Abstract
A series of new RS−, RS−CH2− and R2N−CH2-functionalized сatechols with heterocyclic fragments such as 1,3,4-oxadiazole, 1,2,4-triazole, thiazole, or pyridine were synthesized by the reaction of 3,5-di-tert-butyl-o-benzoquinone or 3,5-di-tert-butyl-6-methoxymethylcatechol with different heterocyclic thiols. The S-functionalized catechols were prepared by the Michael reaction from 3,5-di-tert-butyl-o-benzoquinone and the corresponding thiols. The starting reagents such as substituted 1,3,4-oxadiazole-2-thiols and 4H-triazole-3-thiols are characterized by thiol–thione tautomerism, therefore their reactions with 3,5-di-tert-butyl-6-methoxymethylcatechol can proceed at the sulfur or nitrogen atom. In the case of mercapto-derivatives of thiazole or pyridine, this process leads to the formation of the corresponding thioethers with a methylene linker. At the same time, thiolated 1,3,4-oxadiazole or 1,2,4-triazole undergo alkylation at the nitrogen atom in the reaction with 3,5-di-tert-butyl-6-methoxymethylcatechol to form the corresponding thiones. The yield of reaction products ranges from 42 to 80%. The crystal structures of catechols with 3-nitropyridine or 1,3,4-oxadiazole-2(3H)-thione moieties were established by single-crystal X-ray analysis. The possibility of forming intra- and intermolecular hydrogen bonds has been established for these compounds. The electrochemical behavior of the studied compounds is influenced by several factors: the nature of the heterocycle and its substituents, the presence of a sulfur atom in the catechol ring, or a thione group in the heterocyclic core. The radical scavenging activity and antioxidant properties were determined using the reaction with synthetic radicals, the cupric reducing antioxidant capacity assay, the inhibition process of superoxide radical anion formation by xanthine oxidase, and the process of lipid peroxidation of rat liver (Wistar) homogenates in vitro.

Graphical Abstract
Introduction
Synthetic derivatives of polyphenols, in particular catechol (hydroquinone), represent a promising group of pharmacologically active substances [1,2]. Catechol-containing compounds demonstrate neuroprotective, antihypoxic effects, act as antiparkinsonian agents [3-5], exhibit antitumor and antibacterial activity [6-8], possess antioxidant properties for regulating free radical processes [9-11]. The functionalization of polyphenolic compounds by introducing various substituents or heteroatoms (nitrogen, sulfur, selenium, tellurium, etc.) as well as redox-active functional groups allows one to vary significantly the biological activity of such compounds.
Heterocyclic molecular blocks are widely used in medicinal chemistry [12]. Thiazole, oxadiazole, triazole, imidazole, and other heterocycles are classified as privileged medicinal scaffolds being components of many drugs [13,14]. Thiazole derivatives and their reduced forms exhibit antitumor (thiazofurin, ixabepilone), antibacterial (cefotaxime, ceftaroline, cefiderocol), antifungal (isavuconazole, fosravuconazole), antiviral (simeprevir, ritonavir, cobicistat), and other types of biological activities [15-19]. Besides, among the registered sulfur-containing drugs, 19% contain a thiazole ring, and 15% contain a thioether group [20]. Oxadiazole fragments are found in the structures of anticancer drugs (zibotentan), antituberculosis agents [13], as well as in drugs against arrhythmia (nesapidil) or muscular dystrophy (ataluren) [21]. The 1,3,4- and 1,2,4-oxadiazoles are the most promising from the point of view of bioactivity among the four isomers of oxadiazole. The presence of a 1,3,4-oxadiazole ring in the structure of compounds leads to an increased lipophilicity and, as a result, facilitates the transfer of the molecule through cell membranes to the target site [22]. 1,2,4-Triazole derivatives also play a key role in medicinal chemistry, especially in the development of new antivirals [23,24] and antibacterial agents [25].
A feature of mercapto-substituted heterocycles is the possibility of their existence in two tautomeric forms: thiol or thione [26,27]. The ability to tautomeric transformations affects their biological activity and allows the use of these molecular blocks to design compounds with different properties [28]. Anti-tuberculosis activity was identified for 5-substituted 2-mercapto-1,3,4-oxadiazoles [29], 5-substituted-2-[(3,5-dinitrobenzyl)-sulfanyl]-1,3,4-oxadiazoles and 1,3,4-thiadiazoles [30]. Some derivatives of 5-(4-tert-butylphenyl)-1,3,4-oxadiazole-2(3H)-thione showed antiproliferative activity against the HeLa cancer cell line [22]. 1,2,4-Triazole thioglycoside derivatives have antiviral activity against influenza strains H3N2 and H5N1 [31,32]. The 1,2,4-triazole-3-thione-imidazole hybrids displayed potential antibacterial activity against E. coli, S. aureus [33]. Heterocyclic thione derivatives are used as ligands in the synthesis of metal complexes (Ni(II), Pd(II), Pt(II), Cu(I), Ag(I) etc.) exhibiting antibacterial and antitumor activity [34].
In most cases, polyfunctional catechol thioethers were obtained by Michael reaction via the interaction of o-, p-benzoquinone and the corresponding thiol [35-38], in the nucleophilic substitution reaction in the aromatic ring of catechol [39,40] or under electrochemical conditions [41-43]. An anodic activation of catechols in the presence of a thiol leads to S-functionalized catechols with triazole, triazine, pyrimidine, imidazole, thiadiazole, or other fragments [44-48].
Previously, we obtained a series of sterically hindered catechols linked through a sulfide bridge with various polar or low-polar groups [36,49-51] and heterocyclic fragments [52] via the Michael addition reaction. Also, we have previously found that the reaction between 3,5-di-tert-butyl-6-methoxymethylcatechol and different functionalized thiols occurs via an acid-catalyzed mechanism and leads to corresponding thioethers with methylene linker [53,54]. Hybrid structures of this type are of particular interest from the point of view of studying their potential biological activity.
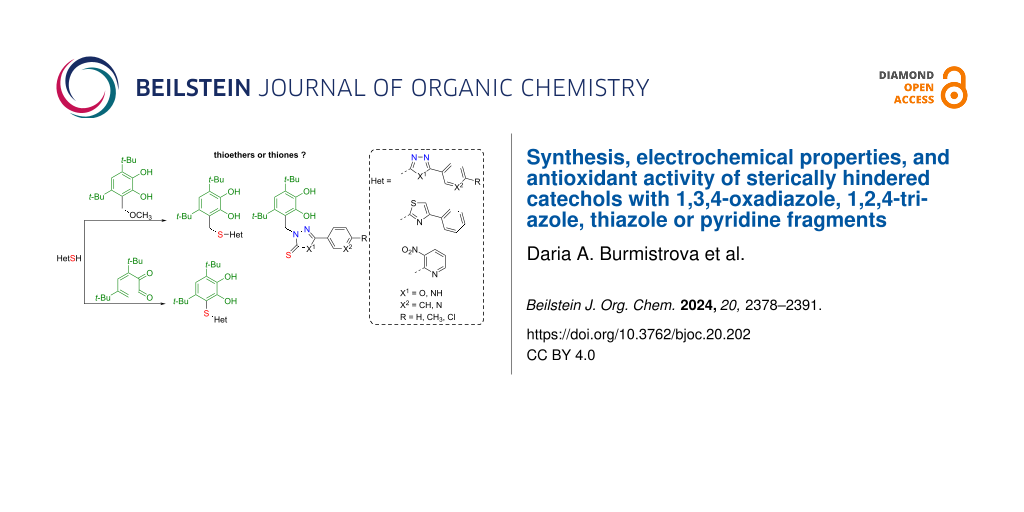
This work aimed to obtain new multifunctional catechols containing pharmacologically active heterocyclic fragments such as thiazole, 1,3,4-oxadiazole, 1,2,4-triazole, and pyridine. We decided to use a non-hydrolyzable thioether group as a bridging moiety. A feature of heterocyclic thiols used as starting reagents is the possibility of thiol–thione tautomerism which leads to the appearance of two nucleophilic centers: a sulfur or nitrogen atom. Therefore, in the case of their interaction with 3,5-di-tert-butyl-6-methoxymethylcatechol, the possibility of alkylation of the nitrogen atom in the heterocycle cannot be excluded. This alternative pathway is similar to the previously described interaction of the aforementioned catechol with 3,5-dimethylpyrazole or benzimidazole [55]. A study of the structure, electrochemical properties, anti/prooxidant, and antiradical activity was carried out for the catechols synthesized in this work.
Results and Discussion
Synthesis
The interaction of 3,5-di-tert-butyl-o-benzoquinone with the corresponding thiols in ethanol at room temperature under argon leads to the formation of catechol thioethers 1–3 (69–80%) (Scheme 1a). The reaction between 3,5-di-tert-butyl-6-methoxymethylcatechol and 2-mercapto-4-phenylthiazole or 3-nitropyridine-2-thiol occurs via an acid-catalyzed mechanism and leads to corresponding thioethers 4, 5 with methylene linker in a good yield (80% and 63%, correspondingly) (Scheme 1b). At the same time, the interaction of 2-mercapto-substituted derivatives of 1,3,4-oxadiazole or 1,2,4-triazole with the starting catechol proceeds at the nitrogen atom of the five-membered heterocycle due to the possibility of the thione–thiol tautomerism in the starting thiol [26,27]. Consequently, compounds 6–9 are products of alkylation of the nitrogen atom of the heterocycle. Thiones 6–9 were obtained in 40–79% yield (Scheme 1,b).
![[1860-5397-20-202-i1]](/bjoc/content/inline/1860-5397-20-202-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of catechol-containing compounds 1–9.
Scheme 1: Synthesis of catechol-containing compounds 1–9.
