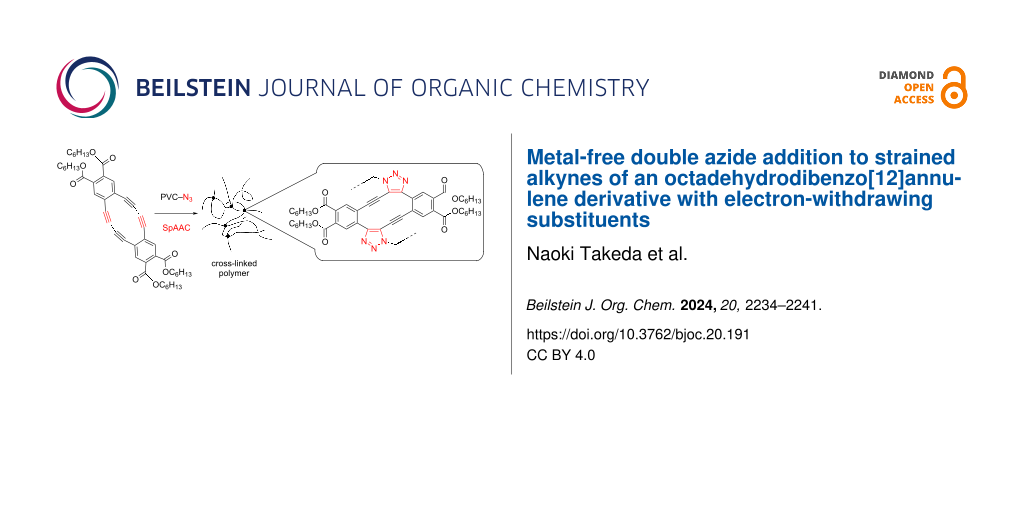
Abstract
Strain-promoted azide–alkyne cycloaddition (SpAAC) is a powerful tool in the field of bioconjugation and materials research. We previously reported a regioselective double addition of organic azides to octadehydrodibenzo[12]annulene derivatives with electron-rich alkyloxy substituents. In order to increase the reaction rate, electron-withdrawing substituents were introduced into octadehydrodibenzo[12]annulene. In this report, the synthesis of new octadehydrodibenzo[12]annulene derivatives, regioselective double addition of organic azides, and an application to crosslinking polymers are described.

Graphical Abstract
Introduction
The strain-promoted azide–alkyne cycloaddition (SpAAC) is one of the most representative metal-free click chemistry reactions [1-5]. SpAAC has been mainly employed in bioconjugation in the fields of chemical biology and medicinal chemistry due to its high efficiency under physiologically active conditions and the absence of any toxic metal ions. SpAAC is biorthogonal, which allows for the specific labeling and imaging of biomolecules even in living cells and organisms. Recently, a more rapid click reaction was desired and the strain-promoted oxidation-controlled cyclooctyne-1,2-quinone cycloaddition (SPOCQ) was developed and employed in the same fields of chemical biology [6,7]. On the other hand, the use of the SpAAC in materials science was slow. We developed another class of metal-free click chemistry reactions, such as the [2 + 2] cycloaddition–retroelectrocyclization (CA-RE) between electron-rich alkynes and electron-deficient olefins [8]. The [2 + 2] CA-RE click reactions were employed to produce a variety of functional materials, such as nonlinear optical chromophores [9,10], super acceptors [11,12], ion sensing D–A systems [13], and crosslinked polymers [14-17]. Since crosslinking polymers requires high reaction efficiency under mild conditions, developing such reactions is crucial.
We previously reported the regioselective double azide addition to octadehydrodibenzo[12]annulene with hexyloxy substituents (DBA-OHex), which are readily accessible by the oxidative acetylenic coupling of a 1,2-diethynylbenzene derivative (Figure 1) [18]. The chemical stability of DBAs depends on the electronic character of the substituents. Electron-donating alkyloxy groups are known to enhance the stability of DBAs. On the other hand, DBAs substituted with electron-withdrawing groups have been little studied [19], and their chemical stability and physical properties are not well understood. In this paper, a new DBA substituted with ester groups was synthesized, and the double azide addition was comprehensively investigated. Finally, the double azide addition reaction was applied to polymer crosslinking and the mechanical properties of the self-standing polymer films were compared.
![[1860-5397-20-191-1]](/bjoc/content/figures/1860-5397-20-191-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Previously reported regioselective double azide addition to DBA with hexyloxy substituents and molecular design in this study.
