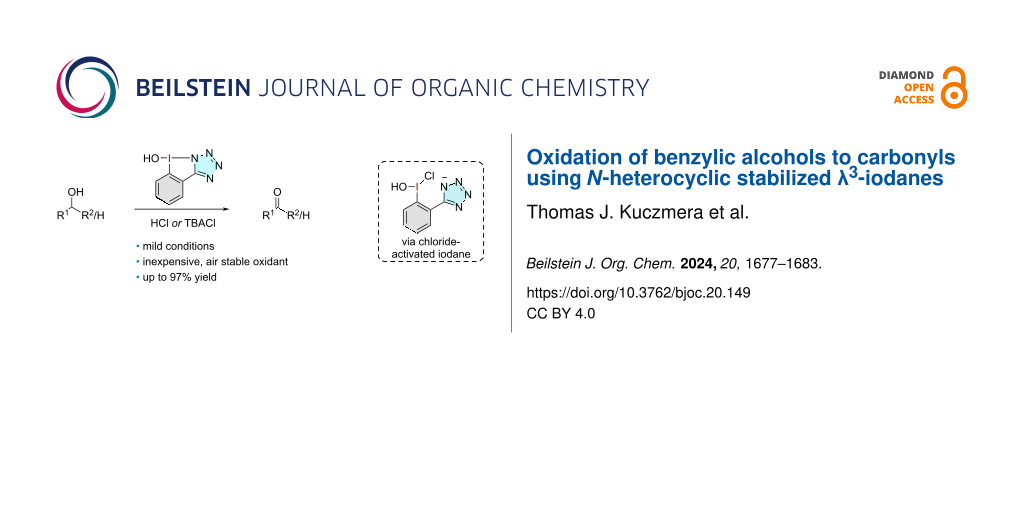
Abstract
We present N-heterocycle-stabilized iodanes (NHIs) as suitable reagents for the mild oxidation of activated alcohols. Two different protocols, both involving activation by chloride additives, were used to synthesize benzylic ketones and aldehydes without overoxidation in up to 97% yield. Based on MS experiments an activated hydroxy(chloro)iodane is proposed as the reactive intermediate.

Graphical Abstract
Introduction
The oxidation of alcohols to aldehydes and ketones is an essential transformation in organic chemistry [1,2]. Generating aldehydes is particularly challenging as they are easily overoxidized to carboxylic acids. Over the past decades a variety of methods have been developed, utilizing toxic heavy metals such as pyridinium dichromate (PDC) [3-5] or manganese dioxide (Figure 1) [6,7]. Molecular oxygen [8] and peroxides [9,10] can also be used as inexpensive terminal oxidants in combination with transition-metal catalysts. Metal-free methods employ chlorodimethylsulfonium compounds as the reactive species and have gained great popularity under the name Swern oxidation or the Corey–Kim oxidation [11]. Hypervalent iodine compounds have also been studied and are well established in several oxidative transformations including the synthesis of complex molecules and drugs [12,13]. The most prominent examples are the pentavalent derivatives 2-iodoxybenzoic acid (IBX) and Dess–Martin periodinane (DMP) [14,15]. Although mild and selective oxidants, these highly oxidized λ5-iodanes have drawbacks, in particular low solubility and moisture sensitivity [11]. Hypervalent iodine compounds in a lower oxidation state (λ3-iodanes), such as iodosobenzene (PhIO)n or phenyliodine(III) diacetate (PIDA) have been reported in alcohol oxidations but they often result in overoxidation to the corresponding carboxylic acids [16]. Additives such as bromide salts or Al2O3 can eliminate this problem and allow selective oxidation to some extent [17-20].
![[1860-5397-20-149-1]](/bjoc/content/figures/1860-5397-20-149-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Overview of common non-iodine-based (left) and iodine-based (right) oxidizing reagents for the generation of carbonyls from the corresponding alcohols.
Figure 1: Overview of common non-iodine-based (left) and iodine-based (right) oxidizing reagents for the gene...
