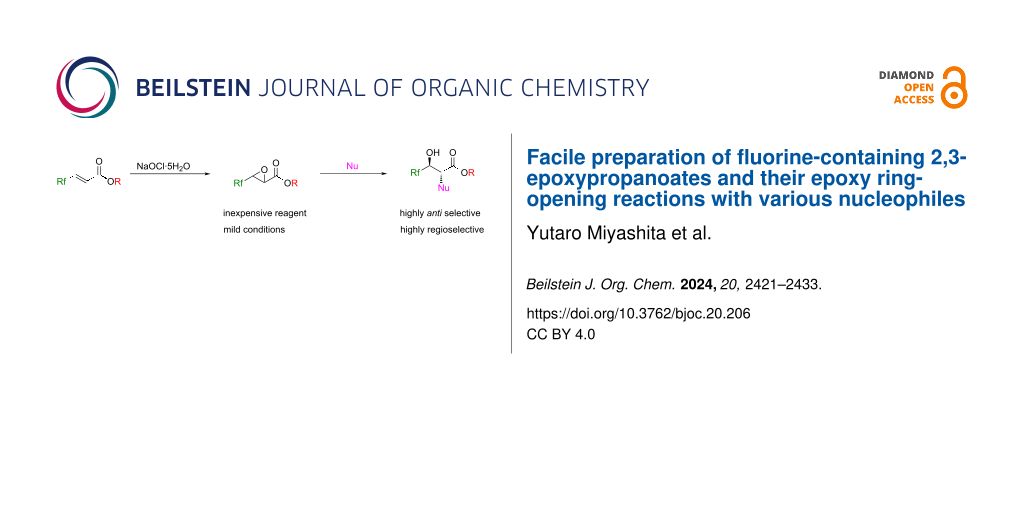
Abstract
We describe herein a facile method to access 2,3-epoxyesters with fluorine-containing substituents at their 3-position starting from the corresponding enoates by utilization of the low-costed and easy-to-handle reagent, NaOCl·5H2O. Because very little has been disclosed about the reactivity of such 2,3-epoxyesters, their epoxy ring opening by a variety of nucleophiles was carried out and we succeeded in clarifying these chemo- as well as regioselective processes proceeding via the SN2 mechanism to mainly afford 2-substituted 3-hydroxyesters usually in a highly anti selective manner.

Graphical Abstract
Introduction
Fluorine-containing compounds have been utilized in diverse fields due to their special character originating from unique fluorine atoms or fluorinated groups [1-7]. During our study in this area, ethyl 4,4,4-trifluorobut-2-enoate (1a) has been frequently employed as a potent and convenient Michael acceptor towards a variety of enolates [8-15] as well as organometallic species [16-19]. At least in part, its high reactivity was considered to be due to the significantly lower-lying LUMO energy level by the attachment of electron-withdrawing trifluoromethyl (CF3) and ethoxycarbonyl groups [20]. As we previously pointed out [10,21], the effective intramolecular interaction between fluorine and metals would also facilitate the smooth progress of these reactions. Such high potential of 1a allowed us to apply it to nucleophilic epoxidation because the resultant epoxyester 2a is recognized as an intriguing building block (Scheme 1).
![[1860-5397-20-206-i1]](/bjoc/content/inline/1860-5397-20-206-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Expectation of the regio- as well as stereoselective reactions of 2.
Scheme 1: Expectation of the regio- as well as stereoselective reactions of 2.
Another expectation to 2a is the high regio- and stereoselectivities of its epoxy ring opening specifically occurring at the 2 position in an SN2 manner, when it is treated with appropriate nucleophiles (Nu), leading to the formation of the 2-substituted 3-hydroxyesters with 2,3-anti stereochemistry. These characteristic outcomes would stem from a result of the electronically repulsive interaction between the incoming nucleophiles and an electronically strongly negative CF3 group, and the anticipated clean SN2 mechanism of epoxides in general, respectively. This is interestingly compared with the case of 2a with nonfluorinated Rf groups which sometimes suffered from the contamination of the regioisomers as a consequence of the regiorandom attack of nucleophiles [22-26].
Despite such significant advantage, compound 2a was previously prepared only by 1) the LDA-mediated iodination-intramolecular ring closure sequence from the corresponding chiral 4,4,4-trifluoro-3-hydroxybutyrate at low temperature [27-30], and 2) t-BuO2Li-mediated transformation of the enoates like 1g at −78 °C [31,32] and, to the best of our knowledge, no report has appeared on the convenient methods applicable to the larger scale synthesis to get access to the synthetically quite useful compounds like 2a [33,34].
Under such situations, we envisaged that the high electrophilicity of compound 1a would permit the usage of the extraordinarily convenient and mild reagent NaOCl [35-38] which opens the promising route for the preparation of 2a. Moreover, the fact that only very limited examples are known for their synthetic application except for the synthesis of 4,4,4-trifluorothreonine [29,33], stereoselective ring opening with organometallic species [29], and so on [32] also stimulated our interest. In this article, we would like to describe in detail the results of the preparation of epoxyesters 2 with various Rf groups as well as their reactivity with diverse nucleophiles [39].
Results and Discussion
Preparation of (E)-2,3-epoxypropanoates 2 with Rf groups at the 3 position
Because the urea·H2O2 complex proved its usefulness for the epoxidation of the β-CF3-α,β-unsaturated ketones [40], we applied this method at first for the epoxidation of 1b. However, contrary to our anticipation, only a total recovery of the substrate was observed, and further search for an oxidant reached the usage of a NaClO aqueous solution with its convenient handling and availability at a low cost. Following to the reported protocol [41], although a catalytic amount of Al2O3 and MgO worked nicely (entries 1 and 2 in Table 1), it was clarified that these additives were not necessary for the attainment of the same level of chemical yields (entries 3 vs 1 and 2). The drawback of this sequence was the isolated yield of 2b no more than 70% which was, at least in part, due to the production of the undesired hydrolyzed products from 1b and/or 2b under the alkaline conditions of this epoxidation reagent. This was experimentally proved by the detection of benzaldehyde which was considered to be formed by the NaClO-mediated oxidation of benzyl alcohol generated by hydrolysis. Changing the oxidizing reagent to crystalline NaClO·5H2O nicely solved the problem with the realization of 86% isolated yield of 2b by the utilization of this oxidant (2 equiv) at 0 °C with 6 h stirring (entry 8 in Table 1). We also tried to apply these conditions to other fluorine-containing substrates 1c–f and successfully obtained good to high yields of the desired products 2c–f, respectively (entries 10–13 in Table 1). The requirement of longer reaction time and higher temperature especially in the case of compounds 1e and 1f as well as the high loading of the oxidant in the latter might be due to their higher oleophobicity by possessing longer Rf chains. For all instances, epoxyesters 2 were obtained as single E-isomers, and based on the result obtained by the t-BuO2Li reagent [31], we speculated that NaClO·5H2O would similarly work for the corresponding Z-1 with retention of stereochemistry.
Table 1: Optimization of epoxidation conditions of 1.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-206-i8.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Sub. | NaClOa | (equiv) | Conditions | Isolated yieldb (%) |
| 1c | 1b | AQ | 1.0 | 25 °C, 6 h | 59 (67) |
| 2d | 1b | AQ | 1.0 | 25 °C, 5 h | (69) |
| 3 | 1b | AQ | 1.0 | 25 °C, 4.5 h | 60 (63) |
| 4 | 1b | S | 1.0 | 20 °C, 3 h | (65) |
| 5 | 1b | S | 1.5 | 20 °C, 3 h | (83) |
| 6 | 1b | S | 1.5 | 20 °C, 6 h | (84) |
| 7 | 1b | S | 1.5 | 0 °C, 6 h | (89) |
| 8 | 1b | S | 2.0 | 0 °C, 6 h | 86 (94) |
| 9 | 1b | S | 3.0 | 0 °C, 6 h | (83) |
| 10 | 1c | S | 2.0 | 0 °C, 6 h | 79 |
| 11 | 1d | S | 2.0 | 0 °C, 6 h | 78 |
| 12 | 1e | S | 2.0 | 0 °C, 6 h: 20 °C, 12 h | 73 |
| 13 | 1f | S | 5.0 | 20 °C, 48 h | 61 |
aAQ: a 5% aqueous solution, S: solid of NaClO·5H2O; bthe yields determined by 19F NMR were described in the parentheses; c10 mol % of Al2O3 was added; d20 mol % of MgO was added.
The procedure found here was also applied to the three representative CF3-containing α,β-unsaturated esters,1h–j [42] with different substitution patterns (Scheme 2).
![[1860-5397-20-206-i2]](/bjoc/content/inline/1860-5397-20-206-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Attempts of the present epoxidation to other α,β-unsaturated esters, 1h–j.
Scheme 2: Attempts of the present epoxidation to other α,β-unsaturated esters, 1h–j.