The structures of synthesized compounds were confirmed by the spectral methods IR-, 1H NMR, 13C{1H} NMR spectroscopy (Figures S1–S18 in Supporting Information File 1), HRMS, and elemental analysis. The molecular structures of compounds 5, 6 and 8 in crystal state were determined by single-crystal X-ray analysis.
X-ray data
The X-ray suitable crystals 5, 6·0.5CH3CN, and 8 were grown by slow recrystallization of the compounds from acetonitrile solutions. The unit cells of complexes 5 and 8 do not contain any solvent molecules, while the unit cell of crystals 6·0.5CH3CN contains one acetonitrile molecule per two catechol molecules. The nitro-substituted pyridine group in 5 is disordered in two positions. The X-ray structure of catechol 5 is shown in Figure 1, and the X-ray structures of catechols 6, and 8 are shown in Figure 2.
![[1860-5397-20-202-1]](/bjoc/content/figures/1860-5397-20-202-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The X-ray structure of catechol 5 (the thermal ellipsoids of 50% probability). The hydrogen atoms except those of hydroxy groups are omitted.
Figure 1: The X-ray structure of catechol 5 (the thermal ellipsoids of 50% probability). The hydrogen atoms e...
![[1860-5397-20-202-2]](/bjoc/content/figures/1860-5397-20-202-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The X-ray structures of catechols 6 (a) and 8 (b) (the thermal ellipsoids of 50% probability). The hydrogen atoms except those of hydroxy groups are omitted.
Figure 2: The X-ray structures of catechols 6 (a) and 8 (b) (the thermal ellipsoids of 50% probability). The ...
The geometrical characteristics of catechol moieties of these compounds, in general, have the standard parameters: the bonds C1–O1, C2–O2 are 1.369(11) and 1.377(9) Å in 5, 1.371(2) and 1.387(2) Å in 6, 1.389(3) and 1.377(3) Å in 8; the average lengths of the С–С bonds of six-membered carbon cycle С(1–6) (1.396 ± 0.013 Å in 5, 1.397 ± 0.009 Å in 6, 1.396 ± 0.012 Å in 8) are typical for catechols [50,55,56].
The presence of complex polyfunctional substituents affects their spatial location regarding a catechol fragment. In most cases, the catechol molecules in the crystal are located in such a way that catechol hydroxy fragments look at each other and, in addition to intra-molecular hydrogen bond O–H···O between the two hydroxy groups, the intermolecular hydrogen interactions are also being realized between the catechol fragments of neighboring molecules [57-61]. The intra- and intermolecular interactions in crystals 5, 6·0.5CH3CN, and 8 lead to different complex structural motifs in crystals of these compounds.
Two intramolecular hydrogen bonds (Figure 1) are observed in the crystal structure of 5: between OH groups (O1–H1···O2: the H1···O2 distance is 1.98(1) Å, the O1···O2 distance is 2.581(3) Å, angle O1–H1–O2 is 117°) and between hydroxy group O2–H2 and nitrogen atom N1 of the pyridine cycle (O2–H2···N1A/B: the H2···N1 distance is 1.75(1)/2.08(1) Å, the O2···N1 distance is 2.560(3)/2.893(3) Å, angle O2–H2–N1 is 160°/164°, here and further, two data sets are given, taking into account the pyridine cycle disorder).
In the crystal of 5, two molecules form pairs due to the π–π interactions between pyridine cycles (the Cg···Cg distance between centroids is 3.855(4) and 3.891(4) Å; the angle between the planes of the cycles corresponds to 5°). These pairs (Figure 3) are bound in a supramolecular chain along the 0c axis owing to two H-bonds between the OH group of catechol and the oxygen atom of nitro-group (O1–H1···O4B (1−x, 1−y, −1+z): the H1···O4 distance is 2.46(1) Å, the O1···O4 distance is 2.886(3) Å, the angle O1–H1–O4 106°; O1–H1···O4A: the H1···O4 distance is 2.82(1) Å, the O1···O4 distance is 3.146(3) Å, the angle O1–H1–O4 101°, Figure 3). Additionally, the packing of these chains in the crystal is caused by the intermolecular C–H···O and N–O···π contact.
![[1860-5397-20-202-3]](/bjoc/content/figures/1860-5397-20-202-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Fragment of the pack of catechol 5 in crystal (the H-bonds and π–π interactions are shown as dotted lines).
Figure 3: Fragment of the pack of catechol 5 in crystal (the H-bonds and π–π interactions are shown as dotted...
Catechol 6 in crystal also forms pairs formed by two crystallographically independent molecules А and В (Figure 4). Intermolecular hydrogen interaction O1–H1···O2 takes place in both molecules of this pair (the distance H1A···O2A is 2.09(1) Å, the angle O1A–H1A–O2A is 122°; the distance H1B···O2B is 2.14(1) Å, the angle O1B–H1B–O2B is 121.6°). In addition, the intermolecular H-bonding is observed between group O2–H2 of molecule A and the thione group C=S of molecule B (the distance H2A···S1B is 2.42(1) Å, the angle O2A–H2A–S1B is 161°).
![[1860-5397-20-202-4]](/bjoc/content/figures/1860-5397-20-202-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The interactions in pair of independent molecules A and B of 6 in crystal 6·0.5CH3CN (the H-bonds are shown as dotted lines).
Figure 4: The interactions in pair of independent molecules A and B of 6 in crystal 6·0.5CH3CN (the H-bonds a...
At the same time, for molecule B, there is a hydrogen bond between the O2B–H2B group with the nitrogen atom N3 of the nitrile group of the solvent acetonitrile molecule presenting in the cell: the corresponding distance H2B···N3 is 2.03(1) Å, the angle O2B–H2B–N3 is 169°. As a result, molecules A and B in this pair are located according to the principle of "chairs inserted into each other."
The intermolecular H-bonding O2–H2···O1 between catechol moieties of two neighbouring molecules leading to pairs was also observed in the crystal of catechol 8 (Figure 5): The H2···O1 distance is 2.06 (1) Å, the O2···O1 distance is 2.87(1) Å, the angle O2–H2–O1 is 151.4°. However, the additional intermolecular contacts O1–H1···N3 (the H1···N3 distance is 1.84(1) Å, the O1···N3 distance is 2.73(1) Å, the angle O1–H1–N3 is 168.3°) with the pyridine group of the catechol moiety of the neighbouring pair lead to the formation of chains. As a result of such interaction, the hydroxy group O1–H1 is turned away from the neighbouring group O2–H2, and there is no intramolecular hydrogen bond between hydroxy groups in molecule 8 (Figure 2(b)). A π-stalking was found between the pyridine groups in molecules belonging to the neighbouring pairs with the corresponding distance of 3.45(1) Å.
![[1860-5397-20-202-5]](/bjoc/content/figures/1860-5397-20-202-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Fragment of the pack of catechol 8 in crystal (the H-bonds and π–π interactions are shown as dotted lines).
Figure 5: Fragment of the pack of catechol 8 in crystal (the H-bonds and π–π interactions are shown as dotted...
Electrochemical properties
The study of the electrochemical properties of functionalized catechols allows one to suggest the mechanism of their electrooxidation, establish electron transfer centers, and predict antioxidant activity based on electrochemical data. To determine electron-transfer centers, the oxidation potentials of the starting mercapto-substituted heterocycles were studied. Based on cyclic voltammetry data, the starting compounds can be arranged in increasing order of the anodic peak potentials: 5-(3-pyridyl)-1,2,4-triazole-3-thiol (0.87 V) < 2-mercapto-4-phenylthiazole (0.94 V) < 3-nitropyridine-2-thiol (1.18 V) < 1,3,4-oxadiazole derivatives (1.37–1.39 V). The redox properties of catechols 1–9 were studied by cyclic voltammetry (CV) (Table 1).
Table 1: The peak potentials of 1–9 obtained by the CV (GC-electrode, CH3CN, ν = 0.2 V∙s−1, 0.1 М n-Bu4NClO4, c = 1–3 mmol·L−1, Ag/AgCl/KCl (sat.)).
| Compound | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9a |
|---|---|---|---|---|---|---|---|---|---|
| Epa1, V | 1.25 | 1.22 | 1.02 | 0.94 | 1.00 | 1.17 | 1.19 | 1.17 | 0.85 |
| Epa2, V | 1.56 | 1.55 | 1.81 | 1.56 | 1.84 | 1.64 | 1.68 | 1.90 | – |
aCompound was dissolved in MeCN/DMSO = 3:1 (v:v).
In the anodic area, the electrochemical profile for catechol thioethers 1–5 represents two successive irreversible oxidative stages. The first two-electron peak is observed in the range of 0.94–1.25 V. It refers to the oxidation of the catechol moiety to the corresponding o-benzoquinone, as previously shown for related compounds [36]. The second redox transition at 1.55–1.84 V characterizes the oxidation of the sulfide fragment (Scheme 2).
![[1860-5397-20-202-i2]](/bjoc/content/inline/1860-5397-20-202-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Electrochemical transformations of compounds 1–3.
Scheme 2: Electrochemical transformations of compounds 1–3.
To confirm the participation of the catechol group in the first redox transition, the microelectrolysis of 3 was carried out at a controlled potential of 1.35 V in MeCN (2 h, 0.8 F/mol). After electrolysis, a decrease in the current intensity of the first oxidation peak is observed on the CVs of this compound (conversion reaches 51%). Besides, the current value of the second peak remains unchanged. In the cathodic region, a one-electron quasi-reversible peak is displayed (Epc = −0.58 V). This process corresponds to the reduction of o-benzoquinone to o-benzosemiquinone. During electrolysis, the color of the solution changes. This is confirmed by the appearance of an absorption maximum in the UV–visible spectrum at λ = 490 nm, which is characteristic of thiolated o-benzoquinones. This absorption band corresponds to intramolecular charge transfer between the thioether group and the o-benzoquinone fragment [36,62-65].
