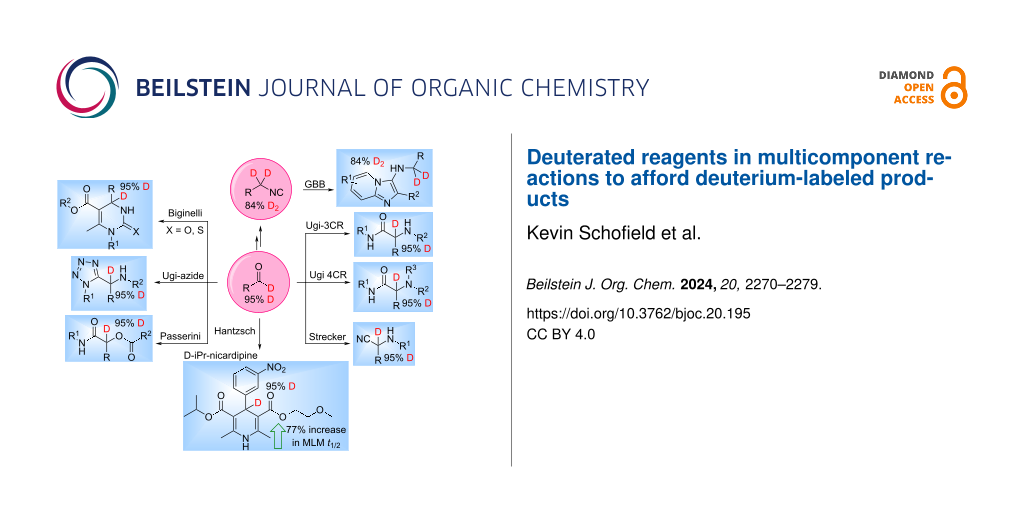
Abstract
The utility of bio-isosteres is broad in drug discovery and methodology herein enables the preparation of deuterium-labeled products is the most fundamental of known bio-isosteric replacements. As such we report the use of both [D1]-aldehydes and [D2]-isonitriles across 8 multicomponent reactions (MCRs) to give diverse arrays of deuterated products. A highlight is the synthesis of several FDA-approved calcium channel blockers, selectively deuterated at a t1/2 limiting metabolic soft-spot via use of [D1]-aldehydes. Surrogate pharmacokinetic analyses of microsomal stability confirm prolongation of t1/2 of the new deuterated analogs. We also report the first preparation of [D2]-isonitriles from [D3]-formamides via a modified Leuckart–Wallach reaction and their use in an MCR to afford products with [D2]-benzylic positions and likely significantly enhanced metabolic stability, a key parameter for property-based design efforts.

Graphical Abstract
Introduction
Multicomponent reactions (MCRs) are one-pot reactions that utilize three or more readily available starting materials [1-4]. Typically, MCRs use reactive functional groups such as ketones or aldehydes, carboxylic acids, amines, and isocyanides where these simple building blocks can be utilized to form large libraries of drug-like compounds with synthetic ease [5,6]. In recent years use of deuterium in drug discovery has expanded beyond mechanistic and tracer studies to deuterium incorporation in small molecules in attempts to hijack the deuterium kinetic isotope effect to induce longer drug t1/2 and greater systemic exposure [7-9]. Herein, we describe applications of deuterium-labeled reagents with MCRs through use of deuterated aldehydes and deuterated isocyanides, an area of study with sparingly few examples.
One example by Srivastava obtained a 65% deuterated Passerini product starting from a 65% deuterated aldehyde [10]. Latterly, Yamamoto utilized a 90% deuterated [D2]-isocyanide in a copper catalyzed [3 + 2] cycloaddition to afford a 60% deuterated [D2]-pyrrole [11]. The utility of the Leuckart–Wallach reaction towards the generation of isocyanides was first explored by Dömling [12], yet the use of such reagents in MCRs and determination of discrepancies in deuterium retention with MCRs has yet to be explored, although one would expect scrambling to be limited. Thus, we began by gathering highly deuterated aldehydes (>95% D) prepared via NHC catalysis [13] and developed a route to deuterated [D2]-benzylic isocyanides with a goal to apply them to the field of isocyanide multicomponent reaction (IMCR) chemistry which enables rapid access to arrays of biologically relevant chemotypes or secondary reactions thereafter [14,15]. Many of these chemotypes have populated corporate collections through in-house production or external purchase and have progressed along the value chain to the clinic and full approval [5]. Literature inspection reveals that an established common method to prepare deuterated benzylic isonitriles is reduction of a nitrile in the presence of a deuterium source (Scheme 1) [16-18].
![[1860-5397-20-195-i1]](/bjoc/content/inline/1860-5397-20-195-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Competitive examples of D2-benzylamine formation via phenyl-nitriles.
Scheme 1: Competitive examples of D2-benzylamine formation via phenyl-nitriles.
Results and Discussion
We hypothesized that [D1]-aldehydes could be converted to [D2]-benzylic isocyanides using [D2]-formic acid via Leuckart–Wallach reaction followed by dehydration. Surprisingly, the Leuckart–Wallach reaction gave [D3]-formamides which are scarce in the primary literature. The common method to prepare [D1]-formamides (D–C=O) is through a Leuckart–Wallach reaction with an amine and [D1]-methyl/ethyl formate or [D1]-dimethylformamide [19,20]. Stockmann and co-workers produced [D2]-formamides (N–D, D–C=O) via acid-catalyzed nitrile hydrolysis with HCl and D2O [21]. Thus, using the Leuckart–Wallach methodology developed herein, deuterated aldehydes can be converted into [D2]-isocyanides. The optimized conditions for this reaction are summarized below (Table 1).