The subjection of the compounds 1h and 1i to the standard conditions described above resulted in high recovery of the substrates, which could be explained by their higher LUMO + 1 energy levels responsible for the epoxidation [43]. Extensive decomposition was observed in the case of 1j even in a shorter period possibly because of its significantly high electrophilicity by the attachment of three strongly electron-withdrawing moieties.
Reactions of (E)-3-Rf-2,3-epoxypropanoates 2 with amines, thiols, and metal halides
Because the epoxide ring opening is known to occur in an SN2 fashion, compounds 2 were recognized as versatile building blocks for the construction of 2-amino-3-hydroxypropanoates with 2,3-anti stereochemistry, if appropriate amines work nicely in a nucleophilic manner [44].
After the brief optimization of the conditions for the reaction of 2b and p-anisidine, good yields with high stereoselectivity were similarly recorded for the other substrates 2c and 2d possessing different Rf groups at the 3 position (Table 2, entries 1–3). Mixing of 2b with different primary (entries 4–7 in Table 2) and secondary (entries 8 and 9) amines led to the formation of the respective products in high to excellent yields without detection of any regio- as well as stereoisomers. The chirality contained in amines did not work efficiently for the stereochemical induction of the products (entries 6 and 7 in Table 2). In the case of secondary amines, the sterically demanding dibenzylamine failed in this transformation and recovery of 2b was observed (Table 2, entry 10). As was pointed out in the introductory section, the highly regioselective epoxy ring opening is well compared with the case when the nonfluorinated substrate (Ph instead of CF3 in 2b) was employed [25,26].
Table 2: Reactions of 2 with a variety of amines.
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-206-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Rf | R1 | R2 | Time (h) | Isolated yield (%) |
|---|---|---|---|---|---|
| 1a | CF3 | 4-MeOC6H4 | H | 19 | 78 (3ba) |
| 2a | CHF2 | 4-MeOC6H4 | H | 19 | 59 (3ca) |
| 3a | CClF2 | 4-MeOC6H4 | H | 19 | 76 (3da) |
| 4 | CF3 | PhCH2 | H | 7 | 86 (3bb) |
| 5 | CF3 | n-Bu | H | 7 | 48 (3bc) |
| 6 | CF3 | PhCH(CH3) | H | 18 | 77c (3bd) |
| 7b | CF3 | EtCH(Me)CH(CO2Bn) | H | 24 | 72c (3be) |
| 8 | CF3 | Et | Et | 7 | 83 (3bf) |
| 9 | CF3 | (CH2)4 | 7 | 56 (3bg) | |
| 10 | CF3 | Bn | Bn | 7 | –d |
aEtOH was used as the solvent and the reaction temperature was 50 °C; breaction was performed with 2.5 equiv of benzyl isoleucinate·TsOH and Et3N; cconsisted of 53:47 diastereomers in both cases; dno reaction was observed.
With the successful employment of amines as nucleophiles for the epoxy ring opening in a highly stereoselective fashion, we next turned our attention to thiols. Optimization of the reaction conditions based on the ones for amines clarified the tendency that the longer reaction time and the higher temperature decreased the chemical yields as well as the diastereomeric ratios (Table 3, entries 1–4). The higher pKa values of the carbonyl α-proton of 4 (for example, the pKa values of the protons of X-CH2C(O)Ph in DMSO were reported to be 17.1 (X: PhS) [45] and 20.3 (X: Ph2N) [46]) would result in the contamination of the stereoisomers when compared with the case of the compounds 3 [47,48]. Because control of the amount of PhCH2SH to 1.0 equiv did not give a positive effect, the conditions in entry 4 (Table 3) were eventually determined as the best.
Table 3: Reactions of 2 with a variety of thiols.
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-206-i10.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Rf | R1 | Time (h) | Isolated yield (%) | dra |
|---|---|---|---|---|---|
| 1b | CF3 | PhCH2 | 3 | 92 (4ba) | 87:13 |
| 2b | CF3 | PhCH2 | 12 | 75 (4ba) | 75:25 |
| 3c | CF3 | PhCH2 | 3 | 80 (4ba) | 61:39 |
| 4 | CF3 | PhCH2 | 5 | 90 (4ba) | 94:6 |
| 5d | CF3 | PhCH2 | 5 | 90 (4ba) | 94:6 |
| 6 | CHF2 | PhCH2 | 48 | 76 (4ca) | >99:1 |
| 7 | CClF2 | PhCH2 | 24 | 87 (4da) | 90:10 |
| 8 | C2F5 | PhCH2 | 81 | 72 (4ea) | 69:31 |
| 9 | CF3 | CH3(CH2)9 | 10 | 59e (4bb) | 95:5 |
| 10 | CF3 | Ph | 5 | 92 (4bc) | 93:7 |
| 11 | CF3 | CH3OC(O)CH2 | 5 | 94 (4bd) | 95:5 |
aDetermined by 19F NMR; breaction at 40 °C; creaction at 60 °C; dutilization of 1.0 equiv of PhCH2SH resulted in the observation of 9% recovery of 2b by 19F NMR; e7% recovery of 2a was observed by 19F NMR.
The different epoxyesters 2c–e were also applied for this ring-opening reaction with the same thiol (entries 6–8 in Table 3). It is interesting to note that a longer reaction time was required for these substrates which would be the major reason for the relatively low diastereomeric ratio (especially in the case of entry 8 in Table 3) while the CHF2-possessing epoxyester 2b furnished a single stereoisomer (entry 6) whose reason was not clear yet. Other thiols like decanethiol, thiophenol, and thioglycolate all worked nicely to furnish the corresponding products 4bb–bd in good to excellent chemical yields with high stereoselectivities (Table 3, entries 9–11).
The stereostructure of the products was confirmed by X-ray crystallographic analysis using the minor diastereomer of 3bd, nicely separated from the major isomer by recrystallization, and the major product 4ba. As was our expectation, these compounds [49] possess the anti relationship between the 2 and 3 positions which clearly proved the epoxy ring opening taking place at the 2 position in an SN2 fashion (Figure 1).
![[1860-5397-20-206-1]](/bjoc/content/figures/1860-5397-20-206-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Crystallographic structure of the epoxy ring-opening products by PhCH(NH2)Me (3bd) and PhCH2SH (4ba).
Figure 1: Crystallographic structure of the epoxy ring-opening products by PhCH(NH2)Me (3bd) and PhCH2SH (4ba...
The introduction of an additional halogen atom was considered to be possible by treatment of 2b with an appropriate metal salt, and actually, similar results to the case of amines and thiols were obtained by using the corresponding MgX2 [23,24]. It was proved that a larger amount of nucleophiles, higher temperature, and longer time all led to a decrease in the diastereomeric ratio of the products 5 (ca 10%) like the case of thiols described above. This is the reason why the three examples shown in Scheme 3 stopped before completion, and, for example, 24 h stirring in the case of the Cl atom entry furnished 67% yield of 5ba [32,34] and 19% recovery of 2b with the diastereomeric ratio of the former of 97:3. Contamination by the deiodinated 3-hydroxyester [50] was noticed during the synthesis of 5bc using LiI.
![[1860-5397-20-206-i3]](/bjoc/content/inline/1860-5397-20-206-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Introduction of additional halogen atoms at the 2-position of the compound 2b.
Scheme 3: Introduction of additional halogen atoms at the 2-position of the compound 2b.
Reactions of (E)-4,4,4-trifluoro-2,3-epoxybutanoate 2b with compounds possessing an acidic proton
It was very interesting to know that there were scarce examples in the literature [51] on the ring opening of 2,3-epoxyesters in general by the stabilized anionic species from, for example, malonate. One reason could be because of the formation of the less stable alkoxide by the progress of the nucleophilic addition. If this is really the case, the presence of the strongly electron-withdrawing fluorine-containing groups in our instance should nicely affect the characteristics of the resultant intermediate which could lead to the realization of the addition of such nucleophilic species.
First of all, as shown in Table 4, we started to investigate the reactivity of 2b toward sodium malonate as the representative nucleophile. Because a brief solvent search indicated DMSO as the best for the attainment of high yields and diastereoselectivity (entries 1–5 vs 6 in Table 4), we further examined bases in this solvent to find out that t-BuOK behaved nicely, and the reaction of 2b with 2.0 equiv of diethyl malonate for 0.5 h at room temperature furnished 93% yield of the product (Table 4, entry 15). During this optimization process, the obtained product was uncovered not to be a single component but a mixture of two compounds, anti,syn-7a and anti,syn-7b, the latter of which seemed to be produced from the former by the attack of the ethoxide ion released during the lactone-forming process. Their close structural resemblance led to a significant peak overlap both in the 1H and 19F NMR spectra which made it difficult to obtain their exact ratio and thus, the combined 19F NMR yields were shown in Table 4. Separation of these two compounds was eventually succeeded by the usual hydrogenolysis to furnish the carboxylic acid anti,syn-8a in 79% isolated yield and the lactone anti,syn-7b was recovered in 13% yield (Scheme 4) which was considered to be the reflection of the original composition of anti,syn-7a and -7b. The relative stereochemistry of anti,syn-8a was confirmed as 2,3-anti-3,4-syn by its X-ray crystallographic analysis [49] (Figure 2) whose construction could be readily understood as the result of a highly stereoselective SN2-type epoxy ring opening of 2a, followed by the intramolecular lactone formation with the pro-R ethoxycarbonyl group possibly due to the higher steric congestion by the selection of the other CO2Et moiety.