The potentials of the first anodic peak of thioethers 1 and 2 with an oxadiazole core are fixed at close values. (Supporting Information File 1, Figure S19 for 1, Figure 6 for 2).
![[1860-5397-20-202-6]](/bjoc/content/figures/1860-5397-20-202-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: The CV curve of 2 at the potential range from −0.50 to 1.60 V (CH3CN, GC electrode, Ag/AgCl/KCl(sat.), 0.15 M n-Bu4NClO4, c = 1 mmol·L−1).
Figure 6: The CV curve of 2 at the potential range from −0.50 to 1.60 V (CH3CN, GC electrode, Ag/AgCl/KCl(sat...
The replacement of the oxadiazole cycle with a thiazole one in catechol thioether 3 leads to a shift in the oxidation peak of the catechol fragment to the cathode region (Figure 7). This is due to the less acceptor nature of the thiazole ring compared to the more electron-deficient oxadiazole cycle, which contains three heteroatoms. In addition, according to NMR spectral data, one proton of the hydroxy group of catechol 3 is shifted to the thiazole nitrogen atom, which also affects the decrease in the oxidation potential. In the case of 3, the second anodic peak corresponding to thioether oxidation is multielectronic and shifted towards more positive values.
![[1860-5397-20-202-7]](/bjoc/content/figures/1860-5397-20-202-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: The CV curves of 3 at the potential ranges from –0.5 to 1.2 V (curve 1); from –0.5 to 2.0 V (curve 2) (CH3CN, GC electrode, Ag/AgCl/KCl(sat.), 0.15 M n-Bu4NClO4, c = 3 mmol·L−1).
Figure 7: The CV curves of 3 at the potential ranges from –0.5 to 1.2 V (curve 1); from –0.5 to 2.0 V (curve ...
More intensive current can be explained by the proximity of the thioether linker and the thiazole moiety oxidation potentials. The appearance of a methylene bridge between the aromatic ring and the sulfur atom in catechol 4 leads to a shift of the catechol oxidation peak to the cathodic area (Supporting Information File 1, Figure S20) as compared to catechol 3. This effect is due to a decrease in the acceptor effect of the thioether group. In turn, the second two-electron stage for 4 is recorded at earlier potential values. On the contrary, this behavior is caused by the presence of CH2-linker which divides electrogenerated quinone form and heterocycle moiety. The formation of intramolecular hydrogen bonds between the OH group and the pyridine ring facilitates the electrooxidation process (1.,0 V) (Supporting Information File 1, Figure S21) for catechol thioether 5. The second oxidation peak is shifted to the anodic region due to the influence of the electron-withdrawing nitro group.
Some differences are observed in the electrochemical behavior of thiones 6–9 compared to thioethers 1–5. In the case of compounds 6–8 with a 1,3,4-oxadiazole-2-thione fragment, the oxidation potential of the first peak is shifted to the cathodic region compared to catechols 1 and 2 which contain a similar heterocycle (Figure 8 for 7, Supporting Information File 1, Figures S22 and S23 for 6 and 8, respectively). This effect is due to the presence of a CH2 linker which separates the catechol and heterocyclic fragments, thereby reducing the acceptor effect of the heterocycle on the catechol ring. The change of substituents in the 1,3,4-oxadiazole-2-thione ring has virtually no effect on the potential of the first redox transition. For compounds 6 and 7, the second anodic peak is single-electron and presumably corresponds to the oxidation of the thione group. This signal is shifted to the anodic region by 0.09–0.13 V compared to the Epa2 values corresponding to the oxidation of the thioether group in catechols 1 and 2 with a similar heterocyclic fragment (Figure 6).
![[1860-5397-20-202-8]](/bjoc/content/figures/1860-5397-20-202-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: The CV curves of 7 at the potential ranges from –0.5 to 1.3 V (curve 1); from –0.5 to 1.8 V (curve 2) (CH3CN, GC electrode, Ag/AgCl/KCl(sat.), 0.15 M n-Bu4NClO4, c = 3 mmol·L−1).
Figure 8: The CV curves of 7 at the potential ranges from –0.5 to 1.3 V (curve 1); from –0.5 to 1.8 V (curve ...
The presence of an acceptor 3-pyridyl ring in the 1,3,4-oxadiazole-2-thione core for compound 8, leads to a shift of Epa2 by 0.22–0.26 V to the anodic region compared to compounds 6 and 7. In addition, the current intensity of the second peak corresponding to the transfer of two electrons increases (Figure S23 in Supporting Information File 1). The electrochemical behavior of catechol 9 with a 1,2,4-triazole-5-thione ring was studied in a MeCN–DMSO mixture to increase its solubility. The first redox transition is detected at early potential values (0.85 V) which coincides with the value obtained for the corresponding starting thiol. However, there is a peak at E = −0.11 V on the reverse CV branch of 9, characteristic of compounds with a catechol fragment (Figure S24 in Supporting Information File 1). The second anodic peak for 9 is not observed due to the narrow electrochemical window of the solvent in the positive CV region. The formation of o-benzoquinones at the first stage of oxidation was confirmed during a controlled potential microelectrolysis of compound 6 at 1.30 V (2 h, 0.8 F/mol). A similar electrochemical picture is observed for electrolysis products of catechol 6 as in the case of catechol 3. The recorded absorption spectra after the electrolysis display an intense maximum at λ = 412 nm which is characteristic for sterically hindered o-benzoquinones. The proposed scheme for the electrooxidation of catechols 6–9 is presented in Scheme 3.
![[1860-5397-20-202-i3]](/bjoc/content/inline/1860-5397-20-202-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism of an electrooxidation of compounds 6–8.
Scheme 3: Proposed mechanism of an electrooxidation of compounds 6–8.
Based on the current intensities of the second anodic peaks for compounds 6–8 with a 1,3,4-oxadiazole-2-thione ring (Figure 8 for 7, Supporting Information File 1, Figures S22 and S23 for 6 and 8, respectively), it can be assumed that the second redox transition can occur in one (for 6 and 7) and a two-electron (for 8) manner.
Thus, the electrochemical behavior of the studied catechols with heterocyclic fragments is influenced by several factors: the nature of the heterocycle and its substituents, the presence/absence of a sulfur atom in the catechol ring or thione sulfur atom in the heterocyclic core. Based on electrochemical data, it can be assumed that the most pronounced antioxidant activity can be exhibited by compounds with minimal first oxidation potentials: catechol 4 containing a 4-phenylthiazole ring and catechol thione 9 with a 1,2,4-triazole ring.
Radical scavenging, anti/prooxidant activity of catechols with heterocyclic fragments
The radical scavenging properties and antioxidant activity of catechols 1–9 were investigated in the reaction with a diphenylpicrylhydrazyl (DPPH) radical, ABTS·+ radical cation, CUPRAC test, and inhibition process of superoxide radical anion formation by xanthine oxidase (NBT assay). The presence of a catechol fragment and thioether or thione groups determines the ability to neutralize different radical species. A comparative evaluation of the antioxidant activity of catechols 1–9 and Trolox was performed in a reaction with DPPH in dichloromethane at 298 K (Table 2).
Table 2: Radical scavenging activity characteristics of catechols 1–9 and Trolox in DPPH (CH2Cl2, 298 K), ABTS•+ and CUPRAC assays expressed as IC50 and TEAC.
| Compound | DPPH | ABTS∙+ | CUPRAC | |
|---|---|---|---|---|
| IC50 (DPPH), µM | TEC50, min | IC50 (ABTS∙+), µM | CUPRACTEAC | |
| 1 | >100 | – | 45.6 ± 1.8 | 0.12a |
| 2 | 34.6 ± 1.3 | 40 | 38.4 ± 1.5 | 0.12 |
| 3 | 13.0 ± 1.0 | 13 | 17.6 ± 1.3 | 1.04 |
| 4 | 14.3 ± 1.5 | 15 | 17.9 ± 1.7 | 1.15 |
| 5 | 14.8 ± 0.8 | 35 | 25.3 ± 0.6 | 0.96 |
| 6 | 24.0 ± 1.1 | 15 | 10.5 ± 0.3 | 0.99 |
| 7 | 19.6 ± 2.0 | 20 | 11.6 ± 0.5 | 1.13 |
| 8 | 16.6 ± 0.8 | 25 | 20.6 ± 2.3 | 0.96 |
| 9 | 18.6 ± 0.7 | 17.0 | 13.9 ± 1.0 | 1.92a |
| Trolox | 12.0 ± 0.5 | 10.3 | 16.0±1.0 | 1.00 |
| CatH2-CH2-S-thiazolylb | 21.5 ± 1.5 | <1.0 | 21.6 ± 1.8 | 0.83 ± 0.06 |
| CatH2-CH2-S-pyridinylb | 11.5 ± 0.4 | 23.0 | 17.6 ± 2.0 | 1.25 ± 0.11 |
aData were observed in DMSO. bData were obtained from [54].
Based on the IC50 value, compound 3 with a 4-phenylthiazole ring linked through a sulfur atom to the catechol ring exhibits the highest activity (13.0 ± 1.0 µM) in the reaction with the DPPH radical. The number of converted DPPH molecules per one molecule of 3 is equal to two. Comparable IC50 values were obtained for catechol thioethers 4 and 5 with methylene bridge.
