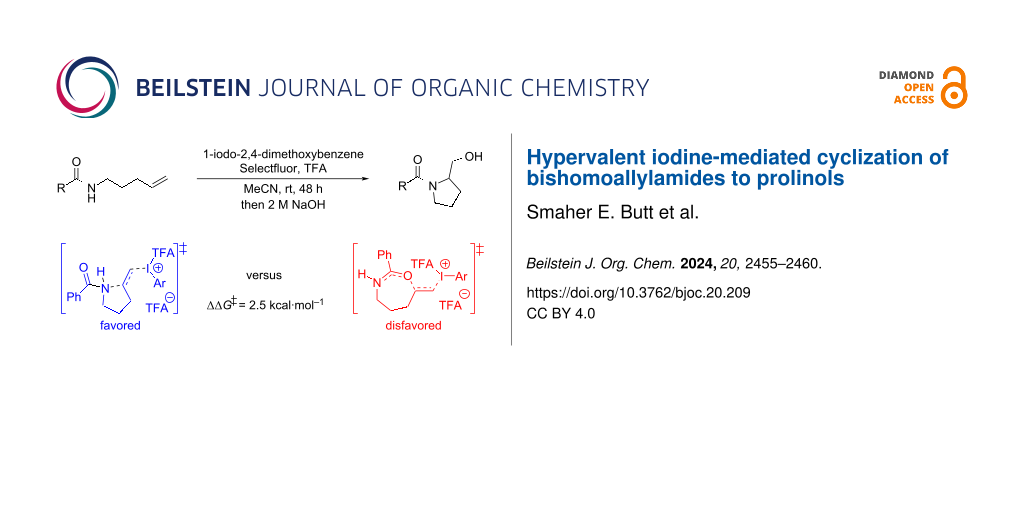
Abstract
A change in mechanism was observed in the hypervalent iodine-mediated cyclization of N-alkenylamides when the carbon chain between the alkene and the amide increased from two to three atoms. In the latter case, cyclization at the amide nitrogen to form the pyrrolidine ring was favored over cyclization at the amide oxygen. A DFT study was undertaken to rationalize the change in mechanism of this cyclization process. In addition, reaction conditions were developed, and the scope of this cyclization studied.

Graphical Abstract
Introduction
Proline is one of the 20 DNA-encoded proteinogenic amino acids that are essential to life [1,2]. In addition, the pyrrolidine core is present in many organocatalysts [3-5], natural products (e.g., the potent α-glucosidase inhibitor (−)-codonopsinol B) [6,7], and pharmaceutical drug molecules such as saxagliptin and ramipril (Figure 1) [8]. Accordingly, the development of methods to access substituted prolines and pyrrolidines is an important area of study as this ring system is prevalent in many useful molecules. Typical literature procedures include multistep derivations of proline itself, e.g., the destruction of the stereocenter and then its reinstallation by an enantioselective conjugate addition [9]. Other methods include the enantioselective conjugate addition to α,β-unsaturated pyroglutamic acid derivatives followed by deoxygenation [10], and the enantioselective organocatalytic reaction between 2-acylaminomalonates and α,β-unsaturated aldehydes [11,12].
![[1860-5397-20-209-1]](/bjoc/content/figures/1860-5397-20-209-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Functional molecules containing a substituted pyrrolidine core.
Figure 1: Functional molecules containing a substituted pyrrolidine core.
The development of new synthetic methods using hypervalent iodine reagents has become increasingly popular in recent years probably due to their useful reactivity, ease of handling, and low toxicity [13]. In particular, hypervalent iodine compounds have been shown to be effective reagents and catalysts for a range of cyclization reactions [14]. In 2015, we reported the iodoarene-catalyzed cyclization of N-allylamides 1 and N-homoallylamides 2 to 2-oxazolines 4 and dihydrooxazines 5, respectively (Scheme 1A) [15]. We also reported that an N-bishomoallylamide 3 (n = 3) was cyclized under the reaction conditions, but in just 30% yield. It turned out that the product of this reaction was the five-membered prolinol 7a rather than the initially assigned isomeric seven-membered tetrahydrooxazepine 6 [16]. Subsequently, we set out to understand the O- versus N-chemoselectivity by DFT modelling, and to develop an effective synthetic protocol for the preparation of prolinols 7 in high yield (Scheme 1B). Notably, we are unaware of any reported method to achieve this specific transformation in the literature. Although, Tellitu and co-workers have reported a related preparation of indoline derivatives mediated by bis(trifluoroacetoxy)iodobenzene (PIFA) [17].