During the past years, N-heterocycle-stabilized iodanes (NHIs) were demonstrated as suitable tools for various applications among them group transfer reactions [21] and as building blocks [22-24]. The synthetic potential of NHIs has been previously studied in model transformations such as thioanisole oxygenation, oxidative lactonization, or diacetoxylation of alkenes [25-28]. In this work, we want to apply NHIs in a mild oxidation of primary and secondary benzylic alcohols to aldehydes and ketones as an alternative to λ5-iodanes.
Results and Discussion
Initially, we investigated a variety of pyrazole-, triazole-, and oxazole-substituted hydroxy-NHIs previously developed by our group [25]. However, none of them proved to be effective in a model oxidation reaction of n-octanol (2). Since previous investigations have repeatedly shown that the number of heteroatoms in the N-heterocycle correlates with the NHIs activity, a series of tetrazole- and tetrazine-substituted NHIs 1a–e was synthesized (Figure 2) [29,30]. A crystal structure was additionally obtained for tetrazine 1c. Bond lengths and angles were similar to those of known five-membered NHIs [25], including a strong intramolecular interaction between the nitrogen of the tetrazine and the hypervalent iodine atom (I1–N1: 2.44(4) Å; the sum of VdW radii: 3.61 Å [31]).
![[1860-5397-20-149-2]](/bjoc/content/figures/1860-5397-20-149-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: NHIs investigated for the oxidation of benzylic alcohols and the crystal structure (ORTEP drawing) of 1c (CCDC 2321313), showing the coordination of the triflate to two positions of the iodane. Thermal ellipsoids are displayed with 50% probability. Selected bond lengths and angles: I1–N1: 2.44(4) Å; I1–O1: 1.94(9) Å; I1–O2: 3.04(1) Å; C1–I1–N1: 73.5(8)°; O1–I1–N1: 166.6(5)°; N1–I1–C1–O1: 177.8(3)°.
Figure 2: NHIs investigated for the oxidation of benzylic alcohols and the crystal structure (ORTEP drawing) ...
Beginning with the electron-deficient and thereby highly reactive NHIs 1a and 1c, we explored the potential for a ligand-exchange process on the iodane via 1H NMR spectroscopy by combining equimolar quantities of NHI and n-octanol (2). When the tetrazole-substituted hydroxy(aryl)iodane 1a was added, no significant shifts in the NMR spectral signals were detected, probably due to the poor solubility of the iodane. Conversely, with the addition of the red tetrazine salt 1c, a significant downfield shift was observed for the alpha-carbon protons from 3.51 ppm to 4.55 ppm, as illustrated in Figure 3a.
![[1860-5397-20-149-3]](/bjoc/content/figures/1860-5397-20-149-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR spectra of the time-dependent formation of a) an alkoxy-NHI which is causing a significant downfield shift of the protons in alpha-position (orange) compared to the free alcohol 2 (blue) and b) oxidation of p-tolylmethanol (3a, blue) to the aldehyde 4a (green) and carboxylic acid 4a’ (red). Reaction conditions: An equimolar mixture of NHI 1c (10.0 µmol) and alcohol (2 or 3a, 10.0 µmol) were dissolved in CD3CN (600 µL) and 1H NMR spectra were recorded.
Figure 3: 1H NMR spectra of the time-dependent formation of a) an alkoxy-NHI which is causing a significant d...
This indicates a ligand exchange of the hydroxy group resulting in a loss of electron density and the formation of the alkoxy-NHI 2'. The chemical shift is consistent with previously measured alkoxyiodanes [32].
The experiments were repeated using activated p-tolylmethanol (3a), again showing no reaction with iodane 1a. Utilizing the tetrazine 1c, p-methylbenzaldehyde (4a) was observed as a new species at 9.94 ppm (Figure 3b). The reaction reached 31% conversion after 72 h, however, p-methylbenzoic acid (4a’) was formed in 35% as well, showing an undesired overreaction. In this experiment no formation of an alkoxyiodane was observed, indicating that the formation of this ligand-exchanged intermediate is slower than the dehydrogenation. As a consequence, we attempted to accelerate the ligand exchange through the addition of a Lewis acid and the performance of the NHIs was compared with common iodine(III) reagents by 1H NMR spectroscopy (Figure 4). After 60 h the measurements revealed a higher yield of aldehyde 4a using 1a (68%) compared to 1c (30%) under the influence of AlCl3. As a comparison, the use of PIDA (5b) and IBA (5c) with the additive resulted in a significantly lower oxidation of the alcohol. Only small amounts of benzoic acid 4a’ were observed in all reactions with additional AlCl3, suggesting that the additive inhibits the previously observed overoxidation.