As in the case of compound 3, catechol 4 contains a 4-phenylthiazole moiety but is a less active radical scavenger, probably due to the presence of a CH2−linker between the catechol and heterocyclic moieties. At the same time, the appearance of a phenyl substituent in the thiazole ring of 4 reduces the IC50 value (14.3 ± 1.5 µM) compared to the previously studied 3-((thiazol-2-ylthio)methyl)-4,6-di-tert-butylcatechol (CatH2-CH2-S-thiazolyl) (21.5 ± 1.5 µM) [54]. However, for CatH2-CH2-S-thiazolyl, the equilibrium state is reached in less than one minute which increases significantly the antiradical activity of AE compared to catechol 4. On the contrary, the presence of a nitro group in the pyridine ring in catechol 5 reduces its antiradical activity compared to the unsubstituted 3-((pyridin-2-ylthio)methyl)-4,6-di-tert-butylcatechol (CatH2-CH2-S-pyridinyl). In the series of catechols 6–8 with a thione group in the 1,3,4-oxadiazole ring, the IC50 values decrease gradually from 24.0 to 16.6 µM, while an inverse relationship is observed for TEC50. Replacement of the methyl group with a chlorine atom in the phenyl ring of compound 7 leads to a decrease in the IC50 value compared to catechol 6. The appearance of a pyridyl substituent in the oxadiazole core has an even more noticeable effect on reducing the IC50. Catechol thioether 2 containing a 1,3,4-oxadiazole ring showed the least neutralizing activity. Compound 1 was inactive in this test (IC50 >> 100 µM). TEC50 varies in the range of 13–40 min for all studied compounds. Based on the results of the calculation of the AE indicator, which takes into account the parameters EC50 and TEC50 [66], the studied compounds can be arranged in ascending order of neutralizing activity: 2 (0.7) < 5 (1.9) < 8 (2.4) < 7 (2.5) < 6 (2.7) < 9 (3.1) < 4 (4.7) < Trolox (5.1) < 3 (5.9). Thus, in each series of thioethers (1–5) or thiones (6–9), the leading compound can be identified: catechol 9 with 1,2,4-triazole-5-thione core and 3 with 4-phenylthiazole cycle, which is the only one of the studied compounds exceeding Trolox activity in this assay. Consequently, the presence of 1,2,4-triazole-5-thione or 4-phenylthiazole fragment favors the pronounced antiradical activity.
The use of ABTS radical cation to assess the antioxidant capacity of compounds is one of the widely used methods which is based on the transfer of an electron from the studied molecules to the acceptor [67]. The obtained IC50 values for synthesized catechols vary over a wide range from 10.5 to 45.6 μM. As in the case of the reaction with the DPPH radical, thioethers 1 and 2 possess a maximal IC50 value which indicates their low radical scavenging activity. Compounds 3 and 4 containing a 4-phenylthiazole moiety have similar IC50 values. This points out that the methylene bridge separating the catechol ring and the sulfur atom does not influence their neutralizing activity. The presence of a phenyl substituent in the heterocyclic fragment of compound 4 has a positive effect on the antiradical activity in this test in comparison with CatH2-CH2-S-thiazolyl. The electron-withdrawing NO2 group in the pyridine ring of catechol 5 suppresses its antiradical properties in comparison with CatH2-CH2-S-pyridinyl which is similar to the patterns observed in the DPPH test. In general, compounds with thione moieties were more active ABTS·+ scavengers than thioethers 1–5 except catechol 8. The minimum IC50 values in this test were obtained for compounds 6 and 7. Therefore, catechol thiones 6, 7, and 9 were more effective ABTS·+ scavengers than Trolox.
The CUPRAC-assay is used to determine the electronic capacity of an antioxidant relative to Trolox under the action of a soft oxidant a bis-(neo-cuproine) copper(II) complex. The CUPRACTEAC parameter varies in a wide range of values from 0.12 to 1.92 (Table 2). Compounds 1 and 2 containing a 1,3,4-oxadiazole ring and a sulfur atom directly bonded to the catechol ring are practically inactive in this reaction which correlates with the data of the DPPH-test. Compounds 3, 5, 6, and 8 exhibit antioxidant activity comparable to Trolox, while catechols 4 and 7 are 13–15% higher than the activity of this standard. The introduction of an electron-withdrawing nitro-group into the pyridine ring leads to a 29% decrease in the CUPRACTEAC value for compound 5 compared to CatH2-CH2-S-pyridinyl. The presence of a phenyl substituent in the thiazole ring has a positive effect on the antioxidant activity of compound 4 and leads to its increase by 32% compared to the previously studied CatH2-CH2-S-thiazolyl. The highest CUPRACTEAC value (1.92) was determined for 9 with 3-(pyridin-3-yl)-1,2,4-triazole-5-thione fragment. Thus, in this test, thioether 4 and thiones 7 and 9 have the greatest antioxidant capacity.
The neutralizing activity of compounds 1, 3, 4, and 8 concerning the superoxide anion radical was studied in the NBT-test. Based on the IC50 values, compound 8 with the 5-(pyridin-3-yl)-1,3,4-oxadiazole-2-thione fragment showed a pronounced inhibitory effect (31.13 ± 0.84 μM) compared to Trolox (92.55 ± 0.24 μM). Among compounds 3 and 4 containing a thiazole ring, the more effective O2·– scavenger is catechol 3 (40.39 ± 0.89 μM), in which the sulfur atom is directly bonded to the aromatic ring. The IC50 value is 63.48 ± 1.08 μM for compound 4. The appearance of a phenyl substituent in the thiazole ring affects negatively the antiradical ability of compound 4 compared to its unsubstituted analogue CatH2-CH2-S-thiazolyl (34.0 ± 0.10 μM). Compound 1 was the least effective in this test (81.10 ± 1.86 μM) as in the case of the methods described above.
We have previously shown that catechol thioethers can act as anti- or prooxidants on lipid peroxidation processes in vitro depending on a functional group or a heterocycle substitute at the sulfur atom [49,50,54]. Non-enzymatic process of the rat (Wistar) liver homogenate lipid peroxidation (LP) in the presence of additives of target compounds 1–9 was studied in vitro. The concentration of the carbonyl compounds forming in the reaction of the lipid peroxidation was determined by the accumulation of a colored complex with thiobarbituric acid (TBARS) (Figure 9).
![[1860-5397-20-202-9]](/bjoc/content/figures/1860-5397-20-202-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: The level of TBARS in rat liver homogenates in vitro, in the presence of compounds 1–9, Trolox, and without additives (control) during the incubation for 3, 24, and 48 hours (concentration of compounds in the reaction medium is 0.1 mM). The results are expressed as mean ± standard deviation (* p < 0.001; ** p < 0.01; ***p < 0.05).
Figure 9: The level of TBARS in rat liver homogenates in vitro, in the presence of compounds 1–9, Trolox, and...
A decrease in TBARS concentration was observed in the presence of all tested compounds which indicates their antioxidant effect during incubation time. At the initial stage (3 h), catechol thione 8 containing a pyridine substituent in the 1,3,4-oxadiazole-2-thione cycle has the greatest inhibitory effect (36%) on the LP process compared to the control sample. For compounds 2, 4–7, and 9, a less pronounced antioxidant effect (13–19%) is observed comparable to the inhibitory activity of Trolox. Initially (3 h), catechols 1 and 3 exhibit a weak inhibitory effect (7–8%). An increase in the incubation time to 24 hours leads to a rise in antioxidant activity for all compounds. A decrease in TBARS content by 41–52% was noted compared to the control experiment. All the studied compounds exhibit more effective inhibitory activity than Trolox. The inhibitory effect for compounds 2, 5, 6, 8, and 9 increases to 47–64% after 48 hours. This indicates the ability of these catechols to have a prolonged effect on the LP process. In the presence of Trolox, amounts of TBARS decrease by 33% over 48 hours of incubation.
The CTBARS value varies in a narrow range of values and is practically independent of experimental time for 5 and 6. This behavior indicates the occurrence of an induction period during lipid peroxidation when the level of TBARS remains constant. In the case of 1 and 7, a decrease in the inhibitory effect to 17–28% is observed with an increase in incubation time from 24 to 48 hours. Catechol thioether 5 with a 3-nitro-substituted 2-pyridine moiety and 8 with a 3-pyridine substituent in the 1,3,4-oxadiazole-2-thione ring demonstrate the maximum antioxidant activity (63–64%) over 48 hours of the experiment. A comparative assessment of the effect of thioethers 1 and 2 in the LP process points out that the replacement of the methyl group in the phenyl ring with a chlorine atom leads to increased antioxidant activity of 2. In the presence of catechol thioether 3, the TBARS content is slightly lower compared to 4. A phenyl substitute in the thiazole ring in 4 affects negatively its antioxidant activity in the LP process compared to CatH2-CH2-S-thiazolyl reducing the inhibitory effect from 81% to 43% (48 h). Consequently, catechol thioethers 2 and 5 as well as catechol thione 8 are the most effective antioxidants in the lipid peroxidation reaction.
Conclusion
Thus, we have synthesized a series of new sterically hindered catechols with thioether or thione groups containing various heterocyclic fragments with good yields of 42–80%. The interaction of 3,5-di-tert-butyl-o-benzoquinone with heterocyclic thiols leads to the formation of S-functionalized catechols. In the case of the acid-catalyzed reaction between 3,5-di-tert-butyl-6-methoxymethylcatechol and mercapto-substituted thiazole or pyridine derivatives, catechol−thioethers are formed with an additional methylene bridge between the catechol ring and the sulfur atom. In contrast, the mercapto derivatives of 1,3,4-oxadiazole or 1,2,4-triazole undergo alkylation at the nitrogen atom in the reaction with 3,5-di-tert-butyl-6-methoxymethylcatechol which leads to the corresponding catechol thiones. The molecular structures of catechols with a 2-pyridine or 1,3,4-oxadiazole-2-thione ring in the crystalline state were studied by X-ray diffraction.
It has been established that the number of heteroatoms in the cycle which are bonded to the catechol ring through a sulfur atom and/or methylene bridge as well as the presence of thioether or thione groups affect the electrochemical behavior of catechols. The electrochemical data made it possible to determine compounds characterized by minimal anodic potentials (4 and 9) which may exhibit more pronounced antioxidant activity among the studied compounds.