Table 4: Reactions of 2b with the anionic species from diethyl malonate.
![[Graphic 4]](/bjoc/content/inline/1860-5397-20-206-i11.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Base | Solvent | Yielda (%) | dr | Recovery (%) |
| 1b | NaH | THF | 20 | >99:1 | 0 |
| 2b | NaH | Toluene | 6 | >99:1 | 13 |
| 3b | NaH | Et2O | 12 | >99:1 | 13 |
| 4b | NaH | MeCN | 45 | 98:2 | 7 |
| 5b | NaH | DMF | 75 | 96:4 | 0 |
| 6 | NaH | DMSO | 78 | 99:1 | 0 |
| 7 | Et3N | DMSO | 0 | – | 83 |
| 8 | TMG | DMSO | 22 | 14:86 | 3 |
| 9 | DBU | DMSO | 13 | 23:77 | 2 |
| 10 | CsF | DMSO | 34 | 91:9 | 21 |
| 11 | K2CO3 | DMSO | 50 | 98:2 | 13 |
| 12 | t-BuOK | DMSO | 85 | 98:2 | 0 |
| 13c | t-BuOK | DMSO | 91 | 99:1 | 0 |
| 14d | t-BuOK | DMSO | 94 | 98:2 | 0 |
| 15c,e | t-BuOK | DMSO | 93 | 99:1 | 0 |
| 16b,c,e | t-BuONa | DMF | 56 | 98:2 | 5 |
| 17b,c,e | t-BuOLi | DMF | trace | – | 32 |
aCombined yields of anti,syn-7a and -7b were determined by 19F NMR and isolated yield of anti,syn-7a was shown in parentheses; b0 °C for 30 min were employed for the step 1 instead of rt, 15 min; c2.0 equiv of malonate was used; d3.0 equiv of malonate was used; estirring for 0.5 h for step 2.
![[1860-5397-20-206-i4]](/bjoc/content/inline/1860-5397-20-206-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Clarification of the stereochemistry of anti,syn-8a and -7b.
Scheme 4: Clarification of the stereochemistry of anti,syn-8a and -7b.
![[1860-5397-20-206-2]](/bjoc/content/figures/1860-5397-20-206-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystallographic structure of anti,syn-8a.
Figure 2: Crystallographic structure of anti,syn-8a.
As shown in entries 8 or 9 in Table 4, it was proved that the usage of tetramethylguanidine (TMG) or DBU as the base provided a different stereoisomer as the major component. For the confirmation of its stereostructure, the isolated inseparable mixture of anti,syn-7a and -7b by the reaction of 2b and diethyl malonate was treated with an equimolar amount of DBU in DMSO (rt, 3 h) to furnish products which were identical to the ones obtained in entries 8 or 9 (Table 4). The relative stereochemistry of the isomerized products was concluded by the observed NOESY cross peaks between H2-H4 and H3-H4, clearly demonstrating the relationship between these three hydrogen atoms as cis. Formation of syn,syn-7a and -7b by the above tertiary amines would be mechanistically elucidated by the deprotonation of the most acidic H2 from the initially formed anti,syn-7a and -7b, followed by the re-protonation by the sterically bulky [H·amine]+ from the less congested top side.
These results prompted us to further investigate the ring opening of 2b by other nucleophiles with active hydrogen whose results are summarized in Scheme 5. If the in situ conversion of anti,syn-7a to anti,syn-7b follows the above ester alcohol exchange mechanism, employment of dibenzyl malonate should afford a single compound. This is actually the case and the expected dihydrofuran anti,syn-7c was obtained in 53% yield as a 98:2 diastereomer mixture, and stereochemistry of the major isomer was deduced from the above result as anti,syn. Although malononitrile also furnished the dihydrofuran syn-7d in good yield as a sole stereoisomer, a sharp contrast to these results was observed when 2b was subjected to the anionic species from cyanoacetate, allowing to isolate the acyclic hydroxyester anti-9e in 76% yield. Smooth conversion to the dihydrofuran syn-7e was observed from this intermediary compound anti-9e by refluxing the crude solution in AcOEt.
![[1860-5397-20-206-i5]](/bjoc/content/inline/1860-5397-20-206-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of 2b with other stabilized nucleophiles.
Scheme 5: Reaction of 2b with other stabilized nucleophiles.
Different from these outcomes, other possible nucleophilic candidates like acetylacetone (pKa value of the active hydrogen in DMSO: 13.3 [52]), nitromethane (17.2 [53]), ethyl (diethylphosphono)acetate (18.6 [52]), malononitrile (11.1 [53]), ethyl 2-nitroacetate (9.1 [54]), ethyl 2-cyanoacetate (13.1 [55]), and diethyl malonate (16.4 [56]) all failed to afford the desired addition products. From the complex mixture after mixing 2b with t-BuOK and nitroacetate in DMSO at 80 °C, the unexpected compound 2,3-dihydroxybutyrate anti-10a was isolated as a single isomer. Its production was also detected by 19F NMR from the reaction mixture when nitromethane (16%) and ethyl (diethylphosphono)acetate (17%) were employed instead of nitroacetate, while no other compound was separated from these mixtures due to their complexity (Scheme 6).
![[1860-5397-20-206-i6]](/bjoc/content/inline/1860-5397-20-206-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Production of 4,4,4-trifluoro-2,3-dihydroxybutanoate anti-10a.
Scheme 6: Production of 4,4,4-trifluoro-2,3-dihydroxybutanoate anti-10a.
Reactions of (E)-4,4,4-trifluoro-2,3-epoxybutanoate 2b with Grignard-based copper reagents
Despite the previous report by the Seebach group on the intriguing reactivity of the CF3-containing ethyl 2,3-epoxybutanoate 2a towards a variety of organometallic species [27-29], because relatively readily accessible Grignard-based cuprates were not involved, their applicability to 2b as the representative partner was investigated here (Table 5).
Table 5: Optimization of the reaction conditions of 2b with the n-C10H21MgBr-based cuprate.
![[Graphic 5]](/bjoc/content/inline/1860-5397-20-206-i12.svg?max-width=637&scale=1.0)
|
|||||
| Amount (equiv) | 19F NMR yielda (%) | ||||
| Entry | CuI | n-C10H21MgBr | Time (h) | 11a | 12a |
| 1b | 2.0 | 2.0 | 1 | 47 | 4 |
| 2 | 2.0 | 4.0 | 1 | 30 | 40 |
| 3 | 2.0 | 4.0 | 4 | 64 | 12 |
| 4 | 1.6 | 3.2 | 4 | 66 | 16 |
| 5 | 1.2 | 2.4 | 4 | 49 | 19 |
| 6c | 1.6 | 3.2 | 1 | 56 | 7 |
| 7 | – | 3.2 | 0.5 | 0 | 84 (73) |
| 8d | 1.6 | 3.2 | 3 | 3 | 13 |
| 9e | 1.6 | 3.2 | 3 | 7 | 14 |
| 10f | 1.6 | 3.2 | 3 | (79) | 6 |
aIsolated yields are described in parentheses; breaction at 0 °C when the cuprate was added to 2b; c19% of the epoxy ketone 13a was detected by 19F NMR; dthe reaction was carried out in THF; eCuCN was employed instead of CuI; fthe cuprate was prepared at −40 °C.
The 1:2 ratio of CuI and n-C10H21MgBr was selected due to the better material balance than the case of 1:1 (entries 1 and 2 in Table 5), the latter of which afforded an almost equimolar amount of the hydroxyketone 11a and epoxyalcohol 12a. A decrease of the temperature to −40 °C resulted in the better preference of 11a (Table 5, entry 3), and 1.6 and 3.2 equiv of CuI and n-C10H21MgBr, respectively, were concluded as the best amounts for the synthesis of the nucleophilic species (entries 3–5). The shorter reaction time led to a slightly better ratio of 11a to 12a with a lower combined yield along with the detection of the epoxyketone 13a at the same instance (entry 6 in Table 5). We recognized compound 13a as the precursor for the formation of 11a and 12a. The conditions in the absence of CuI afforded 12a as the sole product (entry 7 in Table 5) whose result was nicely compared with the one previously reported [29]. Changing the solvent to THF (Table 5, entry 8) or the Cu species to CuCN (entry 9) both did not have a positive effect on the present reaction, and we eventually found out that the temperature for the preparation of the cuprate was important and lowering it to −40 °C nicely allowed to record 79% isolated yield of 11a with only 6% of the byproduct 12a (entry 10).