![[1860-5397-20-149-4]](/bjoc/content/figures/1860-5397-20-149-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Oxidation of 3a to 4a using different iodine(III) reagents with AlCl3 as an additive. Conditions: The turnover of an equimolar mixture of 3a, iodine(III), and AlCl3 (10.0 µmol, respectively) in CD3CN (500 µL) was monitored via 1H NMR spectroscopy.
Figure 4: Oxidation of 3a to 4a using different iodine(III) reagents with AlCl3 as an additive. Conditions: T...
Surprisingly AlCl3 activated the cyclic tetrazole iodane 1a but had almost no influence on the reactivity of the tetrazine salt 1c. Based on these results, the reaction conditions were further optimized using NHI 1a with the benzyl alcohols 3a (electron-rich) and 3b (electron-poor) as the model substrates. First, the reaction temperature was increased, finding 60 °C to be the optimal value in EtOAc (Table 1, entry 1). At this temperature, the reaction time was significantly reduced to 2.5 h. A variety of other additives were tested next, revealing TsOH or NaOTs inhibiting the reaction (Table 1, entries 2 and 3). The addition of tetrabutylammonium halides showed the chloride salt being superior, giving comparable or even better yields than AlCl3 (Table 1, entries 4–7). Investigation of other chloride sources resulted in a reduced yield in the case of ammonium chloride and an improved yield of 82% of 4a when concentrated aqueous HCl was added (Table 1, entries 8 and 9). Other solvents did not further increase the yield (see the full table in Supporting Information File 1).
Table 1: Varying the additive and solvent in the oxidation of electron-rich and electron-deficient benzylic alcohols with 1a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-149-i2.svg?max-width=637&scale=1.0)
|
||||
| Entry | Additive | Solvent | Yield [%] | |
| 4a | 4b | |||
| 1 | AlCl3 | EtOAc | 65 | 39 |
| 2 | TsOH∙H2O | EtOAc | 1 | 1 |
| 3 | NaOTs | EtOAc | 1 | 1 |
| 4 | TBAF | EtOAc | 9 | 19 |
| 5 | TBACl | EtOAc | 67 | 62 |
| 6 | TBABr | EtOAc | 58 | 47 |
| 7 | TBAI | EtOAc | 40 | 36 |
| 8 | NH4Cl | EtOAc | 37 | 26 |
| 9 | HCl | EtOAc | 82 | 44 |
| 10 | TBACl | MeCN | 64 | 69 |
| 11b | TBACl | MeCN | 74 | 78 |
| 12b | HCl | EtOAc | 90 | 53 |
aReaction conditions: 1a (100 µmol), 3a/3b (100 µmol), and the additive (100 µmol) were stirred in the given solvent (1 mL) at 60 °C for 2.5 h and quenched with Me2S (200 µmol). bOptimum reaction conditions were used: 1a (100 µmol), 3a/3b (100 µmol), and the additive (100 µmol) were stirred in the given solvent (0.5 mL) at 60 °C for 2.5 h and quenched with Me2S (200 µmol). The yield was determined via 1H NMR using tetraethylsilane as an internal standard.
However, when electron-deficient p-chlorobenzyl alcohol (3b) was used the highest yield of 4b (69%) was achieved with TBACl as the chloride source in MeCN (Table 1, entry 10). These optimizations lead to the conclusion that AlCl3, as proposed in the initial experiments is not a Lewis acid activator but just a chloride source. Further optimization studies improved the yield to 78% of 4b using a concentration of 0.20 M of the alcohol and 1.4 equiv of 1a (see Supporting Information File 1). Finally, all NHIs were tested under the optimized conditions, revealing the tetrazole-substituted iodane 1a to be the best oxidant for this reaction (Table 2).