Catechol thioethers 3 and 4 proved to be the most effective scavengers of the DPPH radical. Thiones 6, 7, and 9 showed pronounced antiradical activity in the reaction with ABTS·+. Compounds 3, 4, and 8 showed high inhibitory activity in NBT-test. All studied compounds have antioxidant effects on the process of lipid peroxidation. Thus, in most of the studied assays, compounds 3 and 4 with a 4-phenylthiazole moiety turned out to be effective antioxidants among catechol thioethers, and 8 and 9 among thiones with a catechol fragment, which is consistent with electrochemical data.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data. | ||
| Format: PDF | Size: 1.1 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Augustyniak, A.; Bartosz, G.; Čipak, A.; Duburs, G.; Horáková, L.; Łuczaj, W.; Majekova, M.; Odysseos, A. D.; Rackova, L.; Skrzydlewska, E.; Stefek, M.; Štrosová, M.; Tirzitis, G.; Venskutonis, P. R.; Viskupicova, J.; Vraka, P. S.; Žarković, N. Free Radical Res. 2010, 44, 1216–1262. doi:10.3109/10715762.2010.508495
Return to citation in text: [1] -
Scott, K. A.; Cox, P. B.; Njardarson, J. T. J. Med. Chem. 2022, 65, 7044–7072. doi:10.1021/acs.jmedchem.2c00223
Return to citation in text: [1] -
Müller, T. Drugs 2015, 75, 157–174. doi:10.1007/s40265-014-0343-0
Return to citation in text: [1] -
Arias-Sánchez, R. A.; Torner, L.; Fenton Navarro, B. Molecules 2023, 28, 5415. doi:10.3390/molecules28145415
Return to citation in text: [1] -
Zhang, W.; Dong, X.; Huang, R. Pharmacol. Res. 2023, 193, 106787. doi:10.1016/j.phrs.2023.106787
Return to citation in text: [1] -
Liu, X.; Ou, Y.; Chen, S.; Lu, X.; Cheng, H.; Jia, X.; Wang, D.; Zhou, G.-C. Eur. J. Med. Chem. 2010, 45, 2147–2153. doi:10.1016/j.ejmech.2010.01.051
Return to citation in text: [1] -
Bao, X.-Z.; Dai, F.; Wang, Q.; Jin, X.-L.; Zhou, B. Free Radicals Biol. Med. 2019, 134, 406–418. doi:10.1016/j.freeradbiomed.2019.01.033
Return to citation in text: [1] -
Pagoni, A.; Grabowiecka, A.; Tabor, W.; Mucha, A.; Vassiliou, S.; Berlicki, Ł. J. Med. Chem. 2021, 64, 404–416. doi:10.1021/acs.jmedchem.0c01143
Return to citation in text: [1] -
Villaño, D.; Fernández-Pachón, M. S.; Moyá, M. L.; Troncoso, A. M.; García-Parrilla, M. C. Talanta 2007, 71, 230–235. doi:10.1016/j.talanta.2006.03.050
Return to citation in text: [1] -
Dimić, D.; Milenković, D.; Dimitrić Marković, J.; Marković, Z. Phys. Chem. Chem. Phys. 2017, 19, 12970–12980. doi:10.1039/c7cp01716b
Return to citation in text: [1] -
Haider, K.; Haider, M. R.; Neha, K.; Yar, M. S. Eur. J. Med. Chem. 2020, 204, 112607. doi:10.1016/j.ejmech.2020.112607
Return to citation in text: [1] -
Martinez, A.; Gil, C. Heterocycles Containing Nitrogen and Sulfur as Potent Biologically Active Scaffolds. In Privileged Scaffolds in Medicinal Chemistry; Bräse, S., Ed.; 2015; pp 231–261. doi:10.1039/9781782622246-00231.
Return to citation in text: [1] -
De, S. S.; Khambete, M. P.; Degani, M. S. Bioorg. Med. Chem. Lett. 2019, 29, 1999–2007. doi:10.1016/j.bmcl.2019.06.054
Return to citation in text: [1] [2] -
Othman, A. A.; Kihel, M.; Amara, S. Arabian J. Chem. 2019, 12, 1660–1675. doi:10.1016/j.arabjc.2014.09.003
Return to citation in text: [1] -
Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Eur. J. Med. Chem. 2015, 97, 699–718. doi:10.1016/j.ejmech.2015.04.015
Return to citation in text: [1] -
Chhabria, M. T.; Patel, S.; Modi, P.; Brahmkshatriya, P. S. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2016, 16, 2841–2862. doi:10.2174/1568026616666160506130731
Return to citation in text: [1] -
Ibrahim, N. K. Front. Oncol. 2021, 11, 617874. doi:10.3389/fonc.2021.617874
Return to citation in text: [1] -
Arshad, M. F.; Alam, A.; Alshammari, A. A.; Alhazza, M. B.; Alzimam, I. M.; Alam, M. A.; Mustafa, G.; Ansari, M. S.; Alotaibi, A. M.; Alotaibi, A. A.; Kumar, S.; Asdaq, S. M. B.; Imran, M.; Deb, P. K.; Venugopala, K. N.; Jomah, S. Molecules 2022, 27, 3994. doi:10.3390/molecules27133994
Return to citation in text: [1] -
Niu, Z.-X.; Wang, Y.-T.; Zhang, S.-N.; Li, Y.; Chen, X.-B.; Wang, S.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2023, 250, 115172. doi:10.1016/j.ejmech.2023.115172
Return to citation in text: [1] -
Ilardi, E. A.; Vitaku, E.; Njardarson, J. T. J. Med. Chem. 2014, 57, 2832–2842. doi:10.1021/jm401375q
Return to citation in text: [1] -
Vahora, M. S.; Boruah, J. J.; Das, S. P. Russ. J. Org. Chem. 2023, 59, 846–869. doi:10.1134/s1070428023050160
Return to citation in text: [1] -
Yadav, N.; Kumar, P.; Chhikara, A.; Chopra, M. Biomed. Pharmacother. 2017, 95, 721–730. doi:10.1016/j.biopha.2017.08.110
Return to citation in text: [1] [2] -
Rusinov, V. L.; Sapozhnikova, I. M.; Ulomskii, E. N.; Medvedeva, N. R.; Egorov, V. V.; Kiselev, O. I.; Deeva, E. G.; Vasin, A. V.; Chupakhin, O. N. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2015, 51, 275–280. doi:10.1007/s10593-015-1695-4
Return to citation in text: [1] -
El‐Sebaey, S. A. ChemistrySelect 2020, 5, 11654–11680. doi:10.1002/slct.202002830
Return to citation in text: [1] -
Li, J.; Zhang, J. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2022, 22, 41–63. doi:10.2174/1568026621666211111160332
Return to citation in text: [1] -
Soliman, S. M.; Hagar, M.; Ibid, F.; El Ashry, E. S. H. Spectrochim. Acta, Part A 2015, 145, 270–279. doi:10.1016/j.saa.2015.01.061
Return to citation in text: [1] [2] -
Gökce, H.; Öztürk, N.; Ceylan, Ü.; Alpaslan, Y. B.; Alpaslan, G. Spectrochim. Acta, Part A 2016, 163, 170–180. doi:10.1016/j.saa.2016.03.041
Return to citation in text: [1] [2] -
Das, A.; Ashraf, M. W.; Banik, B. K. ChemistrySelect 2021, 6, 9069–9100. doi:10.1002/slct.202102398
Return to citation in text: [1] -
Makane, V. B.; Krishna, V. S.; Krishna, E. V.; Shukla, M.; Mahizhaveni, B.; Misra, S.; Chopra, S.; Sriram, D.; Azger Dusthackeer, V. N.; Rode, H. B. Future Med. Chem. 2019, 11, 499–510. doi:10.4155/fmc-2018-0378
Return to citation in text: [1] -
Karabanovich, G.; Zemanová, J.; Smutný, T.; Székely, R.; Šarkan, M.; Centárová, I.; Vocat, A.; Pávková, I.; Čonka, P.; Němeček, J.; Stolaříková, J.; Vejsová, M.; Vávrová, K.; Klimešová, V.; Hrabálek, A.; Pávek, P.; Cole, S. T.; Mikušová, K.; Roh, J. J. Med. Chem. 2016, 59, 2362–2380. doi:10.1021/acs.jmedchem.5b00608
Return to citation in text: [1] -
El-Sayed, H. A.; Moustafa, A. H.; Haikal, A. E.-F. Z. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 649–662. doi:10.1080/10426507.2012.668990
Return to citation in text: [1] -
Tolan, H. E. M.; El-Sayed, W. A.; Tawfek, N.; Abdel-Megeid, F. M. E.; Kutkat, O. M. Nucleosides, Nucleotides Nucleic Acids 2020, 39, 649–670. doi:10.1080/15257770.2019.1674331
Return to citation in text: [1] -