The conditions described in entry 10 in Table 5 were applied to the reactions of 2b with other Grignard reagents in the presence of CuI (Table 6).
Table 6: Reactions of Grignard-based cuprates with 2b.
![[Graphic 6]](/bjoc/content/inline/1860-5397-20-206-i13.svg?max-width=637&scale=1.0)
|
||
| Isolated yield (%) | ||
| R | 11 | 12 |
| n-C10H21- (a) | 79 | 6a |
| PhCH2CH2- (b) | 57 | 13a |
| c-Hex- (c) | 55 | 4a |
| Ph- (d) | tracea | 77 |
| 4-MeOC6H4- (e) | 0a | 80 |
aDetermined by 19F NMR.
PhCH2CH2MgBr and c-C6H11MgBr produced the β-hydroxyketones 11b and 11c in 57% and 55% yields, respectively, along with small amounts of the corresponding epoxyalcohols 12b and 12c. On the other hand, 12d and 12e were substantially formed by ArMgBr (Ar: Ph and 4-MeOC6H4, respectively), the former of which was reported to be obtained by the action of PhLi alone [29]. It was intriguing to note that the present method yielded the unprecedented products 11 by the reaction of the epoxyester 2b with other organometallic species.
For the mechanistic clarification of the present reactions, two additional experiments were executed which are shown in Scheme 7. Employment of the epoxyketone 13f (R: n-C6H13), structurally analogous to 13a, to the reaction with (n-C10H21)2CuMgBr furnished a mixture of the hydroxyketone 11f and epoxyalcohol 12f in 32% and 52% yields, respectively. This experimental result clearly indicated that the conversion of 13a to 11a is one of the possible routes.
![[1860-5397-20-206-i7]](/bjoc/content/inline/1860-5397-20-206-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reactions of n-C10H21MgBr-based cuprate with 13f as well as 2b with/without D2O quenching.
Scheme 7: Reactions of n-C10H21MgBr-based cuprate with 13f as well as 2b with/without D2O quenching.
The second reaction was carried out for the verification of the intermediate leading to the product 11. Although we initially assumed that the epoxy ring opening occurred by hydride generated through the β-elimination of the n-C10H21MgBr-based cuprate species, the TLC analysis of the reaction mixture did not show any evidence of the production of the possible olefinic product n-C8H17CH=CH2. Moreover, when the reaction mixture was quenched with D2O, incorporation of deuterium was observed to give 11a-D in a high yield which allowed us to conclude the possible presence of the C-copper species just before quenching. Our result well compares with the one by Alexakis et al. [57]. In their instance, the reaction of t-Bu2CuCNLi2 and cyclohexene oxide afforded a mixture of products in 10 and 50% yields as a result of the epoxy ring opening by t-Bu group and hydride, respectively. Their additional experiment to quench the corresponding intermediate by D2O proved that no deuteration occurred. This result clearly indicated that hydride was released from the t-Bu group of the Cu(III) species formed after the nucleophilic attack of the epoxy ring. In our case, since the strongly electron-withdrawing CF3 group would render the rate of the reductive elimination very slow, the intermediary Cu(III) species safely existed until the addition of D2O. Because the significant overlap of NMR peaks was observed due to the quite similar structure of 11a and 11a-D, quantitative analysis of the deuterium content of 11a-D was not possible. However, the comparison of their specific region of the 13C NMR charts and sharp peaks readily led us to qualitative understanding of the high purity of 11a-D possibly as a single diastereomer (Figure 3).
![[1860-5397-20-206-3]](/bjoc/content/figures/1860-5397-20-206-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: A part of 13C NMR spectra for the compounds 11a and 11a-D.
Figure 3: A part of 13C NMR spectra for the compounds 11a and 11a-D.
Conclusion
As described above, we have succeeded in the facile preparation of 2,3-epoxyesters 2 with a variety of Rf groups at the 3 position starting from the corresponding 3-Rf-acrylates 1 by the action of the low cost and easy-to-handle reagent, NaOCl·5H2O. The special feature of this process is the requirement of a very mild temperature of 0 °C which can be well compared to the previous one executed at −78 °C under the action of LDA [29]. Moreover, by using the epoxyester 2b as the representative substrate, clarification of its reactivity was carried out by mixing with 1) heteronucleophiles like amines, thiols, and magnesium halides, 2) softer carbon nucleophiles such as malonates, and 3) Grignard-based cuprates. These processes usually yielded the addition products along with the epoxy ring opening at the 2 position via the SN2 mechanism, affording 3-Rf-3-hydroxyesters with the incorporation of a variety of substituents at the 2-position in a highly anti-selective fashion. We believe that the facile procedure presented here opens novel routes to the application of these intriguing products in a variety of fields.
Experimental
General procedure for the formation of the epoxyesters (GP-1): Benzyl (E)-2,3-epoxy-4,4,4-trifluorobutanoate (2b)
GP-1A (by use of aqueous NaClO): To a solution of compound 1b [42] (0.23 g, 1.00 mmol) in 3.0 mL of CH3CN was added NaClO aq. (5% in H2O, 1.50 g, 1.00 mmol) and the solution was stirred for 4.5 h at room temperature. This mixture was extracted with CH2Cl2 and the usual workup and purification afforded 0.15 g (0.60 mmol) of the pure title compound in 60% yield.
GP-1B (by use of NaClO·5H2O): To a solution of compound 1b [42] (0.2302 g, 1.00 mmol) in 3.0 mL of CH3CN was added NaClO·5H2O (0.3290 g, 2.00 mmol) at 0 °C, and the solution was stirred for 6 h at the same temperature. After the same workup process and purification with silica gel column chromatography using AcOEt/Hex 1:20 as an eluent, 0.2117 g (0.86 mmol) of the title compound (86% yield) were isolated. Rf 0.52 (Hex/AcOEt 5:1); 1H NMR (300.40 MHz, CDCl3) δ 3.71–3.76 (m, 2H), 5.21 (d, J = 12.3 Hz, 1H), 5.28 (d, J = 12.3 Hz, 1H), 7.34–7.44 (m, 5H); 13C NMR (75.45 MHz, CDCl3) δ 49.4 (q, J = 2.5 Hz), 52.7 (q, J = 42.2 Hz), 68.0, 121.4 (q, J = 276.0 Hz), 128.5, 128.7, 128.8, 134.3, 165.6; 19F NMR (282.65 MHz, CDCl3) δ −75.12 (d, J = 4.5 Hz); IR (neat) ν: 3944, 3689, 3054, 2987, 2685, 2306, 1756, 1456, 1422, 1382, 1341, 1265, 1169, 1089, 988, 929, 896, 664 cm−1; Anal. calcd for C11H9F3O3: C, 53.67; H, 3.68; found: C, 53.54; H, 3.89.
General procedure for the ring opening of epoxides (GP-2). Benzyl 2,3-anti-4,4,4-trifluoro-3-hydroxy-2-(p-methoxyphenyl)amino-butanoate (3ba)
p-Anisidine (0.07 g, 0.60 mmol) was added to an EtOH (3 mL) solution of compound 2b (0.12 g, 0.50 mmol), and the resultant mixture was stirred at 50 °C for 19 h under the open air. After quenching the reaction with 1 M HCl aq., the mixture was extracted with AcOEt three times and the combined organic phase was washed with brine. Evaporation of the volatiles furnished crude materials which were recrystallized by use of Hex/CHCl3 3:2 as a solvent to afford 0.14 g (0.39 mmol) of the title compound 3aa in 78% yield as a sole stereoisomer. Rf 0.30 (Hex/AcOEt 2:1); mp 95–97 °C; 1H NMR (300.40 MHz, CDCl3) δ 3.70 (brs, 1H), 3.76 (s, 3H), 4.31–4.33 (m, 2H), 4.39 (brs, 1H), 5.14 (dd, J = 12.0, 21.3 Hz, 1H), 6.74–6.81 (m, 4H), 7.26–7.36 (m, 5H); 13C NMR (75.45 MHz, acetone-d6) δ 55.5, 59.3, 67.9, 70.0 (q, J = 30.2 Hz), 114.8, 117.7, 124.1 (q, J = 283.5 Hz), 128.5, 128.6, 128.7, 134.4, 139.5, 154.3, 170.2; 19F NMR (282.65 MHz, CDCl3) δ −76.83 (d, J = 9.0 Hz); IR (KBr) ν: 3454, 3315, 2955, 2924, 2854, 2360, 1741, 1519, 1458, 1238, 1204, 1156, 1138, 1097, 1030, 822, 749 cm−1; HRMS–FAB (m/z): [M]+ calcd for C18H18F3NO4, 369.1182; found, 369.1209.
