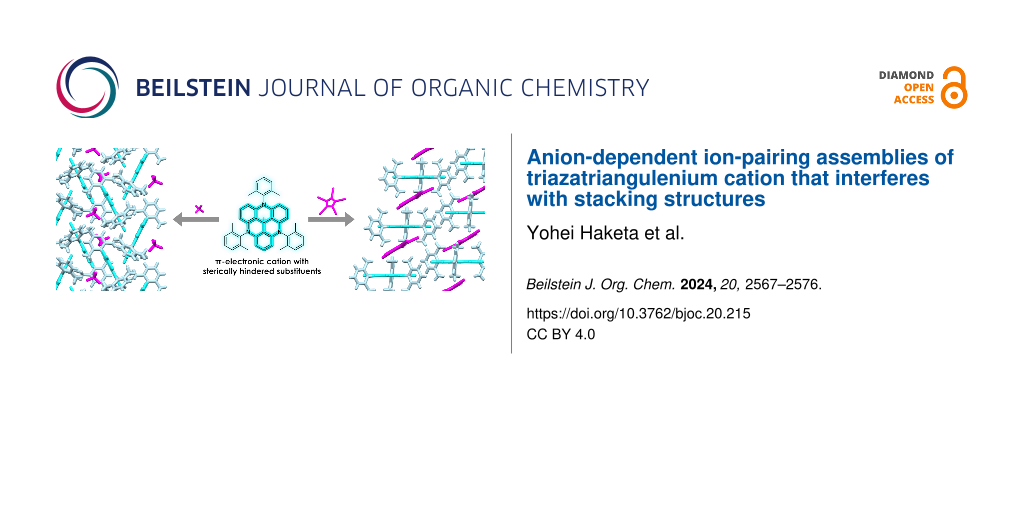
Abstract
Ion pairs of N-(2,6-dimethylphenyl)-substituted triazatriangulenium (TATA+) cation with various counteranions were synthesized to investigate the interactions for the bulky cation. Single-crystal X-ray analysis of the TATA+ ion pairs revealed solid-state ion-pairing assemblies without stacking at the cationic π-planes. The TATA+ cation showed counteranion-dependent assembly structures, with smaller counteranions located at the top of TATA+ and bulkier counteranions displaced from the TATA+ plane to interact with the surrounding TATA+.

Graphical Abstract
Introduction
Triangulenium cations [1,2] have been widely investigated in the past decades for application as fluorescent dyes [3-5], chirality inducers [6-8], acceptors for photoinduced electron transfer (PET) [9,10], and components of supramolecular assemblies [11-14]. Owing to the stable π-electronic systems showing unique electronic and electrooptical properties, various derivatives have been synthesized via peripheral modifications of the triangulenium cations. In particular, various functional units can be introduced at the N-positions of azatriangulenium cations. For example, N-alkyl-substituted triazatriangulenium (TATA+) cations, used as visible light fluorescent dyes, have been synthesized via a nucleophilic aromatic substitution (SNAr) reaction with primary alkylamines (e.g., 1a+; Figure 1) [15,16]. The highly planar geometry of the TATA+ core unit induces π–π stacking structures in single-crystal and film states, as seen in the BF4– ion pairs of N-alkyl-substituted TATA+ cations.
![[1860-5397-20-215-1]](/bjoc/content/figures/1860-5397-20-215-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Triazatriangulenium cations 1a+ and 1b+.
Figure 1: Triazatriangulenium cations 1a+ and 1b+.
The combination of TATA+ cations with counteranions afforded various ion pairs, resulting in characteristic assembled structures [17]. The ion pair comprising N-propyl-substituted 1a+ and pentacyanocyclopentadienide (PCCp−) [18,19] formed a charge-by-charge assembled structure, with alternate stacking of TATA+ and PCCp− through iπ–iπ interactions in the crystal state [20,21]. Charge-by-charge stacking assemblies of 1a+ and porphyrin-based π-electronic anions showed the crystal-state PET between proximally located anions and cations [21]. Moreover, ion-pairing assemblies of 1a+ and the π-electronic receptor–anion complexes as pseudo-π-electronic anions exhibited electric conductive properties [22-25]. The structures and electronic states of ions directly affect the arrangement of constituent ions in the assemblies and resulting properties. Thus, the design and synthesis of ion pairs are important for preparing functional ion-pairing assemblies.