Figure 1: Previously reported regioselective double azide addition to DBA with hexyloxy substituents and mole...
Results and Discussion
Strain-promoted azide–alkyne cycloaddition
Octadehydrodibenzo[12]annulene (DBA) with electron-withdrawing carbonyl substituents 5 was prepared from phthalimide (1, Scheme 1). Iodination followed by hydrolysis afforded 4,5-diiodophthalic acid (2) in 46.7% yield. Esterification with 1-hexanol yielded compound 3 in 56.8% yield and the subsequent Sonogashira coupling with trimethylsilylacetylene provided compound 4 in 80.0%. Silyl deprotection with (n-C4H9)4NF in THF followed by acetylenic oxidative dimerization under Hay conditions produced the desired DBA 5 in 12.7% yield. It should be noted that a thermodynamically more stable trimeric macrocycle was also formed in 17.7%, but it was not isolated and purified because it was outside the scope of this study.
![[1860-5397-20-191-i1]](/bjoc/content/inline/1860-5397-20-191-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Next, DBA 5 was subjected to the SpAAC with benzyl azide. When two equivalents of benzyl azide were added to a solution of 5 in CDCl3 at a controlled temperature of 30 °C, the reaction slowly proceeded (Figure 2). After 22 h, the original peak ascribed to starting compound 5 decreased, while a set of new peaks appeared at 8.40 ppm, 7.79 ppm, and 7.20–7.18 ppm. Considering the high symmetry, no monoadducts were formed and the regioselective double azide addition occurred. In other words, 1,4-disubstituted-5-ethynyltriazole derivative (in-) (6a) or 1,5-disubstituted-4-ethynyltriazole derivative (out-) (6b) adducts were formed. To determine the chemical structure of the product, the NOESY measurement was conducted. The NOESY spectrum suggested the intermolecular through-space coupling between two benzene protons (Figure S10 in Supporting Information File 1). This result indicates that the formed double azide adduct is 6a, which is consistent with our previous report [18].
![[1860-5397-20-191-2]](/bjoc/content/figures/1860-5397-20-191-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) Strain-promoted azide–alkyne cycloaddition between DBA 5 and benzyl azide and (b) 1H NMR spectral change at 30 °C in CDCl3.
Figure 2: (a) Strain-promoted azide–alkyne cycloaddition between DBA 5 and benzyl azide and (b) 1H NMR spectr...
The double azide addition was further investigated by changing the reaction temperature. The rate constant was determined by the temperature-dependent 1H NMR spectra in CDCl3. The reaction kinetics followed a second-order reaction. Since no monoadducts were formed, the rate-determining step is the first azide addition. Based on this fact, the activation energy (Ea) of the reaction between 5 and benzyl azide in CDCl3, determined by the Arrhenius plots, was 60.9 kJ mol−1 (Figure 3). This value was apparently smaller than those previously reported for the reaction between DBA-OHex and benzyl azide (Table S1 in Supporting Information File 1). However, the Ea values were previously measured in DMSO-d6 and C6D6, and it is known that there is a solvent polarity effect on the reactivity of SpAACs. In order to quantitatively compare the reactivity of DBAs with different substituents, the reaction between DBA-OHex and benzyl azide was also monitored in CDCl3. The Ea in CDCl3 was 71.1 kJ mol−1 (Table S1 in Supporting Information File 1). This result clearly suggests that DBA 5 with electron-withdrawing substituents has a lower Ea than DBA with electron-donating substituents.
![[1860-5397-20-191-3]](/bjoc/content/figures/1860-5397-20-191-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Arrhenius plots of the rate constants for the reaction between 5 and benzyl azide in CDCl3.
Figure 3: Arrhenius plots of the rate constants for the reaction between 5 and benzyl azide in CDCl3.
DFT calculations
The reaction mechanism was investigated by computational calculations. The reaction mechanism between 5 and benzyl azide was supported by the ωB97X-D/6-31G(d,p) calculations with the CH2Cl2 polarizable continuum model (PCM) solvent (Figure 4). The reaction initially started with the addition of benzyl azide to one of the internal alkynes of 5. Although the benzyl group is situated on the interior side of DBA, a more energetically unstable transition state (in-ts) is generated. However, the resulting monoadduct (in) is more energetically demanding than the counter monoadduct (out) due to steric factors. The second azide addition follows this step. The alkyne, which is diagonally positioned relative to the triazole group, shows the highest reactivity due to its significant distortion. This finding correlates with the experimental observation that no monotriazoles were obtained. During the second azide addition, the orientation of benzyl azide is once more controlled. The benzyl group positioned on the inner side results in a thermodynamically less stable transition state (in-in-ts) compared to that on the outer side (in-out-ts), but the thermodynamically stable final product is a regioselective “in-in” adduct, i.e., compound 6a.