General procedure for the ring opening of epoxides by enolates (GP-3). 4-Benzyl 5-ethyl anti,syn-tetrahydro-2-oxo-3-(trifluoromethyl)-furan-4,5-dicarboxylate (anti,syn-7a) and 4,5-diethyl anti,syn-tetrahydro-2-oxo-3-(trifluoromethyl)furan-4,5-dicarboxylate (anti,syn-7b)
Diethyl malonate (0.18 mL, 1.20 mmol) was added to a flask containing 0.0673 g (0.60 mmol) of t-BuOK in DMSO (1.8 mL) under an argon atmosphere and the resultant mixture was stirred for 15 min at room temperature. Then, 0.1477 g (0.60 mmol) of 2b in 0.8 mL of DMSO was introduced to the resultant solution and the stirring was continued for 0.5 h. The same workup process and purification furnished 0.1717 g of an inseparable mixture of anti,syn-7a (dr = 99:1) and anti,syn-7b (7a:7b = 83:17). Anti,syn-7a: Rf 0.34 (Hex/AcOEt 4:1); 1H NMR (300.40 MHz, CDCl3) δ 1.32 (t, J = 7.2 Hz, 3H), 4.20–4.27 (m, 2H), 4.21–4.35 (m, 2H), 5.05 (quint, J = 7.2 Hz, 1H), 5.15 (d, J = 12.3 Hz, 1H), 5.23 (d, J = 12.0 Hz, 1H), 7.32–7.40 (m, 5H); 13C NMR (75.45 MHz, CDCl3) δ 13.8, 44.4, 46.4, 63.1, 68.5, 73.5 (q, J = 34.1 Hz), 122.5 (q, J = 282.9 Hz), 128.61, 128.63, 128.8, 134.0, 165.1, 165.6, 167.4; 19F NMR (282.65 MHz, CDCl3) δ −75.84 (d, J = 6.8 Hz); IR (neat) ν: 2987, 1813, 1742, 1457, 1389, 1321,1218, 1182, 1128, 1023, 972, 755 cm−1; HRMS–FAB+ (m/z): [M + H]+ calcd for C16H16F3O6, 361.0893; found, 361.0911. Epimer at the 2 position of anti,syn-7a (syn,syn-7a): 1H NMR (300.40 MHz, CDCl3) δ 1.30 (t, J = 7.2 Hz, 3H), 4.00 (d, J = 8.4 Hz, 1H), 4.08 (dd, J = 6.3, 8.1 Hz, 1H), 4.26–4.34 (m, 2H), 5.00 (quint, J = 5.7 Hz, 1H), 5.22 (d, J = 12.0 Hz, 1H), 5.27 (d, J = 12.3 Hz, 1H), 7.31–7.41 (m, 5H); 13C NMR (75.45 MHz, CDCl3) δ 13.9, 43.2, 48.5, 63.3, 68.6, 74.9 (q, J = 35.4 Hz), 122.4 (q, J = 279.8 Hz), 128.3, 128.8, 128.9, 134.2, 164.5, 167.1, 168.2; 19F NMR (282.65 Hz, CDCl3) δ −79.55 (d, J = 4.8 Hz); HRMS–FAB+ (m/z): [M + H]+ calcd for C16H16F3O6, 361.0893; found, 361.0909.
General procedure for the reaction of the epoxyester 2b with cuprates (GP-4): 1,1,1-Trifluoro-2-hydroxytetradecan-4-one (11a)
1.70 mL of a 0.94 M Et2O solution of decylmagnesium bromide (1.6 mmol) was added to an Et2O (3.0 mL) solution containing 0.1524 g (0.80 mmol) of CuI at −40 °C under an argon atmosphere and the resultant mixture was stirred for 0.5 h at that temperature. A solution of 0.1231 g (0.50 mmol) of 2b in Et2O (1.0 mL) was added and the mixture was stirred for 3 h at the same temperature. After quenching the reaction with a saturated NH4Cl aq, the usual workup afforded 0.1116 g (0.40 mmol) of the title compound in 79% yield after silica gel column chromatography using Hex/AcOEt 6:1 as an eluent. Rf 0.51 (Hex/AcOEt 4:1); 1H NMR (300.40 MHz, CDCl3) δ 0.88 (t, J = 6.9 Hz, 3H), 1.26 (brs, 14H), 1.60 (quint, J = 6.9 Hz, 2H), 2.49 (t, J = 7.5 Hz, 2H), 2.74 (dd, J = 3.6, 17.7 Hz, 1H), 2.83 (dd, J = 9.0, 17.7 Hz, 1H), 3.49 (d, J = 4.2 Hz, 1H), 4.43–4.56 (m, 1H); 13C NMR (75.45 MHz, CDCl3) δ 14.0, 15.0, 22.6, 23.4, 29.26, 29.28, 29.4, 29.5, 31.8, 41.8 (q, J = 1.2 Hz), 43.7, 66.4 (q, J = 32.2 Hz), 124.7 (q, J = 281.1 Hz), 208.9; 19F NMR (282.65 MHz, CDCl3) δ −80.79 (d, J = 7.1 Hz); IR (neat) ν: 3408, 2958, 2927, 2856, 1720, 1469, 1291, 1176, 1146, 899, 841, 719, 643 cm−1; HRMS–FAB+ (m/z): [M + H]+ calcd for C14H26F3O2, 283.1879; found, 283.1893.
Supporting Information
| Supporting Information File 1: Full experimental and analytical details, copies of NMR spectra for new compounds, and crystallographic data. | ||
| Format: PDF | Size: 9.8 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393x
Return to citation in text: [1] -
Uneyama, K., Ed. Organofluorine Chemistry; Blackwell:: Oxford, UK, 2006. doi:10.1002/9780470988589
Return to citation in text: [1] -
Yamazaki, T.; Taguchi, T.; Ojima, I. Unique Properties of Fluorine and their Relevance to Medicinal Chemistry and Chemical Biology; Introduction: Basic Aspects of Fluorine Substituent Effect in Medicinal Chemistry. In Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I., Ed.; Wiley: West Sussex, UK, 2009; pp 3–46. doi:10.1002/9781444312096.ch1
Return to citation in text: [1] -
Brittain, W. D. G.; Lloyd, C. M.; Cobb, S. L. J. Fluorine Chem. 2020, 239, 109630. doi:10.1016/j.jfluchem.2020.109630
Return to citation in text: [1] -
O'Hagan, D. Chem. – Eur. J. 2020, 26, 7981–7997. doi:10.1002/chem.202000178
Return to citation in text: [1] -
Remete, A. M.; Nonn, M.; Escorihuela, J.; Fustero, S.; Kiss, L. Eur. J. Org. Chem. 2021, 5946–5974. doi:10.1002/ejoc.202101094
Return to citation in text: [1] -
Decaens, J.; Couve-Bonnaire, S.; Charette, A. B.; Poisson, T.; Jubault, P. Chem. – Eur. J. 2021, 27, 2935–2962. doi:10.1002/chem.202003822
Return to citation in text: [1] -
Yamazaki, T.; Haga, J.; Kitazume, T.; Nakamura, S. Chem. Lett. 1991, 2171–2174. doi:10.1246/cl.1991.2171
Return to citation in text: [1] -
Yamazaki, T.; Haga, J.; Kitazume, T. Chem. Lett. 1991, 20, 2175–2178. doi:10.1246/cl.1991.2175
Return to citation in text: [1] -
Shinohara, N.; Haga, J.; Yamazaki, T.; Kitazume, T.; Nakamura, S. J. Org. Chem. 1995, 60, 4363–4374. doi:10.1021/jo00119a013
Return to citation in text: [1] [2] -
Yamazaki, T.; Shinohara, N.; Kitazume, T.; Sato, S. J. Org. Chem. 1995, 60, 8140–8141. doi:10.1021/jo00130a012
Return to citation in text: [1] -
Pan, X.; Liu, Z. Tetrahedron 2014, 70, 4602–4610. doi:10.1016/j.tet.2014.05.049
Return to citation in text: [1] -
Shu, C.; Liu, H.; Slawin, A. M. Z.; Carpenter-Warren, C.; Smith, A. D. Chem. Sci. 2020, 11, 241–247. doi:10.1039/c9sc04303a
Return to citation in text: [1] -
Kim, B.-J.; Song, Y.-N.; Lee, S. Y.-M. Chem. Commun. 2021, 57, 11052–11055. doi:10.1039/d1cc04875a
Return to citation in text: [1] -
Wu, J.; Young, C. M.; Watts, A. A.; Slawin, A. M. Z.; Boyce, G. R.; Bühl, M.; Smith, A. D. Org. Lett. 2022, 24, 4040–4045. doi:10.1021/acs.orglett.2c01486
Return to citation in text: [1] -
Yamazaki, T.; Shinohara, N.; Kitazume, T.; Sato, S. J. Fluorine Chem. 1999, 97, 91–96. doi:10.1016/s0022-1139(99)00034-2
Return to citation in text: [1] -
Yamazaki, T.; Taketsugi, M.; Kawasaki-Takasuka, T.; Agou, T.; Kubota, T. Bull. Chem. Soc. Jpn. 2021, 94, 1815–1822. doi:10.1246/bcsj.20210136
Return to citation in text: [1] -
Morigaki, A.; Tanaka, T.; Miyabe, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2013, 11, 586–595. doi:10.1039/c2ob26708j
Return to citation in text: [1] -
Zhang, L.-Y.; Zhou, J.-H.; Xu, Y.-H.; Loh, T.-P. Chem. – Asian J. 2015, 10, 844–848. doi:10.1002/asia.201403303
Return to citation in text: [1] -
Computation of 1b and its non-fluorinated counterpart as the methyl ester clearly showed their LUMO + 1 energy levels as 0.053 and 0.961 eV, respectively, by employment of Gaussian 09W, Revision D.01 using the B3LYP/6-311++G** level of theory.
