinis, N. L.; Ravoo, B. J. J. Am. Chem. Soc. 2016, 138, 4547–4554. doi:10.1021/jacs.6b00484 |
| 9. | Orrego-Hernández, J.; Cobo, J.; Portilla, J. ACS Omega 2019, 4, 16689–16700. doi:10.1021/acsomega.9b02796 |
| 26. | Chowdhury, R.; Kumar, M.; Ghosh, S. K. Org. Biomol. Chem. 2016, 14, 11250–11260. doi:10.1039/c6ob02104b |
| 27. | Vamisetti, G. B.; Chowdhury, R.; Ghosh, S. K. Org. Biomol. Chem. 2017, 15, 3869–3873. doi:10.1039/c7ob00796e |
| 28. | Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. Eur. J. Org. Chem. 2020, 2962–2972. doi:10.1002/ejoc.202000306 |
| 29. | Dubey, A. K.; Chowdhury, R. Beilstein J. Org. Chem. 2021, 17, 2642–2649. doi:10.3762/bjoc.17.177 |
| 4. | Schmidt, A.; Dreger, A. Curr. Org. Chem. 2011, 15, 1423–1463. doi:10.2174/138527211795378263 |
| 5. | Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. Chem. Rev. 2011, 111, 6984–7034. doi:10.1021/cr2000459 |
| 6. | Kumar, V.; Kaur, K.; Gupta, G. K.; Sharma, A. K. Eur. J. Med. Chem. 2013, 69, 735–753. doi:10.1016/j.ejmech.2013.08.053 |
| 7. | Neto, J. S. S.; Zeni, G. Chem. – Eur. J. 2020, 26, 8175–8189. doi:10.1002/chem.201905276 |
| 30. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s |
| 22. | Gogoi, S.; Zhao, C.-G.; Ding, D. Org. Lett. 2009, 11, 2249–2252. doi:10.1021/ol900538q |
| 24. | Sharma, V.; Kaur, A.; Sahoo, S. C.; Chimni, S. S. Org. Biomol. Chem. 2018, 16, 6470–6478. doi:10.1039/c8ob01588k |
| 38. | Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 9748–9770. doi:10.1002/anie.201109036 |
| 39. | Hine, J.; Via, F. A. J. Org. Chem. 1977, 42, 1972–1978. doi:10.1021/jo00431a031 |
| 22. | Gogoi, S.; Zhao, C.-G.; Ding, D. Org. Lett. 2009, 11, 2249–2252. doi:10.1021/ol900538q |
| 25. | Goyal, P.; Dubey, A. K.; Chowdhury, R. Eur. J. Org. Chem. 2024, 27, e202400002. doi:10.1002/ejoc.202400002 |
| 38. | Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 9748–9770. doi:10.1002/anie.201109036 |
| 40. | Moran, A.; Hamilton, A.; Bo, C.; Melchiorre, P. J. Am. Chem. Soc. 2013, 135, 9091–9098. doi:10.1021/ja404784t |
| 10. | Chauhan, P.; Mahajan, S.; Enders, D. Chem. Commun. 2015, 51, 12890–12907. doi:10.1039/c5cc04930j |
| 11. | Liu, S.; Bao, X.; Wang, B. Chem. Commun. 2018, 54, 11515–11529. doi:10.1039/c8cc06196c |
| 12. | Bao, X.; Wang, X.; Tian, J.-M.; Ye, X.; Wang, B.; Wang, H. Org. Biomol. Chem. 2022, 20, 2370–2386. doi:10.1039/d1ob02426d |
| 13. | Rao, K. S.; Ramesh, P.; Trivedi, R.; Kantam, M. L. Tetrahedron Lett. 2016, 57, 1227–1231. doi:10.1016/j.tetlet.2016.02.008 |
| 14. | Kim, Y. H.; Yoon, J. H.; Lee, M. Y.; Kim, D. Y. Bull. Korean Chem. Soc. 2017, 38, 1242–1245. doi:10.1002/bkcs.11241 |
| 15. | Phelan, J. P.; Ellman, J. A. Adv. Synth. Catal. 2016, 358, 1713–1718. doi:10.1002/adsc.201600110 |