![[1860-5397-20-191-4]](/bjoc/content/figures/1860-5397-20-191-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Proposed reaction mechanism for the formation of compound 6a. Free energy profiles (ΔG298 in kJ mol−1) calculated at the ωB97X-D/6-31G(d,p)/PCM (in CH2Cl2).
Figure 4: Proposed reaction mechanism for the formation of compound 6a. Free energy profiles (ΔG298 in kJ mol...
Optical properties
Absorption and emission spectra of 6a were measured in CH2Cl2 (Figure 5). The conjugation is changed and a highly-twisted macrocyle forms by the double azide addition. Thus, compound 6a shows an ultraviolet absorption peak at 249 nm (λmax) and no absorption in the visible region was observed. When excited at 249 nm, an emission band at 513 nm (λem) appeared in the spectrum with a fluorescence quantum yield (Φ) of 7.0%. The absorption and fluorescence spectra were almost independent of solvents. For example, the λem in CHCl3 and THF was 514 nm (Figure S11 in Supporting Information File 1). The double benzyl azide adduct of DBA-OHex displayed similar absorption and emission spectra with a λmax of 247 nm and λem of 539 nm in CH2Cl2. Since the Stokes shift of the double benzyl azide adduct of DBA-OHex was 21900 cm−1 and larger than that of 6a (20700 cm−1), the Φ was 4.3%. This result suggested that replacing the hexyloxy substituents with electron-withdrawing ones enhanced the fluorescence intensity by approximately 1.6 times.
![[1860-5397-20-191-5]](/bjoc/content/figures/1860-5397-20-191-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Absorption (blue) and fluorescence (red) spectra of 6a (2 × 10−5 M) in CH2Cl2.
Figure 5: Absorption (blue) and fluorescence (red) spectra of 6a (2 × 10−5 M) in CH2Cl2.
Crosslinker application
Using the strain-promoted double azide addition feature, DBA 5 was evaluated as a crosslinker for azidated polymers. After 5 (5 mol %) was added to a partially azidated poly(vinyl chloride) (PVC-N3) (x = 0.11, n = 1000) in THF, the solvent was gradually evaporated on a Teflon boat. The resulting self-standing film became insoluble due to the occurrence of strain-promoted double azide–alkyne cycloaddition (Figure 6a). The mechanical properties of the polymer films were then examined to observe the impact of crosslinking.
![[1860-5397-20-191-6]](/bjoc/content/figures/1860-5397-20-191-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: (a) Crosslinking reaction of PVC-N3 (x = 0.11) with compound 5. (b,c) Strain-stress curves of PVC-N3 before (blue) and after (red) crosslinking.
Figure 6: (a) Crosslinking reaction of PVC-N3 (x = 0.11) with compound 5. (b,c) Strain-stress curves of PVC-N3...
A PVC-N3 film was prepared by a solvent-cast method on a Teflon boat. The strain-stress (S–S) curve of the PVC-N3 film exhibited a breakdown at the strain of >300% with a gradual increase in the stress (Figure 6b). This film did not display any yielding points (Figure 6c). Interestingly, the crosslinking with 5 dramatically changed the mechanical features. The crosslinked film showed a clear yield point at the strain of about 4% with the maximum stress of 26.3 MPa (Table 1). When the applied stress gradually increased, the film displayed a ductility up to the strain of about 180 MPa. Young’s moduli were estimated from the initial slopes of the S–S curves. The Young’s modulus significantly increased from 277 MPa to 777 MPa by crosslinking. This result is consistent with the general understanding of covalent crosslinking of linear polymers. The crosslinking points formed by the SpAAC resulted in a rigid film.
Conclusion
Octadehydrodibenzo[12]annulene substituted with electron-withdrawing groups was successfully synthesized. This molecule underwent SpAAC with two equivalents of benzyl azide under mild conditions to quantitatively form the regioselective symmetric bistriazole product. The electron-withdrawing substituents facilitated the reaction progress as compared to the previously reported electron-donating hexyloxy substituents. The addition pattern was experimentally investigated by 2D NMR, which was also supported by DFT calculations. The bistriazole product displayed fluorescence in the visible range with a fluorescence quantum yield of 7.0%. Finally, the developed metal-free click reaction was employed to crosslink a partially azidated poly(vinyl chloride). The crosslinking proceeded by simply mixing the polymer and crosslinker in THF and evaporating, and the formation of the crosslinked polymer film was confirmed by the strain–stress curves. The developed method is straightforward and has broad applicability, extending to other azidated molecules and polymers.