Table 1: Optimization of deuterated Leuckart–Wallach reactiona.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-195-i12.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Aldehyde | C equiv | D equiv | Yield % | Deuteration% D1, D2 |
| 1 | A | 7.2 | 12 | 61 | 51, 42 |
| 2 | A | 6 | 1 | 24 | 70, 88 |
| 3 | A | 20 | 1 | 27 | 83, 94 |
| 4 | A | 12 | 10 | 80 | 27, 61 |
| 5b | B | 20 | 2 | 83 | 78, 40 |
aStandard conditions: 170 °C, 3 h. bReaction was heated to 180 °C for 5 minutes in a microwave reactor. D installed in product from D-aldehyde (A or B), D1 and D2 come from C. % Deuteration was determined via 1H NMR.
It is important to note that 1 equivalent of formamide and excess [D2]-formic acid (Table 1, entry 2) leads to increased deuteration of formamide product while increasing formamide equivalents increases the yield at the cost of deuterated %. Excess reaction time increases side product formation and thermal degradation of the aldehyde starting material. To combat this, microwave irradiation was employed which dramatically increased the overall yield of the reaction (Table 1, entry 5). In summary, minimal formamide (1–2 equiv), excess [D2]-formic acid, and heating to 170 °C in a microwave reactor for 5 minutes is expected to give excellent yields in good deuteration %.
A tentative mechanism to [D3]-formamides is shown in Scheme 2 but is open to debate. Thus, formamide adds to the [D1]-aldehyde A to form hemiaminal B which eliminates D2O to give imine D. Deprotonation of formamide D forms the resonance and zwitterrion-stabilized isocyanate E [22]. We then hypothesize that zwitterion E rearranges with loss of CO2 to form [D3]-formamide.
![[1860-5397-20-195-i2]](/bjoc/content/inline/1860-5397-20-195-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed tentative mechanism of [D3]-formamide formation via modified Leuckart–Wallach reaction with [D2]-formic acid.
Scheme 2: Proposed tentative mechanism of [D3]-formamide formation via modified Leuckart–Wallach reaction wit...
The ability to deuterate at benzylic positions is particularly relevant as benzylic C–H bonds are common in biologically relevant chemotypes and moreover appear in approximately 25% of the top selling 200 pharmaceuticals [23]. Benzyl cation stability is a driver of metabolism at these sites where benzylic C–H bonds readily undergo metabolism driven by cytochrome P450 oxidases via single-electron oxidation [24]. This metabolic lability may be tempered by hydrogen replacement with deuterium, an almost perfect bio-isosteric replacement (C–H to C–D) which maintains 3D surface, shape and flexibility [8]. Indeed, early incorporation of deuterium during hit generation may negate the need for late-stage C–H functionalization which often requires strong external oxidants or affords products with significantly lower biological activity [25-27]. Thus, eight MCRs were evaluated for D-reagent scope of reactivity and determination of deuterium retention using a combination of deuterated aldehydes, [D1]-, and/or [D2]-isocyanides.
We began with the venerable Ugi 4-component reaction (Ugi-4CR), first reported by Ivar Ugi in 1959 [28]. The Ugi-4CR utilizes an amine, carbonyl, carboxylic acid, and isocyanide component to afford α-aminoacyl amide derivatives 1a–g in good yield (Scheme 3). A [D1]-aldehyde and [D1]-isocyanide were independently used in conjunction with supporting reagents to afford 1b–d and 1e and 1f, respectively, with no observation of deuterium scrambling. A post-condensation modification of 1d, representative of the large swath of chemical space accessible by Ugi-deprotect-cyclize (UDC) methodology, gave the dihydroquinoxaline 1g in good yield with high deuterium retention [29-32].
![[1860-5397-20-195-i3]](/bjoc/content/inline/1860-5397-20-195-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Ugi-4CR products: no deuterium scrambling observed.
Scheme 3: Ugi-4CR products: no deuterium scrambling observed.
The catalytic three-component Ugi reaction was first reported by List in 2008 [33,34] and is comprised of reaction of an isonitrile, amine, and aldehyde/ketone, in the presence of phenylphosphinic acid (PPA), to give α-amino amides [35]. Examples of deuterated Ugi-3CR products are represented in Scheme 4. Like the Ugi 4-CR reaction, there was no deuterium scrambling in the Ugi 3-CR. Using a >95% deuterated aldehyde gave a >95% deuterated product. The low yield in 2c is likely due to the difficult preparation of aliphatic deuterated aldehydes which led us to believe that the starting material was partially decomposed. Nonetheless, both aliphatic and aromatic deuterated aldehydes have been demonstrated to work without loss of deuterium in the Ugi-3CR.
![[1860-5397-20-195-i4]](/bjoc/content/inline/1860-5397-20-195-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Ugi-3CR products. No deuterium scrambling observed.
Scheme 4: Ugi-3CR products. No deuterium scrambling observed.