Table 2: Testing different NHIs under the optimum conditions for oxidation of electron-deficient substrate 3b.a
| Iodane | Yield of 4b [%] |
| 1a | 78 |
| 1b | 71 |
| 1c | 46 |
| 1d | 29 |
| 1e | 41 |
aReaction conditions: NHI (1a–d: 140 µmol, 1e: 70.0 µmol), p-chlorobenzyl alcohol (3b, 100 µmol) and TBACl (100 µmol) in MeCN (500 µL) were stirred at 60 °C for 2.5 h and quenched with Me2S (200 µmol). The yield was determined via 1H NMR with tetraethylsilane as an internal standard.
The two suitable methods (A: HCl in EtOAc; B: TBACl in MeCN) were then applied to a variety of activated alcohols. The best option is shown in Figure 5. Model substrate 4a could be isolated in a high yield of 84% with reisolation of the 5-(2-iodophenyl)-1H-tetrazole (6) in 90% yield. Other para-halogenated benzaldehydes 4b–f were isolated in good yields of up to 88%. ortho-Substitution led to a lower yield of the iodinated product 4g (43%) compared to the para-iodinated analogues 4d (75%). The ortho-phenyl-substituted aldehyde 4h was isolated in 85% yield, while the ortho-methoxy substrate did not convert to 4i. The ortho-, meta- and para-permutation of a CF3 group showed lower reactivity for the ortho-substituted 4j (53%), while the meta- and para-derivatives 4k and 4l gave higher yields of 84% and 71%, respectively. The steric inhibition of a doubly substituted phenyl ring was observed in a diminished formation of 2,6-dichlorobenzaldehyde (4m) in 39% yield. Naphthalen-2-ylmethanol gave aldehyde 4n in 44% yield. Pyridines 4o and 4p were also compatible and gave good yields of 87% and 64%, respectively. Unfortunately, the synthesis of vanillin (4q) was unsuccessful due to undesirable oxidation reactions of the electron-rich arene. The cyclopropane derivative 4r was generated from the cyclopropylmethanol in 53% yield. The acetylene derivative 4s could not be isolated due to undesired oxidations of the triple bond. The behavior of secondary benzylic alcohols was tested next, giving 4-methylacetophenone (4t) in an excellent yield of 97% and 1-indanone (4u) in 46%. It is worth noting that for some derivatives oxidized by method A, an acylation of the alcohol was detected as a side reaction via mass spectrometry. Vinyl alcohols were also studied, giving carvone (4v) in 74% yield without oxidation of the double bonds. Finally, other heterocyclic benzylic alcohols were investigated, which led to undesired chlorinations in the case of benzimidazoles 3w and 3x and decomposition for thiophenylmethanol 3y.
![[1860-5397-20-149-5]](/bjoc/content/figures/1860-5397-20-149-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Substrate scope of aldehydes and ketones synthesized from the corresponding alcohols. Isolated yields. Reaction conditions: 1a (700 µmol), alcohol (500 µmol), and method A HCl (37%, 500 µmol) in EtOAc (2.5 mL) or method B TBACl (500 µmol) in MeCN (2.5 mL), respectively, were stirred at 60 °C for 2.5 h and quenched with Me2S (1.40 mmol).
Figure 5: Substrate scope of aldehydes and ketones synthesized from the corresponding alcohols. Isolated yiel...