Return to citation in text: [1] -
Yamazaki, T.; Ando, M.; Kitazume, T.; Kubota, T.; Omura, M. Org. Lett. 1999, 1, 905–908. doi:10.1021/ol990821c
Return to citation in text: [1] -
Gleason, J. G.; Hall, R. F.; Perchonock, C. D.; Erhard, K. F.; Frazee, J. S.; Ku, T. W.; Kondrad, K.; McCarthy, M. E.; Mong, S.; Crooke, S. T.; Chi-Rosso, G.; Wasserman, M. A.; Torphy, T. J.; Muccitelli, R. M.; Hay, D. W.; Tucker, S. S.; Vickery-Clark, L. J. Med. Chem. 1987, 30, 959–961. doi:10.1021/jm00389a001
Return to citation in text: [1] -
Righi, G.; Rumboldt, G.; Bonini, C. J. Org. Chem. 1996, 61, 3557–3560. doi:10.1021/jo951441h
Return to citation in text: [1] [2] -
Righi, G.; Pescatore, G.; Bonadies, F.; Bonini, C. Tetrahedron 2001, 57, 5649–5656. doi:10.1016/s0040-4020(01)00492-6
Return to citation in text: [1] [2] -
Durán Pachón, L.; Gamez, P.; van Brussel, J. J. M.; Reedijk, J. Tetrahedron Lett. 2003, 44, 6025–6027. doi:10.1016/s0040-4039(03)01480-1
Return to citation in text: [1] [2] -
Rackl, D.; Kais, V.; Lutsker, E.; Reiser, O. Eur. J. Org. Chem. 2017, 2130–2138. doi:10.1002/ejoc.201700014
Return to citation in text: [1] [2] -
Seebach, D.; Beck, A. K.; Renaud, P. Angew. Chem., Int. Ed. Engl. 1986, 25, 98–99. doi:10.1002/anie.198600981
Return to citation in text: [1] [2] -
Lin, J. T.; Yamazaki, T.; Takeda, M.; Kitazume, T. J. Fluorine Chem. 1989, 44, 113–120. doi:10.1016/s0022-1139(00)84374-2
Return to citation in text: [1] [2] -
von dem Bussche-Hünnefeld, C.; Seebach, D. Chem. Ber. 1992, 125, 1273–1281. doi:10.1002/cber.19921250538
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Ishii, A.; Kanai, M.; Kuriyama, S.; Yasumoto, M.; Inomiya, N.; Otsuka, T.; Ueda, H. Preparation of 4,4,4-trifluoro-2,3-epoxybutanoic acid esters. Japanese patent JP2005047870, Feb 24, 2005.
See for preparation of compound 2a by the Bayer-Villiger oxidation of epoxyketones.
Return to citation in text: [1] -
Lanier, M.; Haddach, M.; Pastor, R.; Riess, J. G. Tetrahedron Lett. 1993, 34, 2469–2472. doi:10.1016/s0040-4039(00)60443-4
Return to citation in text: [1] [2] -
Lanier, M.; Le Blanc, M.; Pastor, R. Tetrahedron 1996, 52, 14631–14640. doi:10.1016/0040-4020(96)00874-5
Return to citation in text: [1] [2] [3] -
Walborsky, H. M.; Baum, M. E. J. Am. Chem. Soc. 1958, 80, 187–192. doi:10.1021/ja01534a047
Return to citation in text: [1] [2] -
Wakselman, C.; Tordeux, M. J. Fluorine Chem. 1982, 21, 99–106. doi:10.1016/s0022-1139(00)81235-x
Return to citation in text: [1] [2] -
Ooi, T.; Tayama, E.; Doda, K.; Takeuchi, M.; Maruoka, K. Synlett 2000, 1500–1502. doi:10.1055/s-2000-7652
Return to citation in text: [1] -
Moyna, G.; Williams, H. J.; Scott, A. I. Synth. Commun. 1996, 26, 2235–2239. doi:10.1080/00397919608003584
Return to citation in text: [1] -
Yadav, V. K.; Kapoor, K. K. Tetrahedron 1995, 51, 8573–8584. doi:10.1016/0040-4020(95)00472-k
Return to citation in text: [1] -
Semmelhack, M. F.; Jeong, N. Tetrahedron Lett. 1990, 31, 605–608. doi:10.1016/s0040-4039(00)94579-9
Return to citation in text: [1] -
Yamazaki, T.; Someya, S. Method for the preparation of fluorine-containing epoxy ester. Japanese patent JP2010208998, Sept 24, 2010.
Our patent was already published which was on the preparation of 2 by using an aqueous solution of NaOCl.
Return to citation in text: [1] -
Yamazaki, T.; Ichige, T.; Kitazume, T. Org. Lett. 2004, 6, 4073–4076. doi:10.1021/ol048229x
Return to citation in text: [1] -
Foucaud, A.; Bakouetila, M. Synthesis 1987, 854–856. doi:10.1055/s-1987-28104
Return to citation in text: [1] -
Yamazaki, T.; Mano, N.; Hikage, R.; Kaneko, T.; Kawasaki-Takasuka, T.; Yamada, S. Tetrahedron 2015, 71, 8059–8066. doi:10.1016/j.tet.2015.08.048
Return to citation in text: [1] [2] [3] -
Our computation (B3LYP/6-311++G** by the Gaussian 09W (rev. D.01)) clarified that the LUMO+1 energy levels of the compounds of 1b, 1h, and 1i (as the methyl esters) were calculated to be 0.053, 0.429, and 0.405 eV, respectively.
Return to citation in text: [1] -
Davidge, H.; Davies, A. G.; Kenyon, J.; Mason, R. F. J. Chem. Soc. 1958, 4569–4573. doi:10.1039/jr9580004569
Our literature search clarified that only one example was previously reported for the reaction of 2a with amine.
Return to citation in text: [1] -
Bordwell, F. G.; Zhang, X.; Alnajjar, M. S. J. Am. Chem. Soc. 1992, 114, 7623–7629. doi:10.1021/ja00046a003
Return to citation in text: [1] -
Taft, R. W.; Bordwell, F. G. Acc. Chem. Res. 1988, 21, 456–463. doi:10.1021/ar00156a005
Return to citation in text: [1] -
Tang, Z.; Yang, Z.-H.; Chen, X.-H.; Cun, L.-F.; Mi, A.-Q.; Jiang, Y.-Z.; Gong, L.-Z. J. Am. Chem. Soc. 2005, 127, 9285–9289. doi:10.1021/ja0510156
The epimerization for the nonfluorinated epoxysuccinate was noticed by the action of NaN3.
Return to citation in text: [1] -
Sugano, Y.; Naruto, S. Chem. Pharm. Bull. 1988, 36, 4619–4621. doi:10.1248/cpb.36.4619
A similar epimerization was suggested for the non-fluorinated a-sulfenylated esters.
Return to citation in text: [1] -
CCDC 2325464 ((2R*,3S*,2’R*)-3ad), 2325462 (anti,syn-8a), and 2325461 (4aa) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service, https://www.ccdc.cam.ac.uk/structures/.
Return to citation in text: [1] [2] -
Zemtsov, A. A.; Levin, V. V.; Dilman, A. D.; Struchkova, M. I.; Belyakov, P. A.; Tartakovsky, V. A. Tetrahedron Lett. 2009, 50, 2998–3000. doi:10.1016/j.tetlet.2009.03.188
Return to citation in text: [1] -
Takeda, A.; Torii, S. Bull. Chem. Soc. Jpn. 1967, 40, 1261–1263. doi:10.1246/bcsj.40.1261
Return to citation in text: [1] -
Ripin, D. H.; Evans, D. A. http://ccc.chem.pitt.edu/wipf/MechOMs/evans_pKa_table.pdf.