| 16. | Sharma, A.; Sharma, V.; Chimni, S. S. Org. Biomol. Chem. 2019, 17, 9514–9523. doi:10.1039/c9ob01700c |
| 17. | Yang, M.; Zhang, M.; Wang, Z.; Tang, L.; Chen, W.; Ban, S.; Li, Q. Chirality 2018, 30, 1096–1104. doi:10.1002/chir.23003 |
| 18. | Aydin, A. E.; Culha, S. Chirality 2021, 33, 106–114. doi:10.1002/chir.23295 |
| 19. | Sharma, V.; Kaur, J.; Chimni, S. S. Eur. J. Org. Chem. 2018, 3489–3495. doi:10.1002/ejoc.201800589 |
| 20. | Carceller-Ferrer, L.; Vila, C.; Blay, G.; Fernández, I.; Muñoz, M. C.; Pedro, J. R. Org. Biomol. Chem. 2019, 17, 9859–9863. doi:10.1039/c9ob02252j |
| 21. | Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. Asian J. Org. Chem. 2021, 10, 1173–1183. doi:10.1002/ajoc.202100120 |
| 37. | The crystallographic data (CCDC 2234286) for ent-3ba, can be obtained free of charge from the Cambridge crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif. |
| 10. | Chauhan, P.; Mahajan, S.; Enders, D. Chem. Commun. 2015, 51, 12890–12907. doi:10.1039/c5cc04930j |
| 11. | Liu, S.; Bao, X.; Wang, B. Chem. Commun. 2018, 54, 11515–11529. doi:10.1039/c8cc06196c |
| 12. | Bao, X.; Wang, X.; Tian, J.-M.; Ye, X.; Wang, B.; Wang, H. Org. Biomol. Chem. 2022, 20, 2370–2386. doi:10.1039/d1ob02426d |
| 13. | Rao, K. S.; Ramesh, P.; Trivedi, R.; Kantam, M. L. Tetrahedron Lett. 2016, 57, 1227–1231. doi:10.1016/j.tetlet.2016.02.008 |
| 14. | Kim, Y. H.; Yoon, J. H.; Lee, M. Y.; Kim, D. Y. Bull. Korean Chem. Soc. 2017, 38, 1242–1245. doi:10.1002/bkcs.11241 |
| 15. | Phelan, J. P.; Ellman, J. A. Adv. Synth. Catal. 2016, 358, 1713–1718. doi:10.1002/adsc.201600110 |
| 16. | Sharma, A.; Sharma, V.; Chimni, S. S. Org. Biomol. Chem. 2019, 17, 9514–9523. doi:10.1039/c9ob01700c |
| 17. | Yang, M.; Zhang, M.; Wang, Z.; Tang, L.; Chen, W.; Ban, S.; Li, Q. Chirality 2018, 30, 1096–1104. doi:10.1002/chir.23003 |
| 18. | Aydin, A. E.; Culha, S. Chirality 2021, 33, 106–114. doi:10.1002/chir.23295 |
| 19. | Sharma, V.; Kaur, J.; Chimni, S. S. Eur. J. Org. Chem. 2018, 3489–3495. doi:10.1002/ejoc.201800589 |
| 20. | Carceller-Ferrer, L.; Vila, C.; Blay, G.; Fernández, I.; Muñoz, M. C.; Pedro, J. R. Org. Biomol. Chem. 2019, 17, 9859–9863. doi:10.1039/c9ob02252j |
| 21. | Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. Asian J. Org. Chem. 2021, 10, 1173–1183. doi:10.1002/ajoc.202100120 |
| 23. | Wu, B.; Chen, J.; Li, M.-Q.; Zhang, J.-X.; Xu, X.-P.; Ji, S.-J.; Wang, X.-W. Eur. J. Org. Chem. 2012, 1318–1327. doi:10.1002/ejoc.201101529 |
| 38. | Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 9748–9770. doi:10.1002/anie.201109036 |
| 40. | Moran, A.; Hamilton, A.; Bo, C.; Melchiorre, P. J. Am. Chem. Soc. 2013, 135, 9091–9098. doi:10.1021/ja404784t |
© 2024 Goyal et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.