Gao, F.; Wang, T.; Xiao, J.; Huang, G. Eur. J. Med. Chem. 2019, 173, 274–281. doi:10.1016/j.ejmech.2019.04.043
Return to citation in text: [1] -
Lobana, T. S. Coord. Chem. Rev. 2021, 441, 213884. doi:10.1016/j.ccr.2021.213884
Return to citation in text: [1] -
Smolyaninov, I. V.; Pitikova, O. V.; Rychagova, E. S.; Korchagina, E. O.; Poddel’sky, A. I.; Smolyaninova, S. A.; Berberova, N. T. Russ. Chem. Bull. 2016, 65, 2861–2867. doi:10.1007/s11172-016-1669-4
Return to citation in text: [1] -
Smolyaninov, I. V.; Pitikova, O. V.; Poddel’sky, A. I.; Berberova, N. T. Russ. Chem. Bull. 2018, 67, 1857–1867. doi:10.1007/s11172-018-2299-9
Return to citation in text: [1] [2] [3] [4] -
Ukhin, L. Y.; Kuz’mina, L. G.; Alexeenko, D. V.; Belousova, L. V.; Morkovnik, A. S.; Shepelenko, E. N.; Podshibyakin, V. A.; Borodkin, G. S.; Dmitrieva, O. I. Russ. Chem. Bull. 2019, 68, 2290–2297. doi:10.1007/s11172-019-2702-1
Return to citation in text: [1] -
Galustyan, A.; Burmistrova, D. A.; Fokin, V. A.; Simeonova, K. S.; Polovinkina, M. A.; Pomortseva, N. P.; Baranov, E. V.; Almyasheva, N. R.; Smolyaninov, I. V.; Poddel’sky, A. I.; Berberova, N. T. Russ. Chem. Bull. 2024, 73, 714–727. doi:10.1007/s11172-024-4182-1
Return to citation in text: [1] -
Goksel, F. S.; Bayrak, N.; Ibis, C. Phosphorus, Sulfur Silicon Relat. Elem. 2014, 189, 113–123. doi:10.1080/10426507.2013.798787
Return to citation in text: [1] -
Tandon, V. K.; Maurya, H. K.; Kumar, S.; Rashid, A.; Panda, D. RSC Adv. 2014, 4, 12441–12447. doi:10.1039/c3ra47720g
Return to citation in text: [1] -
Davarani, S. S. H.; Ramyar, S.; Masoumi, L.; Sheijooni Fumani, N.; Bayandori Moghaddam, A. J. Electrochem. Soc. 2008, 155, E120. doi:10.1149/1.2948383
Return to citation in text: [1] -
Fakhari, A. R.; Hosseiny Davarani, S. S.; Ahmar, H.; Makarem, S. J. Appl. Electrochem. 2008, 38, 1743–1747. doi:10.1007/s10800-008-9625-0
Return to citation in text: [1] -
Gerken, J. B.; Stamoulis, A.; Suh, S.-E.; Fischer, N. D.; Kim, Y. J.; Guzeia, I. A.; Stahl, S. S. Chem. Commun. 2020, 56, 1199–1202. doi:10.1039/c9cc08878d
Return to citation in text: [1] -
Fotouhi, L.; Asadi, S.; Tammari, E.; Heravi, M. M.; Nematollahi, D. J. Iran. Chem. Soc. 2008, 5, 712–717. doi:10.1007/bf03246154
Return to citation in text: [1] -
Fotouhi, L.; Tammari, E.; Asadi, S.; Heravi, M. M.; Nematollahi, D. Int. J. Chem. Kinet. 2009, 41, 426–431. doi:10.1002/kin.20402
Return to citation in text: [1] -
Nematollahi, D.; Rafiee, M.; Fotouhi, L. J. Iran. Chem. Soc. 2009, 6, 448–476. doi:10.1007/bf03246523
Return to citation in text: [1] -
Toghan, A.; Abo-Bakr, A. M.; Rageh, H. M.; Abd-Elsabour, M. RSC Adv. 2019, 9, 13145–13152. doi:10.1039/c9ra01206k
Return to citation in text: [1] -
Masoumi, H.; Khazalpour, S.; Jamshidi, M. RSC Adv. 2023, 13, 3083–3094. doi:10.1039/d2ra07250e
Return to citation in text: [1] -
Smolyaninov, I.; Pitikova, O.; Korchagina, E.; Poddel’sky, A.; Luzhnova, S.; Berberova, N. Monatsh. Chem. 2018, 149, 1813–1826. doi:10.1007/s00706-018-2264-1
Return to citation in text: [1] [2] -
Smolyaninov, I. V.; Pitikova, O. V.; Korchagina, E. O.; Poddel’sky, A. I.; Fukin, G. K.; Luzhnova, S. A.; Tichkomirov, A. M.; Ponomareva, E. N.; Berberova, N. T. Bioorg. Chem. 2019, 89, 103003. doi:10.1016/j.bioorg.2019.103003
Return to citation in text: [1] [2] [3] -
Burmistrova, D.; Smolyaninov, I.; Berberova, N.; Eremenko, I. ChemistrySelect 2020, 5, 14515–14519. doi:10.1002/slct.202003961
Return to citation in text: [1] -
Smolyaninov, I. V.; Burmistrova, D. A.; Pomortseva, N. P.; Poddel’sky, A. I.; Berberova, N. T. Dokl. Chem. 2022, 504, 100–105. doi:10.1134/s0012500822600134
Return to citation in text: [1] -
Smolyaninov, I. V.; Poddel’sky, A. I.; Smolyaninova, S. A.; Arsenyev, M. V.; Fukin, G. K.; Berberova, N. T. Molecules 2020, 25, 1770. doi:10.3390/molecules25081770
Return to citation in text: [1] -
Smolyaninov, I. V.; Burmistrova, D. A.; Arsenyev, M. V.; Polovinkina, M. A.; Pomortseva, N. P.; Fukin, G. K.; Poddel’sky, A. I.; Berberova, N. T. Molecules 2022, 27, 3169. doi:10.3390/molecules27103169
Return to citation in text: [1] [2] [3] [4] -
Poddel’sky, A. I.; Arsenyev, M. V.; Astaf’eva, T. V.; Chesnokov, S. A.; Fukin, G. K.; Abakumov, G. A. J. Organomet. Chem. 2017, 835, 17–24. doi:10.1016/j.jorganchem.2017.02.035
Return to citation in text: [1] [2] -
Lide, D. R. Handbook of Chemistry and Physics, 85th ed.; CRC Press: Boca Raton, FL, USA, 2004.
Return to citation in text: [1] -
Greatorex, S.; Halcrow, M. A. CrystEngComm 2016, 18, 4695–4698. doi:10.1039/c6ce00966b
Return to citation in text: [1] -
Astaf’eva, T. V.; Arsenyev, M. V.; Rumyantcev, R. V.; Fukin, G. K.; Cherkasov, V. K.; Poddel’sky, A. I. ACS Omega 2020, 5, 22179–22191. doi:10.1021/acsomega.0c02277
Return to citation in text: [1] -
Zherebtsov, M. A.; Zhiganshina, E. R.; Lenshina, N. A.; Kovylin, R. S.; Baranov, E. V.; Shushunova, N. Y.; Shurygina, M. P.; Arsenyev, M. V.; Chesnokov, S. A.; Cherkasov, V. K. Russ. Chem. Bull. 2021, 70, 780–791. doi:10.1007/s11172-021-3151-1
Return to citation in text: [1] -
Alhadid, A.; Jandl, C.; Nasrallah, S.; Kronawitter, S. M.; Mokrushina, L.; Kieslich, G.; Minceva, M. J. Chem. Phys. 2023, 159, 094503. doi:10.1063/5.0158985
Return to citation in text: [1] -
Silva, M. C.; Cunha, G.; Firmino, P.; Sallum, L. O.; Menezes, A.; Dutra, J.; de Araujo-Neto, J.; Batista, A. A.; Ellena, J.; Napolitano, H. B. ACS Omega 2023, 8, 40764–40774. doi:10.1021/acsomega.3c05841
Return to citation in text: [1] -
Norkov, S. V.; Cherkasov, A. V.; Shavyrin, A. S.; Arsenyev, M. V.; Kuropatov, V. A.; Cherkasov, V. K. Beilstein J. Org. Chem. 2021, 17, 273–282. doi:10.3762/bjoc.17.26
Return to citation in text: [1] -
Cherkasov, V. K.; Abakumov, G. A.; Fukin, G. K.; Klementyeva, S. V.; Kuropatov, V. A. Chem. – Eur. J. 2012, 18, 13821–13827. doi:10.1002/chem.201202343
Return to citation in text: [1] -
Kuropatov, V.; Klementieva, S.; Fukin, G.; Mitin, A.; Ketkov, S.; Budnikova, Y.; Cherkasov, V.; Abakumov, G. Tetrahedron 2010, 66, 7605–7611. doi:10.1016/j.tet.2010.07.038
Return to citation in text: [1] -
Abakumov, G. A.; Shurygina, M. P.; Chesnokov, S. A.; Druzhkov, N. O.; Lopatin, M. A.; Chechet, Y. V.; Cherkasov, V. K. High Energy Chem. 2005, 39, 299–303. doi:10.1007/s10733-005-0059-5
Return to citation in text: [1] -
Sánchez‐Moreno, C.; Larrauri, J. A.; Saura‐Calixto, F. J. Sci. Food Agric. 1998, 76, 270–276. doi:10.1002/(sici)1097-0010(199802)76:2<270::aid-jsfa945>3.3.co;2-0
Return to citation in text: [1] -
Ilyasov, I. R.; Beloborodov, V. L.; Selivanova, I. A.; Terekhov, R. P. Int. J. Mol. Sci. 2020, 21, 1131. doi:10.3390/ijms21031131
Return to citation in text: [1]