First reported in 1961, the Ugi-azide reaction differs from the classical Ugi 4-CR in that an azide anion traps out the intermediate nitrilium ion, leading to formation of α-aminotetrazoles [36-39]. Thus, it comprises reaction of an isocyanide, carbonyl, amine and TMSN3 to give tetrazole containing products. Isolated yields for eight analogs are reported with excellent retention of the deuterium label (Scheme 5). Of note, 3i was prepared from combination of a [D2]-isocyanide and a [D1]-aldehyde.
![[1860-5397-20-195-i5]](/bjoc/content/inline/1860-5397-20-195-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Ugi-azide reaction products, no deuterium scrambling observed.
Scheme 5: Ugi-azide reaction products, no deuterium scrambling observed.
Another versatile IMCR is the Passerini reaction [40-43] discovered 60 years prior to the Ugi reaction. It uses the reactivity of isocyanides, aldehydes, and carboxylic acids to yield α-acyloxy amides (Scheme 6). Six deuterated analogs are reported in good yield with no observation of deuterium scrambling. For the preparation of 4d, a deuterated isocyanide was solely employed and for 4a,b and 4e,f, water was used as solvent. For the latter, product precipitates from the aqueous solution which deters undesirable side-reactions whilst also aiding rate of the reaction.
![[1860-5397-20-195-i6]](/bjoc/content/inline/1860-5397-20-195-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Passerini products, no deuterium scrambling observed. aWater was used as solvent.
Scheme 6: Passerini products, no deuterium scrambling observed. aWater was used as solvent.
The fifth MCR employed herein is the ubiquitous Strecker reaction [44,45] where a cyanide, aldehyde, and amine react to afford α-aminonitriles. Compound 5c was converted to the deuterated amino acid 5d under acidic conditions. This finding opens up the possibility of scale-production of deuterium-labeled α-amino acids. Deuterated Strecker products are presented in Scheme 7 in good yield with no observed deuterium scrambling.
![[1860-5397-20-195-i7]](/bjoc/content/inline/1860-5397-20-195-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Strecker reaction products (precursors to [D1]-α-amino acids), no deuterium scrambling was observed. aThe cyano-group was converted to a carboxylic acid via typical saponification conditions without loss of deuterium (See Supporting Information File 1 for conditions).
Scheme 7: Strecker reaction products (precursors to [D1]-α-amino acids), no deuterium scrambling was observed...
The 19th century Biginelli reaction utilizes an arylaldehyde, urea, and acetoacetate component to give 3,4-dihydropyrimidin-2(1H)-ones [46]. Such molecules are widely used as calcium channel blockers and antihypertensive agents (Scheme 8) [47].
![[1860-5397-20-195-i8]](/bjoc/content/inline/1860-5397-20-195-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Biginelli reaction products, no deuterium scrambling was observed. Six site-specific deuterated Biginelli products are revealed in good yield with no deuterium scrambling.
Scheme 8: Biginelli reaction products, no deuterium scrambling was observed. Six site-specific deuterated Big...
The Groebke–Blackburn–Bienaymé (GBB) reaction is an intramolecular variant of the Ugi reaction where the intermediate nitrilium ion is intercepted by heteroatoms from the amino-heterocyclic input. Discovered in 1998, and reported independently by three different research groups, it is a three-component reaction of α-amino-heterocycles, aldehydes, and isocyanides which affords various aza-bicyclic molecules [48-52]. The methodology has been widely used for file enhancement purposes. Three deuterated GBB products are presented, Scheme 9. Unlike the previous MCRs, labeling via a deuterated aldehyde is not feasible as deuterium is removed in favor of the aromatic bicyclic product. Both [D2]- and [D1]-isocyanides are exemplified in 7a–c.
![[1860-5397-20-195-i9]](/bjoc/content/inline/1860-5397-20-195-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: GBB reaction products, no deuterium scrambling was observed. aA 70% [D2]-isocyanide was used in 7a and 7b. Ar = 4-PhPh, Ar1 = 4-MePh.
Scheme 9: GBB reaction products, no deuterium scrambling was observed. aA 70% [D2]-isocyanide was used in 7a ...
Finally, we studied the compatibility of the Hantzsch dihydropyridine synthesis with [D1]-aldehydes. The reaction is a condensation of ethyl acetoacetate with aldehyde and ammonia to give 1,4-dihydropyridine [53]. Such scaffolds are seen in several FDA-approved calcium channel blockers including nifedipine, nicardipine and nimodipine and three site-specific deuterated analogs of approved 1,4-dihydropyridines (DHPs) are presented, Scheme 10. In line with all prior MCRs, good yields were observed with no deuterium scrambling.
![[1860-5397-20-195-i10]](/bjoc/content/inline/1860-5397-20-195-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Modified Hantzsch pyridine synthesis to afford 1,4-dihydropyridines. No deuterium scrambling was observed.
Scheme 10: Modified Hantzsch pyridine synthesis to afford 1,4-dihydropyridines. No deuterium scrambling was ob...
Calcium channel blockers derived from this methodology are heavily metabolized by CYP3A4 via dehydrogenation to afford inactive pyridines, Scheme 11 [54]. As such, bio-isosteric deuterium–hydrogen exchange at this position was thought a reasonable approach to extend drug t1/2 through exploitation of the kinetic isotope effect underpinned by the C–D bond being slightly shorter and stronger than a C–H bond. Such site-specific labeling was hypothesized to slow CYP3A4 metabolism.