Experimental
Materials
All reagents are purchased from TCI, Aldrich, and Kanto Chemical Co. Inc., and used as received. 4,5-Diiodophthalic acid was prepared according to a literature method [20]. A partially azidated poly(vinyl chloride) (PVA-N3) was prepared by stirring poly(vinyl chloride) and NaN3 in DMF at room temperature overnight.
Measurements
NMR spectra were recorded using a JEOL mode Al300 (300 MHz) at room temperature. Deuterated chloroform was used as the solvent unless otherwise stated. Chemical shifts of NMR were reported in ppm relative to the residual solvent peak at 7.26 ppm for 1H NMR spectroscopy and 77.6 ppm for 13C NMR spectroscopy. Coupling constants (J) were given in Hz. The resonance multiplicity was described as s (singlet), t (triplet), and m (multiplet). FTIR spectra were recorded on a JASCO FT/IR-4100 spectrometer in the range from 4000 to 600 cm−1. MALDI–TOF mass spectra were measured on a Shimadzu/Kratos AXIMACFR mass spectrometer equipped with a nitrogen laser (λ = 337 nm) and pulsed ion extraction, which was operated at an accelerating potential of 20 kV. THF solutions containing 1 g L−1 of a sample, 10 g L−1 of dithranol, and 1 g L−1 of sodium trifluoroacetate were mixed at a ratio of 1:1:1, and then 1 μL aliquot of this mixture was deposited onto a sample target plate. UV–vis absorption spectra were recorded on a JASCO V-670 spectrophotometer. Fluorescence spectra were recorded on a JASCO FP-8500.
All calculations were carried out using the Gaussian 16 program [21]. The DFT calculations were carried out using the long-range and dispersion-corrected ωB97X-D functional [22]. The 6-31G(d,p) basis set was used for H, C, O, and S atoms [23,24]. The solvent effect of CH2Cl2 was taken into account by the polarizable continuum model using the integral equation formalism (IEFPCM) [25] for DFT calculations. The optimized molecular structures were verified by vibrational analysis; equilibrium structures did not have imaginary frequencies and transition-state structures had only one imaginary frequency corresponding to the reaction coordinate.
Synthesis of 6a
To a solution of 5 in CH2Cl2, two equivalents of benzyl azide were added and the mixture was stirred at room temperature for 12 h. Evaporation of the solvent quantitatively yielded the target compound. 1H NMR (CDCl3, 300 MHz, 297 K) δ 8.40 (s, 2H), 7.80 (s, 2H), 7.40–7.18 (m, 10H), 5.53 (s, 4H), 4.34–4.29 (m, 8H), 1.74–1.71 (m, 8H), 1.37–1.32 (m, 24H), 0.92–0.88 (m, 12H); 13C NMR (CDCl3, 75 MHz, 297 K) δ 166.26, 147.55, 135.76, 133.90, 133.67, 133.36, 131.92, 130.83, 128.74, 128.70, 128.34, 120.99, 119.57, 103.11, 82.18, 66.30, 53.55, 31.39, 28.42, 25.54, 22.48, 13.98; FTIR ν (cm−1): 2067 (C≡C), 1730 (C=O); MALDI–TOF MS (dithranol, m/z): [M]+ calcd for C62H70N6O8, 1026.53; found, 1026.98.
Supporting Information
| Supporting Information File 1: Experimental section and kinetic study of the reaction of compound 5 and benzyl azide. | ||
| Format: PDF | Size: 1.9 MB | Download |
Data Availability Statement
The data that supports the findings of this study is available from the corresponding author upon reasonable request.