Regarding the reaction mechanism, two plausible pathways can be discussed based on literature examples (Scheme 1, path a [17] and path b [33]). In either path, initial ligand exchange to the hydroxy(chloro)iodane I-OH is proposed. For getting an indication of a chloride-activated iodane of this type, a mixture of NHI 1a and HCl in EtOAc was stirred for 1 h at 60 °C and an ESI(−) mass spectrum was recorded afterward, showing an ion I-OMe with m/z 337.0 [1a – OH + MeO + Cl]− (Scheme 1c). It is known that methanol, which is used as a solvent in the mass spectrometer, can be exchanged with the hydroxy group of the NHI [21]. No such ion was measured in the mixture before heating. This ion therefore indicates an I–Cl bond in the activated iodane. Starting from I-OH, in a potential path a) formation of hypochlorous acid is suggested, which consequently oxidizes the alcohol through the alkyl hypochlorite IIa. The second mechanism (path b) requires a direct ligand exchange of I-OH with the alcohol and subsequent β-elimination of the alkoxy(hydroxy)iodane IIb to form the desired aldehyde 4.
![[1860-5397-20-149-i1]](/bjoc/content/inline/1860-5397-20-149-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Possible reaction mechanisms via the formation of a) a Cl(I) species and b) the formation of an alkoxyiodane IIb. Both are initialized by the activated iodane I-OH, which was observed as c) I-OMe species in the ESI(−) MS.
Scheme 1: Possible reaction mechanisms via the formation of a) a Cl(I) species and b) the formation of an alk...
Conclusion
In conclusion, this study has successfully introduced N-heterocycle-stabilized iodanes (NHIs) as effective λ3-iodane oxidants for the selective synthesis of ketones and aldehydes, avoiding overoxidation to carboxylic acids. The developed protocols proved particularly effective for benzylic alcohols, yielding good to excellent results. The beneficial role of chloride salt additives was investigated, potentially leading to the formation of a hydroxy(chloro)iodane intermediate. This intermediate either liberates hypochlorous acid as the terminal oxidant or undergoes a direct ligand exchange with the alcohol, followed by oxidative elimination to form the aldehyde. Thus, these reagents offer a viable alternative to traditional aryl-λ5-iodane-based oxidants, although further studies are necessary to fully understand their reaction mechanisms.
Experimental
General procedure for oxidation of benzylic alcohols
1a (700 µmol, 201 mg, 1.40 equiv), benzylic alcohol (3, 500 µmol, 1.00 equiv) and method A: aqueous HCl (37%, 500 µmol, 41.6 µL, 1.00 equiv) in EtOAc (2.5 mL) or method B: TBACl (500 µmol, 137 mg, 1.00 equiv) in MeCN (2.5 mL), respectively, were stirred at 60 °C for 2.5 h, quenched with Me2S (2.00 equiv) and the reaction mixture was purified via flash column chromatography on silica.
Supporting Information
| Supporting Information File 1: Experimental part and copies of spectra. | ||