In contrast to N-alkyl-substituted TATA+ cations, only a few N-aryl-substituted TATA+ cations have been reported to date [26-29]. This is probably because the introduction of N-aryl units during the synthesis is rendered difficult owing to steric hindrances. For example, N-phenyl-substituted TATA+ cation 1b+ (Figure 1) [26] exhibited slightly red-shifted absorption and emission, with a higher quantum efficiency compared to those of N-alkyl-substituted TATA+ cations. Moreover, the N-aryl-substituted TATA+ cations have been used as the building units of porous organic polymers for capturing CO2 [27]. Cations with characteristic electron-deficient planar triangular geometries were used as scaffolds for various supramolecular cage structures [28,29]. Although modifications of the N-aryl units as sterically hindered substituents significantly affect the assembling behaviors, their solid-state packing structures are largely unknown. In this study, ion pairs (salts) of an N-aryl-substituted TATA+ as a bulky cation with various counteranions were prepared for investigating the ion-pairing assemblies.
Results and Discussion
The N-(2,6-dimethylphenyl)-substituted triazatriangulenium cation 2+ was synthesized as a BF4− ion pair 2+-BF4− in 19% yield using a modified synthetic procedure for 2+-Cl− [30] (Figure 2). It should be noticed that the SNAr reaction for ring closure with aniline requires a base as reported by Laursen et al. [26], whereas 2,6-dimethylaniline in this study was reacted without the use of a base. This can be explained by the high nucleophilicity of 2,6-dimethylaniline, though it is sterically hindered. As counteranions affect ion-pairing assemblies, 2+-BF4− was further treated with NaPF6, LiB(C6F5)4, and NaPCCp for the ion-pair metathesis to afford ion pairs 2+-X− (X− = PF6−, B(C6F5)4−, and PCCp−) in 44–68% yields. The obtained ion pairs were characterized using 1H, 13C, and 19F nuclear magnetic resonance (NMR) and matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI–TOF MS). The synthesized ion pairs showed similar electronic properties in solution state. In CH3CN, 2+-BF4− exhibited UV–vis absorptions at 273, 350, and 524 nm and fluorescence emission at 607 nm upon excitation at 524 nm. The absorption band at 524 nm was mainly derived from HOMO−1/HOMO-to-LUMO transition (HOMO: highest occupied molecular orbital, LUMO: lowest unoccupied molecular orbital), as revealed by the time-dependent density functional theory (TDDFT) calculation of the optimized structure of 2+ at the B3LYP/6-31+G(d,p) level of theory (Figures S22–25 in Supporting Information File 1) [31]. The absorption band at 524 nm was blue-shifted by 4 nm compared to that of N-phenyl-substituted 1b+-BF4− [15]. This can be ascribed to the lesser conjugation of the core TATA+ unit with the introduced 2,6-dimethylphenyl moieties owing to the more orthogonal arrangement in comparison with the phenyl moieties in 1b+.
![[1860-5397-20-215-2]](/bjoc/content/figures/1860-5397-20-215-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthesis of triazatriangulenium ion pairs 2+-X− (X− = BF4−, PF6−, B(C6F5)4−, and PCCp−).
Figure 2: Synthesis of triazatriangulenium ion pairs 2+-X− (X− = BF4−, PF6−, B(C6F5)4−, and PCCp−).