![[1860-5397-20-209-i1]](/bjoc/content/inline/1860-5397-20-209-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: A) Our previous report on N-alkenylamide cyclizations. B) An overview of the present work.
Scheme 1: A) Our previous report on N-alkenylamide cyclizations. B) An overview of the present work.
Results and Discussion
In 2019, we reported our DFT study on the cyclization of N-allylbenzamide (1a) to the 2-oxazoline 4a, i.e., where n = 1 and Ar = Ph [18]. This work indicated that the alkene is activated by the iodine(III) species and that this triggers cyclization. Intrigued by the change in mechanism from O- to N-cyclization onto the alkene when n = 3, we modelled this reaction using DFT calculations (Scheme 2). Similarly, we concluded that the present reaction commences with activation of the olefin in 3a by the hypervalent iodine species 8, which is generated under the reaction conditions. The activation occurs via an associative pathway where one of the TFA ligands dissociates from 8 upon approaching the substrate and forms the intermediate 9. The calculated ΔG‡ value is quite high here, which could explain the low yield obtained after 16 hours. However, for the cyclization of N-allylbenzamide (1a), we found that the transition state was stabilized by 4.1 kcal·mol−1 by an extra molecule of trifluoroacetic acid. A similar stabilizing interaction was not identified in this case with 3a, despite significant effort, but it cannot be ruled out. The cyclization of 9 was shown to be possible by attack of the amide at both the oxygen and the nitrogen, however, the ΔG‡ value for the latter was lower by 2.5 kcal·mol−1. This demonstrates a clear kinetic preference for formation of the five-membered ring over the seven-membered one [19]. Subsequent deprotonation of 11 leads to tertiary amide 12.
![[1860-5397-20-209-i2]](/bjoc/content/inline/1860-5397-20-209-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Calculated mechanism for the cyclization of amide 3a optimized at the B3LYP/ 6-31+G(d,p) level of theory with SDDall used for iodine. Solvent effects were considered using the CPCM model. All ΔG values are in kcal·mol−1. Optimized structures are shown for the cyclization transition states (hydrogen atoms are omitted for clarity and bond lengths are given in Å).
Scheme 2: Calculated mechanism for the cyclization of amide 3a optimized at the B3LYP/ 6-31+G(d,p) level of t...
Upon cyclization, the iodane moiety in 12 is eliminated by an intramolecular attack by the amide nitrogen to form the aziridinium 13. Finally, ring-opening by SN2 attack of trifluoroacetate leads to the final product 14 [20]. In this case, the kinetic pyrrolidine product is obtained due to the electron-withdrawing benzoyl group on the nitrogen atom preventing equilibration to the thermodynamic piperidine product [21]. Basic workup hydrolyzes the trifluoroacetoxy ester in 14 to alcohol 7a. Consideration of the literature NMR data for the three possible isomeric products (i.e., pyrrolidine, piperidine, and tetrahydrooxazepine) as well as DEPT, HSQC, and HMBC data for 7a support the assignment of the pyrrolidine structure.