| Format: PDF | Size: 2.4 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article
References
-
Arterburn, J. B. Tetrahedron 2001, 57, 9765–9788. doi:10.1016/s0040-4020(01)01009-2
Return to citation in text: [1] -
Caron, S.; Dugger, R. W.; Ruggeri, S. G.; Ragan, J. A.; Ripin, D. H. B. Chem. Rev. 2006, 106, 2943–2989. doi:10.1021/cr040679f
Return to citation in text: [1] -
Guziec, F. S., Jr.; Luzzio, F. A. J. Org. Chem. 1982, 47, 1787–1789. doi:10.1021/jo00348a045
Return to citation in text: [1] -
Collins, J. C.; Hess, W. W.; Frank, F. J. Tetrahedron Lett. 1968, 9, 3363–3366. doi:10.1016/s0040-4039(00)89494-0
Return to citation in text: [1] -
Corey, E. J.; Suggs, J. W. Tetrahedron Lett. 1975, 16, 2647–2650. doi:10.1016/s0040-4039(00)75204-x
Return to citation in text: [1] -
Lou, J.-D.; Xu, Z.-N. Tetrahedron Lett. 2002, 43, 6149–6150. doi:10.1016/s0040-4039(02)01345-x
Return to citation in text: [1] -
Highet, R. J.; Wildman, W. C. J. Am. Chem. Soc. 1955, 77, 4399–4401. doi:10.1021/ja01621a062
Return to citation in text: [1] -
Sigman, M. S.; Jensen, D. R. Acc. Chem. Res. 2006, 39, 221–229. doi:10.1021/ar040243m
Return to citation in text: [1] -
Trost, B. M.; Masuyama, Y. Tetrahedron Lett. 1984, 25, 173–176. doi:10.1016/s0040-4039(00)99832-0
Return to citation in text: [1] -
Bovicelli, P.; Truppa, D.; Sanetti, A.; Bernini, R.; Lupattelli, P. Tetrahedron 1998, 54, 14301–14314. doi:10.1016/s0040-4020(98)00885-0
Return to citation in text: [1] -
Tidwell, T. T. Synthesis 1990, 857–870. doi:10.1055/s-1990-27036
Return to citation in text: [1] [2] -
Tohma, H.; Kita, Y. Adv. Synth. Catal. 2004, 346, 111–124. doi:10.1002/adsc.200303203
Return to citation in text: [1] -
Varala, R.; Seema, V.; Dubasi, N. Organics 2023, 4, 1–40. doi:10.3390/org4010001
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156. doi:10.1021/jo00170a070
Return to citation in text: [1] -
Tohma, H.; Takizawa, S.; Maegawa, T.; Kita, Y. Angew. Chem. 2000, 112, 1362–1364. doi:10.1002/(sici)1521-3757(20000403)112:7<1362::aid-ange1362>3.0.co;2-g
Return to citation in text: [1] -
Salvo, A.; Campisciano, V.; Beejapur, H.; Giacalone, F.; Gruttadauria, M. Synlett 2015, 26, 1179–1184. doi:10.1055/s-0034-1380196
Return to citation in text: [1] [2] -
Takenaga, N.; Goto, A.; Yoshimura, M.; Fujioka, H.; Dohi, T.; Kita, Y. Tetrahedron Lett. 2009, 50, 3227–3229. doi:10.1016/j.tetlet.2009.02.020
Return to citation in text: [1] -
Varma, R. S.; Dahiya, R.; Saini, R. K. Tetrahedron Lett. 1997, 38, 7029–7032. doi:10.1016/s0040-4039(97)01660-2
Return to citation in text: [1] -
Deng, Y.; Lu, S.-C.; Yue, L.-L.; Gong, Y.-L.; Guan, X.-D. Synth. Commun. 2022, 52, 2198–2204. doi:10.1080/00397911.2022.2134799
Return to citation in text: [1] -
Kuczmera, T. J.; Boelke, A.; Nachtsheim, B. J. Eur. J. Org. Chem. 2022, e202200276. doi:10.1002/ejoc.202200276
Return to citation in text: [1] [2] -
Kuczmera, T. J.; Dietz, A.; Boelke, A.; Nachtsheim, B. J. Beilstein J. Org. Chem. 2023, 19, 317–324. doi:10.3762/bjoc.19.27
Return to citation in text: [1] -