![[1860-5397-20-195-i11]](/bjoc/content/inline/1860-5397-20-195-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: CYP3A4 mediated dehydrogenation of dihydropyridines.
Scheme 11: CYP3A4 mediated dehydrogenation of dihydropyridines.
To evaluate the hypothesis, surrogate studies were conducted in mouse liver microsomes to compare deuterated DHPs with their non-deuterated counterparts (Table 2).
Table 2: Microsomal stability package of deuterated and non-deuterated dihydropyridines.
| Sample ID | MLM t1/2 | MLM Cla |
| H-nicardipine | 2.8 min | 1968.2 |
| D-nicardipine | 3.0 min | 1860.8 |
| H-nimodipine | 2.1 min | 2572.3 |
| D-nimodipine | 2.6 min | 2098.5 |
| H-iPr-nicardipine | 8.0 min | 687.0 |
| D-iPr-nicardipine | 14.2 min | 386.6 |
aMouse liver microsomes clearance (MLM Cl), unit: mL/min/kg.
D-Nicardipine saw a marginal increase in stability when compared to its non-deuterated counterpart H-nicardipine (3.0 vs 2.8 min) with both molecules being rapidly metabolized. D-Nimodipine witnessed a 23% improvement in t1/2 over its non-deuterated counterpart although both molecules were quickly metabolized. The most significant improvement was seen with D-iPr-nicardipine – a 77% increase in MLM stability compared to H-iPr-nicardipine. Note that baseline stability of H-iPr-nicardipine was higher (8 min) related to enhanced ester stability with an isopropyl group versus a methyl ester. Collectively these results point to high potential for translation in vivo where novel deuterated analogs exhibit longer t1/2 and by extension oral bioavailability.
Conclusion
In summary we have presented the first method to prepare highly deuterated [D3]-formamides and [D2]-isocyanides from deuterated aldehydes. Furthermore, we have demonstrated that large libraries of deuterated drug-like molecules can be produced rapidly with MCR technology, with particular value for site selective deuteration of often metabolically soft benzylic C–H sites. Lastly and most importantly, preliminary surrogate metabolic stability studies on site selective [D1]-DHPs suggest these novel deuterated analogs may afford increased exposure in an in vivo setting. The methodology is likely to have wide utility for the drug-hunting community at large.
Supporting Information
| Supporting Information File 1: Experimental and analytical data and copies of NMR spectra. | ||
| Format: PDF | Size: 9.6 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Dömling, A.; Wang, W.; Wang, K. Chem. Rev. 2012, 112, 3083–3135. doi:10.1021/cr100233r
Return to citation in text: [1] -
Zhu, J.; Wang, Q.; Wang, M.-X. Multicomponent Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527678174
Return to citation in text: [1] -
Rotstein, B. H.; Zaretsky, S.; Rai, V.; Yudin, A. K. Chem. Rev. 2014, 114, 8323–8359. doi:10.1021/cr400615v
Return to citation in text: [1] -
Brauch, S.; van Berkel, S. S.; Westermann, B. Chem. Soc. Rev. 2013, 42, 4948–4962. doi:10.1039/c3cs35505e
Return to citation in text: [1] -
Hulme, C.; Ayaz, M.; Martinez-Ariza, G.; Medda, F.; Shaw, A. Recent Advances in Multicomponent Reaction Chemistry. In Small Molecule Medicinal Chemistry: Strategies and Technologies; Czechtizky, W.; Hamley, P., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp 145–187. doi:10.1002/9781118771723.ch6
Return to citation in text: [1] [2] -
Hulme, C.; Bienaymé, H.; Nixey, T.; Chenera, B.; Jones, W.; Tempest, P.; Smith, A. L. Methods Enzymol. 2003, 369, 469–496. doi:10.1016/s0076-6879(03)69024-5
Return to citation in text: [1] -
Gant, T. G. J. Med. Chem. 2014, 57, 3595–3611. doi:10.1021/jm4007998
Return to citation in text: [1] -
Pirali, T.; Serafini, M.; Cargnin, S.; Genazzani, A. A. J. Med. Chem. 2019, 62, 5276–5297. doi:10.1021/acs.jmedchem.8b01808
Return to citation in text: [1] [2] -
Meanwell, N. A. J. Med. Chem. 2011, 54, 2529–2591. doi:10.1021/jm1013693
Return to citation in text: [1] -
Ghoshal, A.; Ambule, M. D.; Sravanthi, R.; Taneja, M.; Srivastava, A. K. New J. Chem. 2019, 43, 14459–14474. doi:10.1039/c9nj03533h
Return to citation in text: [1] -
Kamijo, S.; Kanazawa, C.; Yamamoto, Y. J. Am. Chem. Soc. 2005, 127, 9260–9266. doi:10.1021/ja051875m
Return to citation in text: [1] -
Neochoritis, C. G.; Zarganes-Tzitzikas, T.; Stotani, S.; Dömling, A.; Herdtweck, E.; Khoury, K.; Dömling, A. ACS Comb. Sci. 2015, 17, 493–499. doi:10.1021/acscombsci.5b00066