Return to citation in text: [1] [2] -
Matthews, W. S.; Bares, J. E.; Bartmess, J. E.; Bordwell, F. G.; Cornforth, F. J.; Drucker, G. E.; Margolin, Z.; McCallum, R. J.; McCollum, G. J.; Vanier, N. R. J. Am. Chem. Soc. 1975, 97, 7006–7014. doi:10.1021/ja00857a010
Return to citation in text: [1] [2] -
Goumont, R.; Magnier, E.; Kizilian, E.; Terrier, F. J. Org. Chem. 2003, 68, 6566–6570. doi:10.1021/jo034244o
Return to citation in text: [1] -
Bordwell, F. G.; Fried, H. E. J. Org. Chem. 1981, 46, 4327–4331. doi:10.1021/jo00335a001
Return to citation in text: [1] -
Olmstead, W. N.; Bordwell, F. G. J. Org. Chem. 1980, 45, 3299–3305. doi:10.1021/jo01304a033
Return to citation in text: [1] -
Alexakis, A.; Jachiet, D.; Normant, J. F. Tetrahedron 1986, 42, 5607–5619. doi:10.1016/s0040-4020(01)88165-5
Return to citation in text: [1]
| 50. | Zemtsov, A. A.; Levin, V. V.; Dilman, A. D.; Struchkova, M. I.; Belyakov, P. A.; Tartakovsky, V. A. Tetrahedron Lett. 2009, 50, 2998–3000. doi:10.1016/j.tetlet.2009.03.188 |
| 51. | Takeda, A.; Torii, S. Bull. Chem. Soc. Jpn. 1967, 40, 1261–1263. doi:10.1246/bcsj.40.1261 |
| 49. | CCDC 2325464 ((2R*,3S*,2’R*)-3ad), 2325462 (anti,syn-8a), and 2325461 (4aa) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service, https://www.ccdc.cam.ac.uk/structures/. |
| 1. | Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393x |
| 2. | Uneyama, K., Ed. Organofluorine Chemistry; Blackwell:: Oxford, UK, 2006. doi:10.1002/9780470988589 |
| 3. | Yamazaki, T.; Taguchi, T.; Ojima, I. Unique Properties of Fluorine and their Relevance to Medicinal Chemistry and Chemical Biology; Introduction: Basic Aspects of Fluorine Substituent Effect in Medicinal Chemistry. In Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I., Ed.; Wiley: West Sussex, UK, 2009; pp 3–46. doi:10.1002/9781444312096.ch1 |
| 4. | Brittain, W. D. G.; Lloyd, C. M.; Cobb, S. L. J. Fluorine Chem. 2020, 239, 109630. doi:10.1016/j.jfluchem.2020.109630 |
| 5. | O'Hagan, D. Chem. – Eur. J. 2020, 26, 7981–7997. doi:10.1002/chem.202000178 |
| 6. | Remete, A. M.; Nonn, M.; Escorihuela, J.; Fustero, S.; Kiss, L. Eur. J. Org. Chem. 2021, 5946–5974. doi:10.1002/ejoc.202101094 |
| 7. | Decaens, J.; Couve-Bonnaire, S.; Charette, A. B.; Poisson, T.; Jubault, P. Chem. – Eur. J. 2021, 27, 2935–2962. doi:10.1002/chem.202003822 |
| 10. | Shinohara, N.; Haga, J.; Yamazaki, T.; Kitazume, T.; Nakamura, S. J. Org. Chem. 1995, 60, 4363–4374. doi:10.1021/jo00119a013 |
| 21. | Yamazaki, T.; Ando, M.; Kitazume, T.; Kubota, T.; Omura, M. Org. Lett. 1999, 1, 905–908. doi:10.1021/ol990821c |
| 40. | Yamazaki, T.; Ichige, T.; Kitazume, T. Org. Lett. 2004, 6, 4073–4076. doi:10.1021/ol048229x |
| 56. | Olmstead, W. N.; Bordwell, F. G. J. Org. Chem. 1980, 45, 3299–3305. doi:10.1021/jo01304a033 |
| 20. | Computation of 1b and its non-fluorinated counterpart as the methyl ester clearly showed their LUMO + 1 energy levels as 0.053 and 0.961 eV, respectively, by employment of Gaussian 09W, Revision D.01 using the B3LYP/6-311++G** level of theory. |
| 41. | Foucaud, A.; Bakouetila, M. Synthesis 1987, 854–856. doi:10.1055/s-1987-28104 |
| 27. | Seebach, D.; Beck, A. K.; Renaud, P. Angew. Chem., Int. Ed. Engl. 1986, 25, 98–99. doi:10.1002/anie.198600981 |
| 28. | Lin, J. T.; Yamazaki, T.; Takeda, M.; Kitazume, T. J. Fluorine Chem. 1989, 44, 113–120. doi:10.1016/s0022-1139(00)84374-2 |
| 29. | von dem Bussche-Hünnefeld, C.; Seebach, D. Chem. Ber. 1992, 125, 1273–1281. doi:10.1002/cber.19921250538 |
| 16. | Yamazaki, T.; Shinohara, N.; Kitazume, T.; Sato, S. J. Fluorine Chem. 1999, 97, 91–96. doi:10.1016/s0022-1139(99)00034-2 |
| 17. | Yamazaki, T.; Taketsugi, M.; Kawasaki-Takasuka, T.; Agou, T.; Kubota, T. Bull. Chem. Soc. Jpn. 2021, 94, 1815–1822. doi:10.1246/bcsj.20210136 |
| 18. | Morigaki, A.; Tanaka, T.; Miyabe, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2013, 11, 586–595. doi:10.1039/c2ob26708j |
| 19. | Zhang, L.-Y.; Zhou, J.-H.; Xu, Y.-H.; Loh, T.-P. Chem. – Asian J. 2015, 10, 844–848. doi:10.1002/asia.201403303 |
| 32. | Lanier, M.; Le Blanc, M.; Pastor, R. Tetrahedron 1996, 52, 14631–14640. doi:10.1016/0040-4020(96)00874-5 |
| 54. | Goumont, R.; Magnier, E.; Kizilian, E.; Terrier, F. J. Org. Chem. 2003, 68, 6566–6570. doi:10.1021/jo034244o |
| 8. | Yamazaki, T.; Haga, J.; Kitazume, T.; Nakamura, S. Chem. Lett. 1991, 2171–2174. doi:10.1246/cl.1991.2171 |
| 9. | Yamazaki, T.; Haga, J.; Kitazume, T. Chem. Lett. 1991, 20, 2175–2178. doi:10.1246/cl.1991.2175 |
| 10. | Shinohara, N.; Haga, J.; Yamazaki, T.; Kitazume, T.; Nakamura, S. J. Org. Chem. 1995, 60, 4363–4374. doi:10.1021/jo00119a013 |
| 11. | Yamazaki, T.; Shinohara, N.; Kitazume, T.; Sato, S. J. Org. Chem. 1995, 60, 8140–8141. doi:10.1021/jo00130a012 |
| 12. | Pan, X.; Liu, Z. Tetrahedron 2014, 70, 4602–4610. doi:10.1016/j.tet.2014.05.049 |
| 13. | Shu, C.; Liu, H.; Slawin, A. M. Z.; Carpenter-Warren, C.; Smith, A. D. Chem. Sci. 2020, 11, 241–247. doi:10.1039/c9sc04303a |
| 14. | Kim, B.-J.; Song, Y.-N.; Lee, S. Y.-M. Chem. Commun. 2021, 57, 11052–11055. doi:10.1039/d1cc04875a |
| 15. | Wu, J.; Young, C. M.; Watts, A. A.; Slawin, A. M. Z.; Boyce, G. R.; Bühl, M.; Smith, A. D. Org. Lett. 2022, 24, 4040–4045. doi:10.1021/acs.orglett.2c01486 |
| 39. |
Yamazaki, T.; Someya, S. Method for the preparation of fluorine-containing epoxy ester. Japanese patent JP2010208998, Sept 24, 2010.