Vlasenko, Y. A.; Postnikov, P. S.; Trusova, M. E.; Shafir, A.; Zhdankin, V. V.; Yoshimura, A.; Yusubov, M. S. J. Org. Chem. 2018, 83, 12056–12070. doi:10.1021/acs.joc.8b01995
Return to citation in text: [1] -
Tolstaya, T. P.; Egorova, L. D.; Lisichkina, I. N. Chem. Heterocycl. Compd. 1985, 21, 392–396. doi:10.1007/bf00504396
Return to citation in text: [1] -
Boelke, A.; Lork, E.; Nachtsheim, B. J. Chem. – Eur. J. 2018, 24, 18653–18657. doi:10.1002/chem.201804957
Return to citation in text: [1] [2] [3] -
Boelke, A.; Sadat, S.; Lork, E.; Nachtsheim, B. J. Chem. Commun. 2021, 57, 7434–7437. doi:10.1039/d1cc03097c
Return to citation in text: [1] -
Abazid, A. H.; Clamor, N.; Nachtsheim, B. J. ACS Catal. 2020, 10, 8042–8048. doi:10.1021/acscatal.0c02321
Return to citation in text: [1] -
Aertker, K.; Rama, R. J.; Opalach, J.; Muñiz, K. Adv. Synth. Catal. 2017, 359, 1290–1294. doi:10.1002/adsc.201601178
Return to citation in text: [1] -
Kumar, R.; Sayala, K. D.; Camdzic, L.; Siegler, M.; Vaish, A.; Tsarevsky, N. ChemRxiv 2021. doi:10.26434/chemrxiv-2021-gsp7q-v2
Return to citation in text: [1] -
Vaish, A.; Sayala, K. D.; Tsarevsky, N. V. Tetrahedron Lett. 2019, 60, 150995. doi:10.1016/j.tetlet.2019.150995
Return to citation in text: [1] -
Bondi, A. J. Phys. Chem. 1964, 68, 441–451. doi:10.1021/j100785a001
Return to citation in text: [1] -
Sakakibara, Y.; Murakami, K.; Itami, K. Org. Lett. 2022, 24, 602–607. doi:10.1021/acs.orglett.1c04030
Return to citation in text: [1] -
Tohma, H.; Maegawa, T.; Takizawa, S.; Kita, Y. Adv. Synth. Catal. 2002, 344, 328–337. doi:10.1002/1615-4169(200206)344:3/4<328::aid-adsc328>3.0.co;2-s
Return to citation in text: [1]
| 32. | Sakakibara, Y.; Murakami, K.; Itami, K. Org. Lett. 2022, 24, 602–607. doi:10.1021/acs.orglett.1c04030 |
| 25. | Boelke, A.; Lork, E.; Nachtsheim, B. J. Chem. – Eur. J. 2018, 24, 18653–18657. doi:10.1002/chem.201804957 |
| 1. | Arterburn, J. B. Tetrahedron 2001, 57, 9765–9788. doi:10.1016/s0040-4020(01)01009-2 |
| 2. | Caron, S.; Dugger, R. W.; Ruggeri, S. G.; Ragan, J. A.; Ripin, D. H. B. Chem. Rev. 2006, 106, 2943–2989. doi:10.1021/cr040679f |
| 9. | Trost, B. M.; Masuyama, Y. Tetrahedron Lett. 1984, 25, 173–176. doi:10.1016/s0040-4039(00)99832-0 |
| 10. | Bovicelli, P.; Truppa, D.; Sanetti, A.; Bernini, R.; Lupattelli, P. Tetrahedron 1998, 54, 14301–14314. doi:10.1016/s0040-4020(98)00885-0 |
| 25. | Boelke, A.; Lork, E.; Nachtsheim, B. J. Chem. – Eur. J. 2018, 24, 18653–18657. doi:10.1002/chem.201804957 |
| 8. | Sigman, M. S.; Jensen, D. R. Acc. Chem. Res. 2006, 39, 221–229. doi:10.1021/ar040243m |
| 29. | Kumar, R.; Sayala, K. D.; Camdzic, L.; Siegler, M.; Vaish, A.; Tsarevsky, N. ChemRxiv 2021. doi:10.26434/chemrxiv-2021-gsp7q-v2 |
| 30. | Vaish, A.; Sayala, K. D.; Tsarevsky, N. V. Tetrahedron Lett. 2019, 60, 150995. doi:10.1016/j.tetlet.2019.150995 |
| 6. | Lou, J.-D.; Xu, Z.-N. Tetrahedron Lett. 2002, 43, 6149–6150. doi:10.1016/s0040-4039(02)01345-x |