Return to citation in text: [1] -
Geng, H.; Chen, X.; Gui, J.; Zhang, Y.; Shen, Z.; Qian, P.; Chen, J.; Zhang, S.; Wang, W. Nat. Catal. 2019, 2, 1071–1077. doi:10.1038/s41929-019-0370-z
Return to citation in text: [1] -
Ayaz, M.; De Moliner, F.; Dietrich, J.; Hulme, C. Applications of Isocyanides in IMCRs for the Rapid Generation of Molecular Diversity. In Isocyanide Chemistry: Applications in Synthesis and Material Science; Nenajdenko, V. G., Ed.; Wiley-VCH: Weinheim, Germany, 2012; pp 335–384. doi:10.1002/9783527652532.ch10
Return to citation in text: [1] -
Bedard, N.; Fistrovich, A.; Schofield, K.; Shaw, A.; Hulme, C. Recent Applications of Multicomponent Reactions Toward Heterocyclic Drug Discovery. In Multicomponent Reactions towards Heterocycles: Concepts and Applications; Van der Eycken, E. V.; Sharma, U. K., Eds.; Wiley-VCH: Weinheim, Germany, 2022; pp 339–409. doi:10.1002/9783527832439.ch9
Return to citation in text: [1] -
Kim, S. S.; Yang, K. W.; Lee, C. S. J. Org. Chem. 1996, 61, 4827–4829. doi:10.1021/jo940292k
Return to citation in text: [1] -
Kurita, T.; Aoki, F.; Mizumoto, T.; Maejima, T.; Esaki, H.; Maegawa, T.; Monguchi, Y.; Sajiki, H. Chem. – Eur. J. 2008, 14, 3371–3379. doi:10.1002/chem.200701245
Return to citation in text: [1] -
Ding, Y.; Luo, S.; Weng, C.; An, J. J. Org. Chem. 2019, 84, 15098–15105. doi:10.1021/acs.joc.9b02056
Return to citation in text: [1] -
Kesharwani, T.; Verma, A. K.; Emrich, D.; Ward, J. A.; Larock, R. C. Org. Lett. 2009, 11, 2591–2593. doi:10.1021/ol900940k
Return to citation in text: [1] -
Suchý, M.; Elmehriki, A. A. H.; Hudson, R. H. E. Org. Lett. 2011, 13, 3952–3955. doi:10.1021/ol201475j
Return to citation in text: [1] -
Stockmann, V.; Bakke, J. M.; Bruheim, P.; Fiksdahl, A. Tetrahedron 2009, 65, 3668–3672. doi:10.1016/j.tet.2009.02.072
Return to citation in text: [1] -
Kende, A. S.; Brands, K. M. J.; Blass, B. Tetrahedron Lett. 1993, 34, 579–582. doi:10.1016/s0040-4039(00)61624-6
Return to citation in text: [1] -
McGrath, N. A.; Brichacek, M.; Njardarson, J. T. J. Chem. Educ. 2010, 87, 1348–1349. doi:10.1021/ed1003806
Return to citation in text: [1] -
Engst, W.; Landsiedel, R.; Hermersdörfer, H.; Doehmer, J.; Glatt, H. Carcinogenesis 1999, 20, 1777–1785. doi:10.1093/carcin/20.9.1777
Return to citation in text: [1] -
Xu, W.; Wang, W.; Liu, T.; Xie, J.; Zhu, C. Nat. Commun. 2019, 10, 4867. doi:10.1038/s41467-019-12844-9
Return to citation in text: [1] -
Guo, S.; AbuSalim, D. I.; Cook, S. P. J. Am. Chem. Soc. 2018, 140, 12378–12382. doi:10.1021/jacs.8b08547
Return to citation in text: [1] -
Qin, Y.; Zhu, L.; Luo, S. Chem. Rev. 2017, 117, 9433–9520. doi:10.1021/acs.chemrev.6b00657
Return to citation in text: [1] -
Ugi, I. Angew. Chem. 1959, 71, 373–388. doi:10.1002/ange.19590711110
Return to citation in text: [1] -
Tempest, P.; Ma, V.; Thomas, S.; Hua, Z.; Kelly, M. G.; Hulme, C. Tetrahedron Lett. 2001, 42, 4959–4962. doi:10.1016/s0040-4039(01)00919-4
Return to citation in text: [1] -
Dietrich, J.; Kaiser, C.; Meurice, N.; Hulme, C. Tetrahedron Lett. 2010, 51, 3951–3955. doi:10.1016/j.tetlet.2010.05.108
Return to citation in text: [1] -
Hulme, C.; Gore, V. Curr. Med. Chem. 2003, 10, 51–80. doi:10.2174/0929867033368600
Return to citation in text: [1] -
Xu, Z.; De Moliner, F.; Cappelli, A. P.; Ayaz, M.; Hulme, C. Synlett 2014, 25, 225–228. doi:10.1055/s-0033-1340219
Return to citation in text: [1] -
Pan, S. C.; List, B. Angew. Chem., Int. Ed. 2008, 47, 3622–3625. doi:10.1002/anie.200800494
Return to citation in text: [1] -
Ayaz, M.; Martinez-Ariza, G.; Hulme, C. Synlett 2014, 25, 1680–1684. doi:10.1055/s-0033-1339135
Return to citation in text: [1] -
Tripolitsiotis, N. P.; Thomaidi, M.; Neochoritis, C. G. Eur. J. Org. Chem. 2020, 6525–6554. doi:10.1002/ejoc.202001157
Return to citation in text: [1] -
Ugi, I.; Steinbrückner, C. Angew. Chem. 1960, 72, 267–268. doi:10.1002/ange.19600720709
Return to citation in text: [1] -
Ugi, I.; Steinbrückner, C. Chem. Ber. 1961, 94, 734–742. doi:10.1002/cber.19610940323
Return to citation in text: [1] -
Gunawan, S.; Petit, J.; Hulme, C. ACS Comb. Sci. 2012, 14, 160–163. doi:10.1021/co200209a
Return to citation in text: [1] -
Gunawan, S.; Hulme, C. Org. Biomol. Chem. 2013, 11, 6036–6046. doi:10.1039/c3ob40900g
Return to citation in text: [1] -
Passerini, M.; Gazz, S. Gazz. Chim. Ital. 1921, 51, 126–129.