The next stage of the project was to improve the yield of the reaction. We initiated our study by using our initially developed conditions using 20 mol % 2-iodoanisole and found that the reaction outcome led to variable yields of product in the range of 10–25%. Increasing the quantity of 2-iodoanisole to 150 mol % provided a reproducible 30% yield of 7a (Table 1, entry 1). We then varied the iodoarene to see the impact on the reaction outcome. Using iodobenzene, 2-iodobiphenyl, and 3-iodotoluene provided slight improvements in yield (Table 1, entries 2–4). The more electron-rich 2-iodo-1,3-dimethoxybenzene led to a further increase in yield to 44% (Table 1, entry 5). 1,2-Diiodobenzene has been reported to be a superior precatalyst in intermolecular C–H aminations of arenes but only provided 40% yield in the present case [22]. 1-Iodonaphthalene led to an increase in yield to 49% (Table 1, entry 7). 1-Iodo-2,4-dimethoxybenzene afforded the highest yield of all with 59% of tertiary amide being isolated (Table 1, entry 8). Leaving the reaction to stir for an extended period led to a further increase in yield to 68% (Table 1, entry 9). Using the even more electron-rich 2-iodo-1,3,5-trimethoxybenzene only gave 45% yield (Table 1, entry 10). Our previous studies have shown that the oxidized form of 2-iodo-1,3,5-trimethoxybenzene is unstable in solution and decomposes destructively [18]. Iodoethane was also shown to be an effective reagent furnishing the product in up to 56% upon heating to 40 °C (Table 1, entries 11 and 12). It was envisaged that oxidation of iodoethane led to formation of oxidized forms of iodide by C–I-bond cleavage, therefore tetrabutylammonium iodide was utilized to see if the result could be replicated and it was (Table 1, entry 13). Finally, the reaction was shown to occur using PIFA (bistrifluoroacetoxyiodobenzene), which is envisaged to be produced in the reaction mixture when using iodobenzene as reagent, and a similar yield was obtained (Table 1, entry 14 vs entry 2). The cyclization of amide 3a did not occur in the absence of an iodine source. Other oxidants, solvents, and acids were screened but superior conditions were not discovered.
Table 1: Optimization of cyclization reaction.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-209-i5.svg?max-width=637&scale=1.0)
|
||
| Entry | Deviation from conditions | Yield [%]a |
| 1 | none | 30 |
| 2 | iodobenzene | 37 |
| 3 | 2-iodobiphenyl | 37 |
| 4 | 3-iodotoluene | 39 |
| 5 | 2-iodo-1,3-dimethoxybenzene | 44 |
| 6 | 1,2-diiodobenzene | 40 |
| 7 | 1-iodonaphthalene | 49 |
| 8 | 1-iodo-2,4-dimethoxybenzene | 59 |
| 9 | 1-iodo-2,4-dimethoxybenzene, 48 h | 68 |
| 10 | 2-iodo-1,3,5-trimethoxybenzene | 45 |
| 11 | iodoethane | 38 |
| 12 | iodoethane, 40 °C | 56 |
| 13 | Bu4NI | 40 |
| 14 | PIFA, no selectfluor, no TFA | 40 |
aThe yields are for isolated compounds. TFA = trifluoroacetic acid. PIFA = bis(trifluoroacetoxy)iodobenzene.
With the optimized conditions in hand, the scope of the cyclization was investigated (Scheme 3). We examined the cyclization of para-substituted benzamides and chloro- (7b), bromo- (7c), phenyl- (7d), and methoxy- (7e) derivatives were all isolated in moderate yields. The ortho-substituted derivatives 7f, 7g, and 7h were also successfully prepared. Alkylamides were found to be ambiguous substrates as the acetamide 7i and pivalamide 7j were formed in trace quantities whereas the benzyl derivative 7k was isolable. The enamide 7l was not observed. Intriguingly, cyclopropyl and cyclohexyl derivatives 7m and 7n, respectively, were formed and isolated in moderate yields. Furyl derivative 7o was isolated in 63% yield. Installing a geminal dimethyl group on the alkyl linker was anticipated to lead to an improvement in cyclization, however, very low conversion was observed and the product 7p was isolated in 10% yield.
![[1860-5397-20-209-i3]](/bjoc/content/inline/1860-5397-20-209-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
We then investigated the cyclization of cis-disubstituted alkene 3q and were delighted to observe that only one diastereomer of 7q was formed (Scheme 4). This result is in accordance with the calculated mechanism. The more electron-rich trisubstituted alkene 3r reacted directly with Selectfluor leading to a tertiary carbocation which was trapped by acetonitrile in a Ritter-type process to generate bisamide 15 [23]. As would be expected, one regioisomer and a 1:1 mixture of diastereomers was formed.