| 53. | Smolyaninov, I. V.; Poddel’sky, A. I.; Smolyaninova, S. A.; Arsenyev, M. V.; Fukin, G. K.; Berberova, N. T. Molecules 2020, 25, 1770. doi:10.3390/molecules25081770 |
| 54. | Smolyaninov, I. V.; Burmistrova, D. A.; Arsenyev, M. V.; Polovinkina, M. A.; Pomortseva, N. P.; Fukin, G. K.; Poddel’sky, A. I.; Berberova, N. T. Molecules 2022, 27, 3169. doi:10.3390/molecules27103169 |
| 55. | Poddel’sky, A. I.; Arsenyev, M. V.; Astaf’eva, T. V.; Chesnokov, S. A.; Fukin, G. K.; Abakumov, G. A. J. Organomet. Chem. 2017, 835, 17–24. doi:10.1016/j.jorganchem.2017.02.035 |
| 26. | Soliman, S. M.; Hagar, M.; Ibid, F.; El Ashry, E. S. H. Spectrochim. Acta, Part A 2015, 145, 270–279. doi:10.1016/j.saa.2015.01.061 |
| 27. | Gökce, H.; Öztürk, N.; Ceylan, Ü.; Alpaslan, Y. B.; Alpaslan, G. Spectrochim. Acta, Part A 2016, 163, 170–180. doi:10.1016/j.saa.2016.03.041 |
| 1. | Augustyniak, A.; Bartosz, G.; Čipak, A.; Duburs, G.; Horáková, L.; Łuczaj, W.; Majekova, M.; Odysseos, A. D.; Rackova, L.; Skrzydlewska, E.; Stefek, M.; Štrosová, M.; Tirzitis, G.; Venskutonis, P. R.; Viskupicova, J.; Vraka, P. S.; Žarković, N. Free Radical Res. 2010, 44, 1216–1262. doi:10.3109/10715762.2010.508495 |
| 2. | Scott, K. A.; Cox, P. B.; Njardarson, J. T. J. Med. Chem. 2022, 65, 7044–7072. doi:10.1021/acs.jmedchem.2c00223 |
| 12. | Martinez, A.; Gil, C. Heterocycles Containing Nitrogen and Sulfur as Potent Biologically Active Scaffolds. In Privileged Scaffolds in Medicinal Chemistry; Bräse, S., Ed.; 2015; pp 231–261. doi:10.1039/9781782622246-00231. |
| 28. | Das, A.; Ashraf, M. W.; Banik, B. K. ChemistrySelect 2021, 6, 9069–9100. doi:10.1002/slct.202102398 |
| 66. | Sánchez‐Moreno, C.; Larrauri, J. A.; Saura‐Calixto, F. J. Sci. Food Agric. 1998, 76, 270–276. doi:10.1002/(sici)1097-0010(199802)76:2<270::aid-jsfa945>3.3.co;2-0 |
| 9. | Villaño, D.; Fernández-Pachón, M. S.; Moyá, M. L.; Troncoso, A. M.; García-Parrilla, M. C. Talanta 2007, 71, 230–235. doi:10.1016/j.talanta.2006.03.050 |
| 10. | Dimić, D.; Milenković, D.; Dimitrić Marković, J.; Marković, Z. Phys. Chem. Chem. Phys. 2017, 19, 12970–12980. doi:10.1039/c7cp01716b |
| 11. | Haider, K.; Haider, M. R.; Neha, K.; Yar, M. S. Eur. J. Med. Chem. 2020, 204, 112607. doi:10.1016/j.ejmech.2020.112607 |
| 29. | Makane, V. B.; Krishna, V. S.; Krishna, E. V.; Shukla, M.; Mahizhaveni, B.; Misra, S.; Chopra, S.; Sriram, D.; Azger Dusthackeer, V. N.; Rode, H. B. Future Med. Chem. 2019, 11, 499–510. doi:10.4155/fmc-2018-0378 |
| 67. | Ilyasov, I. R.; Beloborodov, V. L.; Selivanova, I. A.; Terekhov, R. P. Int. J. Mol. Sci. 2020, 21, 1131. doi:10.3390/ijms21031131 |
| 6. | Liu, X.; Ou, Y.; Chen, S.; Lu, X.; Cheng, H.; Jia, X.; Wang, D.; Zhou, G.-C. Eur. J. Med. Chem. 2010, 45, 2147–2153. doi:10.1016/j.ejmech.2010.01.051 |
| 7. | Bao, X.-Z.; Dai, F.; Wang, Q.; Jin, X.-L.; Zhou, B. Free Radicals Biol. Med. 2019, 134, 406–418. doi:10.1016/j.freeradbiomed.2019.01.033 |
| 8. | Pagoni, A.; Grabowiecka, A.; Tabor, W.; Mucha, A.; Vassiliou, S.; Berlicki, Ł. J. Med. Chem. 2021, 64, 404–416. doi:10.1021/acs.jmedchem.0c01143 |
| 25. | Li, J.; Zhang, J. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2022, 22, 41–63. doi:10.2174/1568026621666211111160332 |
| 54. | Smolyaninov, I. V.; Burmistrova, D. A.; Arsenyev, M. V.; Polovinkina, M. A.; Pomortseva, N. P.; Fukin, G. K.; Poddel’sky, A. I.; Berberova, N. T. Molecules 2022, 27, 3169. doi:10.3390/molecules27103169 |
| 3. | Müller, T. Drugs 2015, 75, 157–174. doi:10.1007/s40265-014-0343-0 |
| 4. | Arias-Sánchez, R. A.; Torner, L.; Fenton Navarro, B. Molecules 2023, 28, 5415. doi:10.3390/molecules28145415 |
| 5. | Zhang, W.; Dong, X.; Huang, R. Pharmacol. Res. 2023, 193, 106787. doi:10.1016/j.phrs.2023.106787 |
| 26. | Soliman, S. M.; Hagar, M.; Ibid, F.; El Ashry, E. S. H. Spectrochim. Acta, Part A 2015, 145, 270–279. doi:10.1016/j.saa.2015.01.061 |
| 27. | Gökce, H.; Öztürk, N.; Ceylan, Ü.; Alpaslan, Y. B.; Alpaslan, G. Spectrochim. Acta, Part A 2016, 163, 170–180. doi:10.1016/j.saa.2016.03.041 |
| 54. | Smolyaninov, I. V.; Burmistrova, D. A.; Arsenyev, M. V.; Polovinkina, M. A.; Pomortseva, N. P.; Fukin, G. K.; Poddel’sky, A. I.; Berberova, N. T. Molecules 2022, 27, 3169. doi:10.3390/molecules27103169 |
| 13. | De, S. S.; Khambete, M. P.; Degani, M. S. Bioorg. Med. Chem. Lett. 2019, 29, 1999–2007. doi:10.1016/j.bmcl.2019.06.054 |
| 22. | Yadav, N.; Kumar, P.; Chhikara, A.; Chopra, M. Biomed. Pharmacother. 2017, 95, 721–730. doi:10.1016/j.biopha.2017.08.110 |
| 36. | Smolyaninov, I. V.; Pitikova, O. V.; Poddel’sky, A. I.; Berberova, N. T. Russ. Chem. Bull. 2018, 67, 1857–1867. doi:10.1007/s11172-018-2299-9 |
| 20. | Ilardi, E. A.; Vitaku, E.; Njardarson, J. T. J. Med. Chem. 2014, 57, 2832–2842. doi:10.1021/jm401375q |
| 23. | Rusinov, V. L.; Sapozhnikova, I. M.; Ulomskii, E. N.; Medvedeva, N. R.; Egorov, V. V.; Kiselev, O. I.; Deeva, E. G.; Vasin, A. V.; Chupakhin, O. N. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2015, 51, 275–280. doi:10.1007/s10593-015-1695-4 |
| 24. | El‐Sebaey, S. A. ChemistrySelect 2020, 5, 11654–11680. doi:10.1002/slct.202002830 |
| 36. | Smolyaninov, I. V.; Pitikova, O. V.; Poddel’sky, A. I.; Berberova, N. T. Russ. Chem. Bull. 2018, 67, 1857–1867. doi:10.1007/s11172-018-2299-9 |
| 62. | Norkov, S. V.; Cherkasov, A. V.; Shavyrin, A. S.; Arsenyev, M. V.; Kuropatov, V. A.; Cherkasov, V. K. Beilstein J. Org. Chem. 2021, 17, 273–282. doi:10.3762/bjoc.17.26 |
| 63. | Cherkasov, V. K.; Abakumov, G. A.; Fukin, G. K.; Klementyeva, S. V.; Kuropatov, V. A. Chem. – Eur. J. 2012, 18, 13821–13827. doi:10.1002/chem.201202343 |
| 64. | Kuropatov, V.; Klementieva, S.; Fukin, G.; Mitin, A.; Ketkov, S.; Budnikova, Y.; Cherkasov, V.; Abakumov, G. Tetrahedron 2010, 66, 7605–7611. doi:10.1016/j.tet.2010.07.038 |
| 65. | Abakumov, G. A.; Shurygina, M. P.; Chesnokov, S. A.; Druzhkov, N. O.; Lopatin, M. A.; Chechet, Y. V.; Cherkasov, V. K. High Energy Chem. 2005, 39, 299–303. doi:10.1007/s10733-005-0059-5 |
| 15. | Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Eur. J. Med. Chem. 2015, 97, 699–718. doi:10.1016/j.ejmech.2015.04.015 |
| 16. | Chhabria, M. T.; Patel, S.; Modi, P.; Brahmkshatriya, P. S. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2016, 16, 2841–2862. doi:10.2174/1568026616666160506130731 |
| 17. | Ibrahim, N. K. Front. Oncol. 2021, 11, 617874. doi:10.3389/fonc.2021.617874 |
| 18. | Arshad, M. F.; Alam, A.; Alshammari, A. A.; Alhazza, M. B.; Alzimam, I. M.; Alam, M. A.; Mustafa, G.; Ansari, M. S.; Alotaibi, A. M.; Alotaibi, A. A.; Kumar, S.; Asdaq, S. M. B.; Imran, M.; Deb, P. K.; Venugopala, K. N.; Jomah, S. Molecules 2022, 27, 3994. doi:10.3390/molecules27133994 |
| 19. | Niu, Z.-X.; Wang, Y.-T.; Zhang, S.-N.; Li, Y.; Chen, X.-B.; Wang, S.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2023, 250, 115172. doi:10.1016/j.ejmech.2023.115172 |