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Lambruschini, C.; Moni, L.; Riva, R. Chem. Sci. 2021, 12, 15445–15472. doi:10.1039/d1sc03810a
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Guanti, G.; Riva, R. Mol. Diversity 2003, 6, 227–235. doi:10.1023/b:modi.0000006778.42751.7f
Return to citation in text: [1] -
Shaw, A. Y.; Medda, F.; Hulme, C. Tetrahedron Lett. 2012, 53, 1313–1315. doi:10.1016/j.tetlet.2011.12.073
Return to citation in text: [1] -
Strecker, A. Justus Liebigs Ann. Chem. 1850, 75, 27–45. doi:10.1002/jlac.18500750103
Return to citation in text: [1] -
Ayaz, M.; De Moliner, F.; Morales, G. A.; Hulme, C. Sci. Synth. 2014, 99–122. doi:10.1055/sos-sd-210-00029
Return to citation in text: [1] -
Biginelli, P. Ber. Dtsch. Chem. Ges. 1891, 24, 1317–1319. doi:10.1002/cber.189102401228
Return to citation in text: [1] -
Rovnyak, G. C.; Atwal, K. S.; Hedberg, A.; Kimball, S. D.; Moreland, S.; Gougoutas, J. Z.; O'Reilly, B. C.; Schwartz, J.; Malley, M. F. J. Med. Chem. 1992, 35, 3254–3263. doi:10.1021/jm00095a023
Return to citation in text: [1] -
Groebke, K.; Weber, L.; Mehlin, F. Synlett 1998, 661–663. doi:10.1055/s-1998-1721
Return to citation in text: [1] -
Blackburn, C.; Guan, B.; Fleming, P.; Shiosaki, K.; Tsai, S. Tetrahedron Lett. 1998, 39, 3635–3638. doi:10.1016/s0040-4039(98)00653-4
Return to citation in text: [1] -
Bienaymé, H.; Bouzid, K. Angew. Chem., Int. Ed. 1998, 37, 2234–2237. doi:10.1002/(sici)1521-3773(19980904)37:16<2234::aid-anie2234>3.3.co;2-i
Return to citation in text: [1] -
Hulme, C.; Lee, Y.-S. Mol. Diversity 2008, 12, 1–15. doi:10.1007/s11030-008-9072-1
Return to citation in text: [1] -
Umkehrer, M.; Ross, G.; Jäger, N.; Burdack, C.; Kolb, J.; Hu, H.; Alvim-Gaston, M.; Hulme, C. Tetrahedron Lett. 2007, 48, 2213–2216. doi:10.1016/j.tetlet.2007.01.061
Return to citation in text: [1] -
Hantzsch, A. Ber. Dtsch. Chem. Ges. 1881, 14, 1637–1638. doi:10.1002/cber.18810140214
Return to citation in text: [1] -
Zhu, Y.; Wang, F.; Li, Q.; Zhu, M.; Du, A.; Tang, W.; Chen, W. Drug Metab. Dispos. 2014, 42, 245–249. doi:10.1124/dmd.113.055400
Return to citation in text: [1]
| 53. | Hantzsch, A. Ber. Dtsch. Chem. Ges. 1881, 14, 1637–1638. doi:10.1002/cber.18810140214 |
| 54. | Zhu, Y.; Wang, F.; Li, Q.; Zhu, M.; Du, A.; Tang, W.; Chen, W. Drug Metab. Dispos. 2014, 42, 245–249. doi:10.1124/dmd.113.055400 |
| 1. | Dömling, A.; Wang, W.; Wang, K. Chem. Rev. 2012, 112, 3083–3135. doi:10.1021/cr100233r |
| 2. | Zhu, J.; Wang, Q.; Wang, M.-X. Multicomponent Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527678174 |
| 3. | Rotstein, B. H.; Zaretsky, S.; Rai, V.; Yudin, A. K. Chem. Rev. 2014, 114, 8323–8359. doi:10.1021/cr400615v |
| 4. | Brauch, S.; van Berkel, S. S.; Westermann, B. Chem. Soc. Rev. 2013, 42, 4948–4962. doi:10.1039/c3cs35505e |
| 11. | Kamijo, S.; Kanazawa, C.; Yamamoto, Y. J. Am. Chem. Soc. 2005, 127, 9260–9266. doi:10.1021/ja051875m |
| 24. | Engst, W.; Landsiedel, R.; Hermersdörfer, H.; Doehmer, J.; Glatt, H. Carcinogenesis 1999, 20, 1777–1785. doi:10.1093/carcin/20.9.1777 |
| 10. | Ghoshal, A.; Ambule, M. D.; Sravanthi, R.; Taneja, M.; Srivastava, A. K. New J. Chem. 2019, 43, 14459–14474. doi:10.1039/c9nj03533h |