![[1860-5397-20-209-i4]](/bjoc/content/inline/1860-5397-20-209-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reactions of di- and trisubstituted alkene substrates.
Scheme 4: Reactions of di- and trisubstituted alkene substrates.
Conclusion
We have demonstrated that a change in mechanism occurs in the cyclization of N-alkenylamides when increasing the chain length between the amide and the alkene. When there are one or two carbon atoms separating the functional groups, cyclization at the amide oxygen occurs to generate five- and six-membered rings, respectively. However, when there is a three-carbon atom link, the corresponding seven-membered ring is not formed. Instead, cyclization at nitrogen occurs to generate a five-membered ring. We have performed DFT calculations to support the proposed change in mechanism and developed superior reaction conditions to effect this transformation. Finally, we have explored the substrate scope of this cyclization.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data, copies of NMR spectra, cartesian coordinates and energies of calculated structures. | ||
| Format: PDF | Size: 4.6 MB | Download |
Acknowledgements
We thank Professor Mark Heron for his useful discussions and Dr Neil McLay for his NMR expertise. We also acknowledge Joshua Gauntlett for assistance with synthesis of some intermediates.
Funding
This study was supported by an EPSRC Doctoral Training Partnership (grant number EP/T51813X/1) held at the University of Huddersfield (K.K.), a University of Huddersfield funded Ph.D. studentship (S.B.), and by the Université de Pau et des Pays de l’Adour for access to their computer cluster.
Data Availability Statement
The data that supports the findings of this study is available from the corresponding author upon reasonable request.
References
-
Patriarca, E. J.; Cermola, F.; D’Aniello, C.; Fico, A.; Guardiola, O.; De Cesare, D.; Minchiotti, G. Front. Cell Dev. Biol. 2021, 9, 728576. doi:10.3389/fcell.2021.728576
Return to citation in text: [1] -
Then, A.; Mácha, K.; Ibrahim, B.; Schuster, S. Sci. Rep. 2020, 10, 15321. doi:10.1038/s41598-020-72174-5
Return to citation in text: [1] -
Vachan, B. S.; Karuppasamy, M.; Vinoth, P.; Vivek Kumar, S.; Perumal, S.; Sridharan, V.; Menéndez, J. C. Adv. Synth. Catal. 2020, 362, 87–110. doi:10.1002/adsc.201900558
Return to citation in text: [1] -
Cobb, A. J. A.; Shaw, D. M.; Longbottom, D. A.; Gold, J. B.; Ley, S. V. Org. Biomol. Chem. 2005, 3, 84–96. doi:10.1039/b414742a
Return to citation in text: [1] -
Okino, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2003, 125, 12672–12673. doi:10.1021/ja036972z
Return to citation in text: [1] -
Mauger, A. B. J. Nat. Prod. 1996, 59, 1205–1211. doi:10.1021/np9603479
Return to citation in text: [1] -
Wakana, D.; Kawahara, N.; Goda, Y. Chem. Pharm. Bull. 2013, 61, 1315–1317. doi:10.1248/cpb.c13-00516