Our patent was already published which was on the preparation of 2 by using an aqueous solution of NaOCl. |
| 55. | Bordwell, F. G.; Fried, H. E. J. Org. Chem. 1981, 46, 4327–4331. doi:10.1021/jo00335a001 |
| 33. | Walborsky, H. M.; Baum, M. E. J. Am. Chem. Soc. 1958, 80, 187–192. doi:10.1021/ja01534a047 |
| 34. | Wakselman, C.; Tordeux, M. J. Fluorine Chem. 1982, 21, 99–106. doi:10.1016/s0022-1139(00)81235-x |
| 29. | von dem Bussche-Hünnefeld, C.; Seebach, D. Chem. Ber. 1992, 125, 1273–1281. doi:10.1002/cber.19921250538 |
| 33. | Walborsky, H. M.; Baum, M. E. J. Am. Chem. Soc. 1958, 80, 187–192. doi:10.1021/ja01534a047 |
| 52. | Ripin, D. H.; Evans, D. A. http://ccc.chem.pitt.edu/wipf/MechOMs/evans_pKa_table.pdf. |
| 31. | Lanier, M.; Haddach, M.; Pastor, R.; Riess, J. G. Tetrahedron Lett. 1993, 34, 2469–2472. doi:10.1016/s0040-4039(00)60443-4 |
| 32. | Lanier, M.; Le Blanc, M.; Pastor, R. Tetrahedron 1996, 52, 14631–14640. doi:10.1016/0040-4020(96)00874-5 |
| 29. | von dem Bussche-Hünnefeld, C.; Seebach, D. Chem. Ber. 1992, 125, 1273–1281. doi:10.1002/cber.19921250538 |
| 53. | Matthews, W. S.; Bares, J. E.; Bartmess, J. E.; Bordwell, F. G.; Cornforth, F. J.; Drucker, G. E.; Margolin, Z.; McCallum, R. J.; McCollum, G. J.; Vanier, N. R. J. Am. Chem. Soc. 1975, 97, 7006–7014. doi:10.1021/ja00857a010 |
| 27. | Seebach, D.; Beck, A. K.; Renaud, P. Angew. Chem., Int. Ed. Engl. 1986, 25, 98–99. doi:10.1002/anie.198600981 |
| 28. | Lin, J. T.; Yamazaki, T.; Takeda, M.; Kitazume, T. J. Fluorine Chem. 1989, 44, 113–120. doi:10.1016/s0022-1139(00)84374-2 |
| 29. | von dem Bussche-Hünnefeld, C.; Seebach, D. Chem. Ber. 1992, 125, 1273–1281. doi:10.1002/cber.19921250538 |
| 30. |
Ishii, A.; Kanai, M.; Kuriyama, S.; Yasumoto, M.; Inomiya, N.; Otsuka, T.; Ueda, H. Preparation of 4,4,4-trifluoro-2,3-epoxybutanoic acid esters. Japanese patent JP2005047870, Feb 24, 2005.
See for preparation of compound 2a by the Bayer-Villiger oxidation of epoxyketones. |
| 52. | Ripin, D. H.; Evans, D. A. http://ccc.chem.pitt.edu/wipf/MechOMs/evans_pKa_table.pdf. |
| 22. | Gleason, J. G.; Hall, R. F.; Perchonock, C. D.; Erhard, K. F.; Frazee, J. S.; Ku, T. W.; Kondrad, K.; McCarthy, M. E.; Mong, S.; Crooke, S. T.; Chi-Rosso, G.; Wasserman, M. A.; Torphy, T. J.; Muccitelli, R. M.; Hay, D. W.; Tucker, S. S.; Vickery-Clark, L. J. Med. Chem. 1987, 30, 959–961. doi:10.1021/jm00389a001 |
| 23. | Righi, G.; Rumboldt, G.; Bonini, C. J. Org. Chem. 1996, 61, 3557–3560. doi:10.1021/jo951441h |
| 24. | Righi, G.; Pescatore, G.; Bonadies, F.; Bonini, C. Tetrahedron 2001, 57, 5649–5656. doi:10.1016/s0040-4020(01)00492-6 |
| 25. | Durán Pachón, L.; Gamez, P.; van Brussel, J. J. M.; Reedijk, J. Tetrahedron Lett. 2003, 44, 6025–6027. doi:10.1016/s0040-4039(03)01480-1 |
| 26. | Rackl, D.; Kais, V.; Lutsker, E.; Reiser, O. Eur. J. Org. Chem. 2017, 2130–2138. doi:10.1002/ejoc.201700014 |
| 35. | Ooi, T.; Tayama, E.; Doda, K.; Takeuchi, M.; Maruoka, K. Synlett 2000, 1500–1502. doi:10.1055/s-2000-7652 |
| 36. | Moyna, G.; Williams, H. J.; Scott, A. I. Synth. Commun. 1996, 26, 2235–2239. doi:10.1080/00397919608003584 |
| 37. | Yadav, V. K.; Kapoor, K. K. Tetrahedron 1995, 51, 8573–8584. doi:10.1016/0040-4020(95)00472-k |
| 38. | Semmelhack, M. F.; Jeong, N. Tetrahedron Lett. 1990, 31, 605–608. doi:10.1016/s0040-4039(00)94579-9 |
| 53. | Matthews, W. S.; Bares, J. E.; Bartmess, J. E.; Bordwell, F. G.; Cornforth, F. J.; Drucker, G. E.; Margolin, Z.; McCallum, R. J.; McCollum, G. J.; Vanier, N. R. J. Am. Chem. Soc. 1975, 97, 7006–7014. doi:10.1021/ja00857a010 |
| 43. | Our computation (B3LYP/6-311++G** by the Gaussian 09W (rev. D.01)) clarified that the LUMO+1 energy levels of the compounds of 1b, 1h, and 1i (as the methyl esters) were calculated to be 0.053, 0.429, and 0.405 eV, respectively. |
| 31. | Lanier, M.; Haddach, M.; Pastor, R.; Riess, J. G. Tetrahedron Lett. 1993, 34, 2469–2472. doi:10.1016/s0040-4039(00)60443-4 |
| 29. | von dem Bussche-Hünnefeld, C.; Seebach, D. Chem. Ber. 1992, 125, 1273–1281. doi:10.1002/cber.19921250538 |
| 42. | Yamazaki, T.; Mano, N.; Hikage, R.; Kaneko, T.; Kawasaki-Takasuka, T.; Yamada, S. Tetrahedron 2015, 71, 8059–8066. doi:10.1016/j.tet.2015.08.048 |
| 29. | von dem Bussche-Hünnefeld, C.; Seebach, D. Chem. Ber. 1992, 125, 1273–1281. doi:10.1002/cber.19921250538 |
| 57. | Alexakis, A.; Jachiet, D.; Normant, J. F. Tetrahedron 1986, 42, 5607–5619. doi:10.1016/s0040-4020(01)88165-5 |
| 23. | Righi, G.; Rumboldt, G.; Bonini, C. J. Org. Chem. 1996, 61, 3557–3560. doi:10.1021/jo951441h |
| 24. | Righi, G.; Pescatore, G.; Bonadies, F.; Bonini, C. Tetrahedron 2001, 57, 5649–5656. doi:10.1016/s0040-4020(01)00492-6 |
| 32. | Lanier, M.; Le Blanc, M.; Pastor, R. Tetrahedron 1996, 52, 14631–14640. doi:10.1016/0040-4020(96)00874-5 |
| 34. | Wakselman, C.; Tordeux, M. J. Fluorine Chem. 1982, 21, 99–106. doi:10.1016/s0022-1139(00)81235-x |
| 47. |
Tang, Z.; Yang, Z.-H.; Chen, X.-H.; Cun, L.-F.; Mi, A.-Q.; Jiang, Y.-Z.; Gong, L.-Z. J. Am. Chem. Soc. 2005, 127, 9285–9289. doi:10.1021/ja0510156
The epimerization for the nonfluorinated epoxysuccinate was noticed by the action of NaN3. |
| 48. |
Sugano, Y.; Naruto, S. Chem. Pharm. Bull. 1988, 36, 4619–4621. doi:10.1248/cpb.36.4619
A similar epimerization was suggested for the non-fluorinated a-sulfenylated esters. |
| 49. | CCDC 2325464 ((2R*,3S*,2’R*)-3ad), 2325462 (anti,syn-8a), and 2325461 (4aa) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service, https://www.ccdc.cam.ac.uk/structures/. |
| 45. | Bordwell, F. G.; Zhang, X.; Alnajjar, M. S. J. Am. Chem. Soc. 1992, 114, 7623–7629. doi:10.1021/ja00046a003 |
| 42. | Yamazaki, T.; Mano, N.; Hikage, R.; Kaneko, T.; Kawasaki-Takasuka, T.; Yamada, S. Tetrahedron 2015, 71, 8059–8066. doi:10.1016/j.tet.2015.08.048 |
| 46. | Taft, R. W.; Bordwell, F. G. Acc. Chem. Res. 1988, 21, 456–463. doi:10.1021/ar00156a005 |
| 44. |
Davidge, H.; Davies, A. G.; Kenyon, J.; Mason, R. F. J. Chem. Soc. 1958, 4569–4573. doi:10.1039/jr9580004569
Our literature search clarified that only one example was previously reported for the reaction of 2a with amine. |
| 29. | von dem Bussche-Hünnefeld, C.; Seebach, D. Chem. Ber. 1992, 125, 1273–1281. doi:10.1002/cber.19921250538 |
| 25. | Durán Pachón, L.; Gamez, P.; van Brussel, J. J. M.; Reedijk, J. Tetrahedron Lett. 2003, 44, 6025–6027. doi:10.1016/s0040-4039(03)01480-1 |
| 26. | Rackl, D.; Kais, V.; Lutsker, E.; Reiser, O. Eur. J. Org. Chem. 2017, 2130–2138. doi:10.1002/ejoc.201700014 |
| 42. | Yamazaki, T.; Mano, N.; Hikage, R.; Kaneko, T.; Kawasaki-Takasuka, T.; Yamada, S. Tetrahedron 2015, 71, 8059–8066. doi:10.1016/j.tet.2015.08.048 |
© 2024 Miyashita et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.