| 8. | Pirali, T.; Serafini, M.; Cargnin, S.; Genazzani, A. A. J. Med. Chem. 2019, 62, 5276–5297. doi:10.1021/acs.jmedchem.8b01808 |
| 7. | Gant, T. G. J. Med. Chem. 2014, 57, 3595–3611. doi:10.1021/jm4007998 |
| 8. | Pirali, T.; Serafini, M.; Cargnin, S.; Genazzani, A. A. J. Med. Chem. 2019, 62, 5276–5297. doi:10.1021/acs.jmedchem.8b01808 |
| 9. | Meanwell, N. A. J. Med. Chem. 2011, 54, 2529–2591. doi:10.1021/jm1013693 |
| 22. | Kende, A. S.; Brands, K. M. J.; Blass, B. Tetrahedron Lett. 1993, 34, 579–582. doi:10.1016/s0040-4039(00)61624-6 |
| 5. | Hulme, C.; Ayaz, M.; Martinez-Ariza, G.; Medda, F.; Shaw, A. Recent Advances in Multicomponent Reaction Chemistry. In Small Molecule Medicinal Chemistry: Strategies and Technologies; Czechtizky, W.; Hamley, P., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp 145–187. doi:10.1002/9781118771723.ch6 |
| 6. | Hulme, C.; Bienaymé, H.; Nixey, T.; Chenera, B.; Jones, W.; Tempest, P.; Smith, A. L. Methods Enzymol. 2003, 369, 469–496. doi:10.1016/s0076-6879(03)69024-5 |
| 23. | McGrath, N. A.; Brichacek, M.; Njardarson, J. T. J. Chem. Educ. 2010, 87, 1348–1349. doi:10.1021/ed1003806 |
| 5. | Hulme, C.; Ayaz, M.; Martinez-Ariza, G.; Medda, F.; Shaw, A. Recent Advances in Multicomponent Reaction Chemistry. In Small Molecule Medicinal Chemistry: Strategies and Technologies; Czechtizky, W.; Hamley, P., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp 145–187. doi:10.1002/9781118771723.ch6 |
| 19. | Kesharwani, T.; Verma, A. K.; Emrich, D.; Ward, J. A.; Larock, R. C. Org. Lett. 2009, 11, 2591–2593. doi:10.1021/ol900940k |
| 20. | Suchý, M.; Elmehriki, A. A. H.; Hudson, R. H. E. Org. Lett. 2011, 13, 3952–3955. doi:10.1021/ol201475j |
| 14. | Ayaz, M.; De Moliner, F.; Dietrich, J.; Hulme, C. Applications of Isocyanides in IMCRs for the Rapid Generation of Molecular Diversity. In Isocyanide Chemistry: Applications in Synthesis and Material Science; Nenajdenko, V. G., Ed.; Wiley-VCH: Weinheim, Germany, 2012; pp 335–384. doi:10.1002/9783527652532.ch10 |
| 15. | Bedard, N.; Fistrovich, A.; Schofield, K.; Shaw, A.; Hulme, C. Recent Applications of Multicomponent Reactions Toward Heterocyclic Drug Discovery. In Multicomponent Reactions towards Heterocycles: Concepts and Applications; Van der Eycken, E. V.; Sharma, U. K., Eds.; Wiley-VCH: Weinheim, Germany, 2022; pp 339–409. doi:10.1002/9783527832439.ch9 |
| 21. | Stockmann, V.; Bakke, J. M.; Bruheim, P.; Fiksdahl, A. Tetrahedron 2009, 65, 3668–3672. doi:10.1016/j.tet.2009.02.072 |
| 13. | Geng, H.; Chen, X.; Gui, J.; Zhang, Y.; Shen, Z.; Qian, P.; Chen, J.; Zhang, S.; Wang, W. Nat. Catal. 2019, 2, 1071–1077. doi:10.1038/s41929-019-0370-z |
| 12. | Neochoritis, C. G.; Zarganes-Tzitzikas, T.; Stotani, S.; Dömling, A.; Herdtweck, E.; Khoury, K.; Dömling, A. ACS Comb. Sci. 2015, 17, 493–499. doi:10.1021/acscombsci.5b00066 |
| 16. | Kim, S. S.; Yang, K. W.; Lee, C. S. J. Org. Chem. 1996, 61, 4827–4829. doi:10.1021/jo940292k |
| 17. | Kurita, T.; Aoki, F.; Mizumoto, T.; Maejima, T.; Esaki, H.; Maegawa, T.; Monguchi, Y.; Sajiki, H. Chem. – Eur. J. 2008, 14, 3371–3379. doi:10.1002/chem.200701245 |
| 18. | Ding, Y.; Luo, S.; Weng, C.; An, J. J. Org. Chem. 2019, 84, 15098–15105. doi:10.1021/acs.joc.9b02056 |