| 7. | Highet, R. J.; Wildman, W. C. J. Am. Chem. Soc. 1955, 77, 4399–4401. doi:10.1021/ja01621a062 |
| 22. | Kuczmera, T. J.; Dietz, A.; Boelke, A.; Nachtsheim, B. J. Beilstein J. Org. Chem. 2023, 19, 317–324. doi:10.3762/bjoc.19.27 |
| 23. | Vlasenko, Y. A.; Postnikov, P. S.; Trusova, M. E.; Shafir, A.; Zhdankin, V. V.; Yoshimura, A.; Yusubov, M. S. J. Org. Chem. 2018, 83, 12056–12070. doi:10.1021/acs.joc.8b01995 |
| 24. | Tolstaya, T. P.; Egorova, L. D.; Lisichkina, I. N. Chem. Heterocycl. Compd. 1985, 21, 392–396. doi:10.1007/bf00504396 |
| 3. | Guziec, F. S., Jr.; Luzzio, F. A. J. Org. Chem. 1982, 47, 1787–1789. doi:10.1021/jo00348a045 |
| 4. | Collins, J. C.; Hess, W. W.; Frank, F. J. Tetrahedron Lett. 1968, 9, 3363–3366. doi:10.1016/s0040-4039(00)89494-0 |
| 5. | Corey, E. J.; Suggs, J. W. Tetrahedron Lett. 1975, 16, 2647–2650. doi:10.1016/s0040-4039(00)75204-x |
| 25. | Boelke, A.; Lork, E.; Nachtsheim, B. J. Chem. – Eur. J. 2018, 24, 18653–18657. doi:10.1002/chem.201804957 |
| 26. | Boelke, A.; Sadat, S.; Lork, E.; Nachtsheim, B. J. Chem. Commun. 2021, 57, 7434–7437. doi:10.1039/d1cc03097c |
| 27. | Abazid, A. H.; Clamor, N.; Nachtsheim, B. J. ACS Catal. 2020, 10, 8042–8048. doi:10.1021/acscatal.0c02321 |
| 28. | Aertker, K.; Rama, R. J.; Opalach, J.; Muñiz, K. Adv. Synth. Catal. 2017, 359, 1290–1294. doi:10.1002/adsc.201601178 |
| 17. | Salvo, A.; Campisciano, V.; Beejapur, H.; Giacalone, F.; Gruttadauria, M. Synlett 2015, 26, 1179–1184. doi:10.1055/s-0034-1380196 |
| 18. | Takenaga, N.; Goto, A.; Yoshimura, M.; Fujioka, H.; Dohi, T.; Kita, Y. Tetrahedron Lett. 2009, 50, 3227–3229. doi:10.1016/j.tetlet.2009.02.020 |
| 19. | Varma, R. S.; Dahiya, R.; Saini, R. K. Tetrahedron Lett. 1997, 38, 7029–7032. doi:10.1016/s0040-4039(97)01660-2 |
| 20. | Deng, Y.; Lu, S.-C.; Yue, L.-L.; Gong, Y.-L.; Guan, X.-D. Synth. Commun. 2022, 52, 2198–2204. doi:10.1080/00397911.2022.2134799 |
| 21. | Kuczmera, T. J.; Boelke, A.; Nachtsheim, B. J. Eur. J. Org. Chem. 2022, e202200276. doi:10.1002/ejoc.202200276 |
| 14. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 15. | Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156. doi:10.1021/jo00170a070 |
| 21. | Kuczmera, T. J.; Boelke, A.; Nachtsheim, B. J. Eur. J. Org. Chem. 2022, e202200276. doi:10.1002/ejoc.202200276 |
| 12. | Tohma, H.; Kita, Y. Adv. Synth. Catal. 2004, 346, 111–124. doi:10.1002/adsc.200303203 |
| 13. | Varala, R.; Seema, V.; Dubasi, N. Organics 2023, 4, 1–40. doi:10.3390/org4010001 |
| 17. | Salvo, A.; Campisciano, V.; Beejapur, H.; Giacalone, F.; Gruttadauria, M. Synlett 2015, 26, 1179–1184. doi:10.1055/s-0034-1380196 |
| 16. | Tohma, H.; Takizawa, S.; Maegawa, T.; Kita, Y. Angew. Chem. 2000, 112, 1362–1364. doi:10.1002/(sici)1521-3757(20000403)112:7<1362::aid-ange1362>3.0.co;2-g |
| 33. | Tohma, H.; Maegawa, T.; Takizawa, S.; Kita, Y. Adv. Synth. Catal. 2002, 344, 328–337. doi:10.1002/1615-4169(200206)344:3/4<328::aid-adsc328>3.0.co;2-s |
© 2024 Kuczmera et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.