References
-
Xie, S.; Sundhoro, M.; Houk, K. N.; Yan, M. Acc. Chem. Res. 2020, 53, 937–948. doi:10.1021/acs.accounts.0c00046
Return to citation in text: [1] -
Krell, K.; Harijan, D.; Ganz, D.; Doll, L.; Wagenknecht, H.-A. Bioconjugate Chem. 2020, 31, 990–1011. doi:10.1021/acs.bioconjchem.0c00072
Return to citation in text: [1] -
Kang, X.; Cai, X.; Yi, L.; Xi, Z. Chem. – Asian J. 2020, 15, 1420–1429. doi:10.1002/asia.202000005
Return to citation in text: [1] -
Li, K.; Fong, D.; Meichsner, E.; Adronov, A. Chem. – Eur. J. 2021, 27, 5057–5073. doi:10.1002/chem.202003386
Return to citation in text: [1] -
Li, X.; Xiong, Y. ACS Omega 2022, 7, 36918–36928. doi:10.1021/acsomega.2c03931
Return to citation in text: [1] -
Sen, R.; Escorihuela, J.; van Delft, F.; Zuilhof, H. Angew. Chem., Int. Ed. 2017, 56, 3299–3303. doi:10.1002/anie.201612037
Return to citation in text: [1] -
Gahtory, D.; Sen, R.; Kuzmyn, A. R.; Escorihuela, J.; Zuilhof, H. Angew. Chem., Int. Ed. 2018, 57, 10118–10122. doi:10.1002/anie.201800937
Return to citation in text: [1] -
Michinobu, T.; Diederich, F. Angew. Chem., Int. Ed. 2018, 57, 3552–3577. doi:10.1002/anie.201711605
Return to citation in text: [1] -
Biaggio, I. Chem. – Eur. J. 2022, 28, e202103168. doi:10.1002/chem.202103168
Return to citation in text: [1] -
Li, Y.; Tsuboi, K.; Michinobu, T. Macromolecules 2010, 43, 5277–5286. doi:10.1021/ma100869m
Return to citation in text: [1] -
Kivala, M.; Boudon, C.; Gisselbrecht, J.-P.; Enko, B.; Seiler, P.; Müller, I. B.; Langer, N.; Jarowski, P. D.; Gescheidt, G.; Diederich, F. Chem. – Eur. J. 2009, 15, 4111–4123. doi:10.1002/chem.200802563
Return to citation in text: [1] -
Tu, K.-H.; Wang, Y.; Kiyota, Y.; Iwahashi, T.; Ouchi, Y.; Mori, T.; Michinobu, T. Org. Electron. 2020, 87, 105978. doi:10.1016/j.orgel.2020.105978
Return to citation in text: [1] -
Li, Y.; Washino, Y.; Hyakutake, T.; Michinobu, T. Anal. Sci. 2017, 33, 599–604. doi:10.2116/analsci.33.599
Return to citation in text: [1] -
Washino, Y.; Michinobu, T. Phys. Chem. Chem. Phys. 2016, 18, 2288–2291. doi:10.1039/c5cp05180k
Return to citation in text: [1] -
Fujita, H.; Michinobu, T. ACS Macro Lett. 2018, 7, 716–719. doi:10.1021/acsmacrolett.8b00365
Return to citation in text: [1] -
Fujita, H.; Michinobu, T. Soft Matter 2018, 14, 9055–9060. doi:10.1039/c8sm01986j
Return to citation in text: [1] -
Fujita, H.; Nihei, N.; Bito, M.; Michinobu, T. Macromol. Biosci. 2018, 18, 1800336. doi:10.1002/mabi.201800336
Return to citation in text: [1] -
Fukushima, S.; Ashizawa, M.; Kawauchi, S.; Michinobu, T. Helv. Chim. Acta 2019, 102, e1900016. doi:10.1002/hlca.201900016
Return to citation in text: [1] [2] -
Zimmermann, B.; Baranović, G.; Štefanić, Z.; Rožman, M. J. Mol. Struct. 2006, 794, 115–124. doi:10.1016/j.molstruc.2006.01.049
Return to citation in text: [1] -
Jiang, J.; Kaafarani, B. R.; Neckers, D. C. J. Org. Chem. 2006, 71, 2155–2158. doi:10.1021/jo0522198
Return to citation in text: [1] -
Gaussian 16; Gaussian, Inc.: Wallingford, CT, 2016.