Return to citation in text: [1] -
Lenci, E.; Trabocchi, A. Symmetry 2019, 11, 558. doi:10.3390/sym11040558
Return to citation in text: [1] -
Huy, P.; Neudörfl, J.-M.; Schmalz, H.-G. Org. Lett. 2011, 13, 216–219. doi:10.1021/ol102613z
Return to citation in text: [1] -
Perni, R. B.; Farmer, L. J.; Cottrell, K. M.; Court, J. J.; Courtney, L. F.; Deininger, D. D.; Gates, C. A.; Harbeson, S. L.; Kim, J. L.; Lin, C.; Lin, K.; Luong, Y.-P.; Maxwell, J. P.; Murcko, M. A.; Pitlik, J.; Rao, B. G.; Schairer, W. C.; Tung, R. D.; Van Drie, J. H.; Wilson, K.; Thomson, J. A. Bioorg. Med. Chem. Lett. 2004, 14, 1939–1942. doi:10.1016/j.bmcl.2004.01.078
Return to citation in text: [1] -
Rios, R.; Ibrahem, I.; Vesely, J.; Sundén, H.; Córdova, A. Tetrahedron Lett. 2007, 48, 8695–8699. doi:10.1016/j.tetlet.2007.10.028
Return to citation in text: [1] -
Chung, J. Y. L.; Wasicak, J. T.; Arnold, W. A.; May, C. S.; Nadzan, A. M.; Holladay, M. W. J. Org. Chem. 1990, 55, 270–275. doi:10.1021/jo00288a045
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Sihag, M.; Soni, R.; Rani, N.; Kinger, M.; Kumar Aneja, D. Org. Prep. Proced. Int. 2023, 55, 1–62. doi:10.1080/00304948.2022.2113964
Return to citation in text: [1] -
Alhalib, A.; Kamouka, S.; Moran, W. J. Org. Lett. 2015, 17, 1453–1456. doi:10.1021/acs.orglett.5b00333
Return to citation in text: [1] -
Verbraeken, B.; Hullaert, J.; van Guyse, J.; Van Hecke, K.; Winne, J.; Hoogenboom, R. J. Am. Chem. Soc. 2018, 140, 17404–17408. doi:10.1021/jacs.8b10918
Return to citation in text: [1] -
Correa, A.; Tellitu, I.; Domínguez, E.; SanMartin, R. J. Org. Chem. 2006, 71, 8316–8319. doi:10.1021/jo061486q
Return to citation in text: [1] -
Butt, S. E.; Das, M.; Sotiropoulos, J.-M.; Moran, W. J. J. Org. Chem. 2019, 84, 15605–15613. doi:10.1021/acs.joc.9b02623
Return to citation in text: [1] [2] -
Casadei, M. A.; Galli, C.; Mandolini, L. J. Am. Chem. Soc. 1984, 106, 1051–1056. doi:10.1021/ja00316a039
Return to citation in text: [1] -
O'Brien, P.; Towers, T. D. J. Org. Chem. 2002, 67, 304–307. doi:10.1021/jo010824e
Return to citation in text: [1] -
Hayashi, K.; Kujime, E.; Katayama, H.; Sano, S.; Shiro, M.; Nagao, Y. Chem. Pharm. Bull. 2009, 57, 1142–1146. doi:10.1248/cpb.57.1142
Return to citation in text: [1] -
Lucchetti, N.; Scalone, M.; Fantasia, S.; Muñiz, K. Adv. Synth. Catal. 2016, 358, 2093–2099. doi:10.1002/adsc.201600191
Return to citation in text: [1] -
Stavber, S.; Pecan, T. S.; Papež, M.; Zupan, M. Chem. Commun. 1996, 2247–2248. doi:10.1039/cc9960002247
Return to citation in text: [1]
| 23. | Stavber, S.; Pecan, T. S.; Papež, M.; Zupan, M. Chem. Commun. 1996, 2247–2248. doi:10.1039/cc9960002247 |
| 22. | Lucchetti, N.; Scalone, M.; Fantasia, S.; Muñiz, K. Adv. Synth. Catal. 2016, 358, 2093–2099. doi:10.1002/adsc.201600191 |
| 18. | Butt, S. E.; Das, M.; Sotiropoulos, J.-M.; Moran, W. J. J. Org. Chem. 2019, 84, 15605–15613. doi:10.1021/acs.joc.9b02623 |