| 50. | Smolyaninov, I. V.; Pitikova, O. V.; Korchagina, E. O.; Poddel’sky, A. I.; Fukin, G. K.; Luzhnova, S. A.; Tichkomirov, A. M.; Ponomareva, E. N.; Berberova, N. T. Bioorg. Chem. 2019, 89, 103003. doi:10.1016/j.bioorg.2019.103003 |
| 55. | Poddel’sky, A. I.; Arsenyev, M. V.; Astaf’eva, T. V.; Chesnokov, S. A.; Fukin, G. K.; Abakumov, G. A. J. Organomet. Chem. 2017, 835, 17–24. doi:10.1016/j.jorganchem.2017.02.035 |
| 56. | Lide, D. R. Handbook of Chemistry and Physics, 85th ed.; CRC Press: Boca Raton, FL, USA, 2004. |
| 13. | De, S. S.; Khambete, M. P.; Degani, M. S. Bioorg. Med. Chem. Lett. 2019, 29, 1999–2007. doi:10.1016/j.bmcl.2019.06.054 |
| 14. | Othman, A. A.; Kihel, M.; Amara, S. Arabian J. Chem. 2019, 12, 1660–1675. doi:10.1016/j.arabjc.2014.09.003 |
| 21. | Vahora, M. S.; Boruah, J. J.; Das, S. P. Russ. J. Org. Chem. 2023, 59, 846–869. doi:10.1134/s1070428023050160 |
| 57. | Greatorex, S.; Halcrow, M. A. CrystEngComm 2016, 18, 4695–4698. doi:10.1039/c6ce00966b |
| 58. | Astaf’eva, T. V.; Arsenyev, M. V.; Rumyantcev, R. V.; Fukin, G. K.; Cherkasov, V. K.; Poddel’sky, A. I. ACS Omega 2020, 5, 22179–22191. doi:10.1021/acsomega.0c02277 |
| 59. | Zherebtsov, M. A.; Zhiganshina, E. R.; Lenshina, N. A.; Kovylin, R. S.; Baranov, E. V.; Shushunova, N. Y.; Shurygina, M. P.; Arsenyev, M. V.; Chesnokov, S. A.; Cherkasov, V. K. Russ. Chem. Bull. 2021, 70, 780–791. doi:10.1007/s11172-021-3151-1 |
| 60. | Alhadid, A.; Jandl, C.; Nasrallah, S.; Kronawitter, S. M.; Mokrushina, L.; Kieslich, G.; Minceva, M. J. Chem. Phys. 2023, 159, 094503. doi:10.1063/5.0158985 |
| 61. | Silva, M. C.; Cunha, G.; Firmino, P.; Sallum, L. O.; Menezes, A.; Dutra, J.; de Araujo-Neto, J.; Batista, A. A.; Ellena, J.; Napolitano, H. B. ACS Omega 2023, 8, 40764–40774. doi:10.1021/acsomega.3c05841 |
| 31. | El-Sayed, H. A.; Moustafa, A. H.; Haikal, A. E.-F. Z. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 649–662. doi:10.1080/10426507.2012.668990 |
| 32. | Tolan, H. E. M.; El-Sayed, W. A.; Tawfek, N.; Abdel-Megeid, F. M. E.; Kutkat, O. M. Nucleosides, Nucleotides Nucleic Acids 2020, 39, 649–670. doi:10.1080/15257770.2019.1674331 |
| 30. | Karabanovich, G.; Zemanová, J.; Smutný, T.; Székely, R.; Šarkan, M.; Centárová, I.; Vocat, A.; Pávková, I.; Čonka, P.; Němeček, J.; Stolaříková, J.; Vejsová, M.; Vávrová, K.; Klimešová, V.; Hrabálek, A.; Pávek, P.; Cole, S. T.; Mikušová, K.; Roh, J. J. Med. Chem. 2016, 59, 2362–2380. doi:10.1021/acs.jmedchem.5b00608 |
| 49. | Smolyaninov, I.; Pitikova, O.; Korchagina, E.; Poddel’sky, A.; Luzhnova, S.; Berberova, N. Monatsh. Chem. 2018, 149, 1813–1826. doi:10.1007/s00706-018-2264-1 |
| 50. | Smolyaninov, I. V.; Pitikova, O. V.; Korchagina, E. O.; Poddel’sky, A. I.; Fukin, G. K.; Luzhnova, S. A.; Tichkomirov, A. M.; Ponomareva, E. N.; Berberova, N. T. Bioorg. Chem. 2019, 89, 103003. doi:10.1016/j.bioorg.2019.103003 |
| 54. | Smolyaninov, I. V.; Burmistrova, D. A.; Arsenyev, M. V.; Polovinkina, M. A.; Pomortseva, N. P.; Fukin, G. K.; Poddel’sky, A. I.; Berberova, N. T. Molecules 2022, 27, 3169. doi:10.3390/molecules27103169 |
| 22. | Yadav, N.; Kumar, P.; Chhikara, A.; Chopra, M. Biomed. Pharmacother. 2017, 95, 721–730. doi:10.1016/j.biopha.2017.08.110 |
| 36. | Smolyaninov, I. V.; Pitikova, O. V.; Poddel’sky, A. I.; Berberova, N. T. Russ. Chem. Bull. 2018, 67, 1857–1867. doi:10.1007/s11172-018-2299-9 |
| 49. | Smolyaninov, I.; Pitikova, O.; Korchagina, E.; Poddel’sky, A.; Luzhnova, S.; Berberova, N. Monatsh. Chem. 2018, 149, 1813–1826. doi:10.1007/s00706-018-2264-1 |
| 50. | Smolyaninov, I. V.; Pitikova, O. V.; Korchagina, E. O.; Poddel’sky, A. I.; Fukin, G. K.; Luzhnova, S. A.; Tichkomirov, A. M.; Ponomareva, E. N.; Berberova, N. T. Bioorg. Chem. 2019, 89, 103003. doi:10.1016/j.bioorg.2019.103003 |
| 51. | Burmistrova, D.; Smolyaninov, I.; Berberova, N.; Eremenko, I. ChemistrySelect 2020, 5, 14515–14519. doi:10.1002/slct.202003961 |
| 52. | Smolyaninov, I. V.; Burmistrova, D. A.; Pomortseva, N. P.; Poddel’sky, A. I.; Berberova, N. T. Dokl. Chem. 2022, 504, 100–105. doi:10.1134/s0012500822600134 |
| 41. | Davarani, S. S. H.; Ramyar, S.; Masoumi, L.; Sheijooni Fumani, N.; Bayandori Moghaddam, A. J. Electrochem. Soc. 2008, 155, E120. doi:10.1149/1.2948383 |
| 42. | Fakhari, A. R.; Hosseiny Davarani, S. S.; Ahmar, H.; Makarem, S. J. Appl. Electrochem. 2008, 38, 1743–1747. doi:10.1007/s10800-008-9625-0 |
| 43. | Gerken, J. B.; Stamoulis, A.; Suh, S.-E.; Fischer, N. D.; Kim, Y. J.; Guzeia, I. A.; Stahl, S. S. Chem. Commun. 2020, 56, 1199–1202. doi:10.1039/c9cc08878d |
| 44. | Fotouhi, L.; Asadi, S.; Tammari, E.; Heravi, M. M.; Nematollahi, D. J. Iran. Chem. Soc. 2008, 5, 712–717. doi:10.1007/bf03246154 |
| 45. | Fotouhi, L.; Tammari, E.; Asadi, S.; Heravi, M. M.; Nematollahi, D. Int. J. Chem. Kinet. 2009, 41, 426–431. doi:10.1002/kin.20402 |
| 46. | Nematollahi, D.; Rafiee, M.; Fotouhi, L. J. Iran. Chem. Soc. 2009, 6, 448–476. doi:10.1007/bf03246523 |
| 47. | Toghan, A.; Abo-Bakr, A. M.; Rageh, H. M.; Abd-Elsabour, M. RSC Adv. 2019, 9, 13145–13152. doi:10.1039/c9ra01206k |
| 48. | Masoumi, H.; Khazalpour, S.; Jamshidi, M. RSC Adv. 2023, 13, 3083–3094. doi:10.1039/d2ra07250e |
| 35. | Smolyaninov, I. V.; Pitikova, O. V.; Rychagova, E. S.; Korchagina, E. O.; Poddel’sky, A. I.; Smolyaninova, S. A.; Berberova, N. T. Russ. Chem. Bull. 2016, 65, 2861–2867. doi:10.1007/s11172-016-1669-4 |
| 36. | Smolyaninov, I. V.; Pitikova, O. V.; Poddel’sky, A. I.; Berberova, N. T. Russ. Chem. Bull. 2018, 67, 1857–1867. doi:10.1007/s11172-018-2299-9 |
| 37. | Ukhin, L. Y.; Kuz’mina, L. G.; Alexeenko, D. V.; Belousova, L. V.; Morkovnik, A. S.; Shepelenko, E. N.; Podshibyakin, V. A.; Borodkin, G. S.; Dmitrieva, O. I. Russ. Chem. Bull. 2019, 68, 2290–2297. doi:10.1007/s11172-019-2702-1 |
| 38. | Galustyan, A.; Burmistrova, D. A.; Fokin, V. A.; Simeonova, K. S.; Polovinkina, M. A.; Pomortseva, N. P.; Baranov, E. V.; Almyasheva, N. R.; Smolyaninov, I. V.; Poddel’sky, A. I.; Berberova, N. T. Russ. Chem. Bull. 2024, 73, 714–727. doi:10.1007/s11172-024-4182-1 |
| 39. | Goksel, F. S.; Bayrak, N.; Ibis, C. Phosphorus, Sulfur Silicon Relat. Elem. 2014, 189, 113–123. doi:10.1080/10426507.2013.798787 |
| 40. | Tandon, V. K.; Maurya, H. K.; Kumar, S.; Rashid, A.; Panda, D. RSC Adv. 2014, 4, 12441–12447. doi:10.1039/c3ra47720g |
| 33. | Gao, F.; Wang, T.; Xiao, J.; Huang, G. Eur. J. Med. Chem. 2019, 173, 274–281. doi:10.1016/j.ejmech.2019.04.043 |
| 34. | Lobana, T. S. Coord. Chem. Rev. 2021, 441, 213884. doi:10.1016/j.ccr.2021.213884 |
© 2024 Burmistrova et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.