Return to citation in text: [1] -
Chai, J.-D.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. doi:10.1039/b810189b
Return to citation in text: [1] -
Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650–654. doi:10.1063/1.438955
Return to citation in text: [1] -
McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639–5648. doi:10.1063/1.438980
Return to citation in text: [1] -
Tomasi, J.; Mennucci, B.; Cammi, R. Chem. Rev. 2005, 105, 2999–3094. doi:10.1021/cr9904009
Return to citation in text: [1]
| 1. | Xie, S.; Sundhoro, M.; Houk, K. N.; Yan, M. Acc. Chem. Res. 2020, 53, 937–948. doi:10.1021/acs.accounts.0c00046 |
| 2. | Krell, K.; Harijan, D.; Ganz, D.; Doll, L.; Wagenknecht, H.-A. Bioconjugate Chem. 2020, 31, 990–1011. doi:10.1021/acs.bioconjchem.0c00072 |
| 3. | Kang, X.; Cai, X.; Yi, L.; Xi, Z. Chem. – Asian J. 2020, 15, 1420–1429. doi:10.1002/asia.202000005 |
| 4. | Li, K.; Fong, D.; Meichsner, E.; Adronov, A. Chem. – Eur. J. 2021, 27, 5057–5073. doi:10.1002/chem.202003386 |
| 5. | Li, X.; Xiong, Y. ACS Omega 2022, 7, 36918–36928. doi:10.1021/acsomega.2c03931 |
| 11. | Kivala, M.; Boudon, C.; Gisselbrecht, J.-P.; Enko, B.; Seiler, P.; Müller, I. B.; Langer, N.; Jarowski, P. D.; Gescheidt, G.; Diederich, F. Chem. – Eur. J. 2009, 15, 4111–4123. doi:10.1002/chem.200802563 |
| 12. | Tu, K.-H.; Wang, Y.; Kiyota, Y.; Iwahashi, T.; Ouchi, Y.; Mori, T.; Michinobu, T. Org. Electron. 2020, 87, 105978. doi:10.1016/j.orgel.2020.105978 |
| 25. | Tomasi, J.; Mennucci, B.; Cammi, R. Chem. Rev. 2005, 105, 2999–3094. doi:10.1021/cr9904009 |
| 9. | Biaggio, I. Chem. – Eur. J. 2022, 28, e202103168. doi:10.1002/chem.202103168 |
| 10. | Li, Y.; Tsuboi, K.; Michinobu, T. Macromolecules 2010, 43, 5277–5286. doi:10.1021/ma100869m |
| 8. | Michinobu, T.; Diederich, F. Angew. Chem., Int. Ed. 2018, 57, 3552–3577. doi:10.1002/anie.201711605 |
| 22. | Chai, J.-D.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. doi:10.1039/b810189b |
| 6. | Sen, R.; Escorihuela, J.; van Delft, F.; Zuilhof, H. Angew. Chem., Int. Ed. 2017, 56, 3299–3303. doi:10.1002/anie.201612037 |
| 7. | Gahtory, D.; Sen, R.; Kuzmyn, A. R.; Escorihuela, J.; Zuilhof, H. Angew. Chem., Int. Ed. 2018, 57, 10118–10122. doi:10.1002/anie.201800937 |
| 23. | Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650–654. doi:10.1063/1.438955 |
| 24. | McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639–5648. doi:10.1063/1.438980 |
| 19. | Zimmermann, B.; Baranović, G.; Štefanić, Z.; Rožman, M. J. Mol. Struct. 2006, 794, 115–124. doi:10.1016/j.molstruc.2006.01.049 |
| 20. | Jiang, J.; Kaafarani, B. R.; Neckers, D. C. J. Org. Chem. 2006, 71, 2155–2158. doi:10.1021/jo0522198 |
| 18. | Fukushima, S.; Ashizawa, M.; Kawauchi, S.; Michinobu, T. Helv. Chim. Acta 2019, 102, e1900016. doi:10.1002/hlca.201900016 |
| 14. | Washino, Y.; Michinobu, T. Phys. Chem. Chem. Phys. 2016, 18, 2288–2291. doi:10.1039/c5cp05180k |
| 15. | Fujita, H.; Michinobu, T. ACS Macro Lett. 2018, 7, 716–719. doi:10.1021/acsmacrolett.8b00365 |
| 16. | Fujita, H.; Michinobu, T. Soft Matter 2018, 14, 9055–9060. doi:10.1039/c8sm01986j |
| 17. | Fujita, H.; Nihei, N.; Bito, M.; Michinobu, T. Macromol. Biosci. 2018, 18, 1800336. doi:10.1002/mabi.201800336 |
| 13. | Li, Y.; Washino, Y.; Hyakutake, T.; Michinobu, T. Anal. Sci. 2017, 33, 599–604. doi:10.2116/analsci.33.599 |
| 18. | Fukushima, S.; Ashizawa, M.; Kawauchi, S.; Michinobu, T. Helv. Chim. Acta 2019, 102, e1900016. doi:10.1002/hlca.201900016 |
© 2024 Takeda et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.