| 1. | Patriarca, E. J.; Cermola, F.; D’Aniello, C.; Fico, A.; Guardiola, O.; De Cesare, D.; Minchiotti, G. Front. Cell Dev. Biol. 2021, 9, 728576. doi:10.3389/fcell.2021.728576 |
| 2. | Then, A.; Mácha, K.; Ibrahim, B.; Schuster, S. Sci. Rep. 2020, 10, 15321. doi:10.1038/s41598-020-72174-5 |
| 9. | Huy, P.; Neudörfl, J.-M.; Schmalz, H.-G. Org. Lett. 2011, 13, 216–219. doi:10.1021/ol102613z |
| 20. | O'Brien, P.; Towers, T. D. J. Org. Chem. 2002, 67, 304–307. doi:10.1021/jo010824e |
| 21. | Hayashi, K.; Kujime, E.; Katayama, H.; Sano, S.; Shiro, M.; Nagao, Y. Chem. Pharm. Bull. 2009, 57, 1142–1146. doi:10.1248/cpb.57.1142 |
| 6. | Mauger, A. B. J. Nat. Prod. 1996, 59, 1205–1211. doi:10.1021/np9603479 |
| 7. | Wakana, D.; Kawahara, N.; Goda, Y. Chem. Pharm. Bull. 2013, 61, 1315–1317. doi:10.1248/cpb.c13-00516 |
| 18. | Butt, S. E.; Das, M.; Sotiropoulos, J.-M.; Moran, W. J. J. Org. Chem. 2019, 84, 15605–15613. doi:10.1021/acs.joc.9b02623 |
| 3. | Vachan, B. S.; Karuppasamy, M.; Vinoth, P.; Vivek Kumar, S.; Perumal, S.; Sridharan, V.; Menéndez, J. C. Adv. Synth. Catal. 2020, 362, 87–110. doi:10.1002/adsc.201900558 |
| 4. | Cobb, A. J. A.; Shaw, D. M.; Longbottom, D. A.; Gold, J. B.; Ley, S. V. Org. Biomol. Chem. 2005, 3, 84–96. doi:10.1039/b414742a |
| 5. | Okino, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2003, 125, 12672–12673. doi:10.1021/ja036972z |
| 19. | Casadei, M. A.; Galli, C.; Mandolini, L. J. Am. Chem. Soc. 1984, 106, 1051–1056. doi:10.1021/ja00316a039 |
| 14. | Sihag, M.; Soni, R.; Rani, N.; Kinger, M.; Kumar Aneja, D. Org. Prep. Proced. Int. 2023, 55, 1–62. doi:10.1080/00304948.2022.2113964 |
| 16. | Verbraeken, B.; Hullaert, J.; van Guyse, J.; Van Hecke, K.; Winne, J.; Hoogenboom, R. J. Am. Chem. Soc. 2018, 140, 17404–17408. doi:10.1021/jacs.8b10918 |
| 13. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 17. | Correa, A.; Tellitu, I.; Domínguez, E.; SanMartin, R. J. Org. Chem. 2006, 71, 8316–8319. doi:10.1021/jo061486q |
| 11. | Rios, R.; Ibrahem, I.; Vesely, J.; Sundén, H.; Córdova, A. Tetrahedron Lett. 2007, 48, 8695–8699. doi:10.1016/j.tetlet.2007.10.028 |
| 12. | Chung, J. Y. L.; Wasicak, J. T.; Arnold, W. A.; May, C. S.; Nadzan, A. M.; Holladay, M. W. J. Org. Chem. 1990, 55, 270–275. doi:10.1021/jo00288a045 |
| 10. | Perni, R. B.; Farmer, L. J.; Cottrell, K. M.; Court, J. J.; Courtney, L. F.; Deininger, D. D.; Gates, C. A.; Harbeson, S. L.; Kim, J. L.; Lin, C.; Lin, K.; Luong, Y.-P.; Maxwell, J. P.; Murcko, M. A.; Pitlik, J.; Rao, B. G.; Schairer, W. C.; Tung, R. D.; Van Drie, J. H.; Wilson, K.; Thomson, J. A. Bioorg. Med. Chem. Lett. 2004, 14, 1939–1942. doi:10.1016/j.bmcl.2004.01.078 |
| 15. | Alhalib, A.; Kamouka, S.; Moran, W. J. Org. Lett. 2015, 17, 1453–1456. doi:10.1021/acs.orglett.5b00333 |
© 2024 Butt et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.