| 29. | Tempest, P.; Ma, V.; Thomas, S.; Hua, Z.; Kelly, M. G.; Hulme, C. Tetrahedron Lett. 2001, 42, 4959–4962. doi:10.1016/s0040-4039(01)00919-4 |
| 30. | Dietrich, J.; Kaiser, C.; Meurice, N.; Hulme, C. Tetrahedron Lett. 2010, 51, 3951–3955. doi:10.1016/j.tetlet.2010.05.108 |
| 31. | Hulme, C.; Gore, V. Curr. Med. Chem. 2003, 10, 51–80. doi:10.2174/0929867033368600 |
| 32. | Xu, Z.; De Moliner, F.; Cappelli, A. P.; Ayaz, M.; Hulme, C. Synlett 2014, 25, 225–228. doi:10.1055/s-0033-1340219 |
| 25. | Xu, W.; Wang, W.; Liu, T.; Xie, J.; Zhu, C. Nat. Commun. 2019, 10, 4867. doi:10.1038/s41467-019-12844-9 |
| 26. | Guo, S.; AbuSalim, D. I.; Cook, S. P. J. Am. Chem. Soc. 2018, 140, 12378–12382. doi:10.1021/jacs.8b08547 |
| 27. | Qin, Y.; Zhu, L.; Luo, S. Chem. Rev. 2017, 117, 9433–9520. doi:10.1021/acs.chemrev.6b00657 |
| 47. | Rovnyak, G. C.; Atwal, K. S.; Hedberg, A.; Kimball, S. D.; Moreland, S.; Gougoutas, J. Z.; O'Reilly, B. C.; Schwartz, J.; Malley, M. F. J. Med. Chem. 1992, 35, 3254–3263. doi:10.1021/jm00095a023 |
| 48. | Groebke, K.; Weber, L.; Mehlin, F. Synlett 1998, 661–663. doi:10.1055/s-1998-1721 |
| 49. | Blackburn, C.; Guan, B.; Fleming, P.; Shiosaki, K.; Tsai, S. Tetrahedron Lett. 1998, 39, 3635–3638. doi:10.1016/s0040-4039(98)00653-4 |
| 50. | Bienaymé, H.; Bouzid, K. Angew. Chem., Int. Ed. 1998, 37, 2234–2237. doi:10.1002/(sici)1521-3773(19980904)37:16<2234::aid-anie2234>3.3.co;2-i |
| 51. | Hulme, C.; Lee, Y.-S. Mol. Diversity 2008, 12, 1–15. doi:10.1007/s11030-008-9072-1 |
| 52. | Umkehrer, M.; Ross, G.; Jäger, N.; Burdack, C.; Kolb, J.; Hu, H.; Alvim-Gaston, M.; Hulme, C. Tetrahedron Lett. 2007, 48, 2213–2216. doi:10.1016/j.tetlet.2007.01.061 |
| 44. | Strecker, A. Justus Liebigs Ann. Chem. 1850, 75, 27–45. doi:10.1002/jlac.18500750103 |
| 45. | Ayaz, M.; De Moliner, F.; Morales, G. A.; Hulme, C. Sci. Synth. 2014, 99–122. doi:10.1055/sos-sd-210-00029 |
| 46. | Biginelli, P. Ber. Dtsch. Chem. Ges. 1891, 24, 1317–1319. doi:10.1002/cber.189102401228 |
| 36. | Ugi, I.; Steinbrückner, C. Angew. Chem. 1960, 72, 267–268. doi:10.1002/ange.19600720709 |
| 37. | Ugi, I.; Steinbrückner, C. Chem. Ber. 1961, 94, 734–742. doi:10.1002/cber.19610940323 |
| 38. | Gunawan, S.; Petit, J.; Hulme, C. ACS Comb. Sci. 2012, 14, 160–163. doi:10.1021/co200209a |
| 39. | Gunawan, S.; Hulme, C. Org. Biomol. Chem. 2013, 11, 6036–6046. doi:10.1039/c3ob40900g |
| 40. | Passerini, M.; Gazz, S. Gazz. Chim. Ital. 1921, 51, 126–129. |
| 41. | Banfi, L.; Basso, A.; Lambruschini, C.; Moni, L.; Riva, R. Chem. Sci. 2021, 12, 15445–15472. doi:10.1039/d1sc03810a |
| 42. | Banfi, L.; Basso, A.; Guanti, G.; Riva, R. Mol. Diversity 2003, 6, 227–235. doi:10.1023/b:modi.0000006778.42751.7f |
| 43. | Shaw, A. Y.; Medda, F.; Hulme, C. Tetrahedron Lett. 2012, 53, 1313–1315. doi:10.1016/j.tetlet.2011.12.073 |
| 33. | Pan, S. C.; List, B. Angew. Chem., Int. Ed. 2008, 47, 3622–3625. doi:10.1002/anie.200800494 |
| 34. | Ayaz, M.; Martinez-Ariza, G.; Hulme, C. Synlett 2014, 25, 1680–1684. doi:10.1055/s-0033-1339135 |
| 35. | Tripolitsiotis, N. P.; Thomaidi, M.; Neochoritis, C. G. Eur. J. Org. Chem. 2020, 6525–6554. doi:10.1002/ejoc.202001157 |
© 2024 Schofield et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.