Solid-state ion-pairing assemblies of the ion pairs 2+-X− (X− = Cl−, BF4−, PF6−, B(C6F5)4−, and PCCp−) were investigated by X-ray analysis of the orange rod-shaped single crystals, which were obtained by vapour diffusion using CHCl3/n-heptane for 2+-Cl−, 2+-BF4−, and 2+-PCCp−, CH2Cl2/n-heptane with small amounts of MeOH and toluene for 2+-PF6−, and toluene/n-hexane for 2+-B(C6F5)4− (Figure 3 and Figures S11–15 in Supporting Information File 1) [32]. In the single crystals, 2+ showed highly planar core structures with mean-plane deviations of 0.014–0.071 Å for the 22 core atoms. 2,6-Dimethylphenyl units were orthogonally arranged with dihedral angles of 81.0°–89.9°, which are similar to those of N-phenyl-substituted 1b+ [26]. In the packing structure, the 2+-Cl− units were aligned in a columnar structure with an interplane distance of 9.26 Å for the mean planes of the 22 core atoms. Such a long interplane distance is mainly derived from the bulky 2,6-dimethylphenyl units that hinder the close stacking of the TATA+ core units, as observed in N-alkyl-substituted 1a+ [11,12,15,16,22] and N-phenyl-substituted 1b+ [26]. Columnar structures with alternately arranged 2+ and counteranions were also formed for 2+-B(C6F5)4− and 2+-PCCp− with interplane distances of 15.7 and 8.46 Å, respectively. On the other hand, in the packing structures of ion pairs 2+-BF4– and 2+-PF6–, cation 2+ formed herringbone structures, which were not observed in the other TATA+ cations that form π–π stacking structures [11,12,15,16,22]. The angles between the two staggered TATA+ planes were 53.7° and 51.4° for 2+-BF4− and 2+-PF6−, respectively. The orthogonal arrangement of TATA+ and the o-CH3-substituted aryl units was suitable for forming intermolecular C–H···π interactions between the proximally located CH3 and TATA+ units, as obtained by the Hirshfeld surface analysis (Figure 4) [33-36].
![[1860-5397-20-215-3]](/bjoc/content/figures/1860-5397-20-215-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Single-crystal X-ray structures of (a) 2+-Cl−, (b) 2+-BF4−, (c) 2+-PF6−, (d) 2+-B(C6F5)4−, and (e) 2+-PCCp− as (i) packing structures and (ii) enlarged views for the columnar structures. In (i), cation and anion are represented in cyan and magenta colors, respectively. In (ii), brown, pink, yellow, blue, yellow green, orange, and green (spherical) refer to carbon, hydrogen, boron, nitrogen, fluorine, phosphorus, and chlorine, respectively.
Figure 3: Single-crystal X-ray structures of (a) 2+-Cl−, (b) 2+-BF4−, (c) 2+-PF6−, (d) 2+-B(C6F5)4−, and (e) 2...
![[1860-5397-20-215-4]](/bjoc/content/figures/1860-5397-20-215-4.png?scale=2.2&max-width=1024&background=FFFFFF)
Figure 4: Hirshfeld surface analysis mapped with dnorm of closely contacted two 2+ in (a) 2+-BF4− and (b) 2+-PF6−. Atom color code: brown, pink, and blue refer to carbon, hydrogen, and nitrogen, respectively.
Figure 4: Hirshfeld surface analysis mapped with dnorm of closely contacted two 2+ in (a) 2+-BF4− and (b) 2+-...
The arrangement of the ion pairs depended on the counteranions, which formed different interactions. Smaller counteranions were located above the electron-deficient TATA+ core through the interactions of the methyl units. In fact, in 2+-Cl−, Cl− was located 3.43 Å above the TATA+ core and was interacted with the surrounding CH3 units, with C(–H)···Cl− distances of 3.74, 3.85, and 3.96 Å (Figure 3a, Figure 5a and Figure S17 in Supporting Information File 1). As a crystallization solvent, three CHCl3 molecules were located between the 2,6-dimethylphenyl units by forming hydrogen bonding with Cl− with C(–H)···Cl− distances of 3.36, 3.36, and 3.37 Å. A similar arrangement of the counteranions was observed for 2+-BF4− and 2+-PF6−, with the BF4− and PF6− anions located above the TATA+ plane (Figure 3b,c and Figure 5b,c). Three of the fluorine atoms of BF4− and PF6− were pointed toward the TATA+ plane. The offset angles for Cl−, BF4−, and PF6− to the TATA+ central carbon were 87.7°, 78.1°, and 84.3°, respectively. The distances between fluorine and the TATA+ mean plane in 2+-BF4− were 2.94, 3.09, and 3.14 Å, while those in 2+-PF6− were 2.90, 3.17, and 3.20 Å. Hirshfeld surface analysis of the proximally located ion pairs mapped with dnorm clearly indicated close contacts between the anions and the TATA+ plane (Figure 5b,c and Figures S18 and S19 in Supporting Information File 1).
![[1860-5397-20-215-5]](/bjoc/content/figures/1860-5397-20-215-5.png?scale=2.2&max-width=1024&background=FFFFFF)
Figure 5: Hirshfeld surface analysis mapped with dnorm of closely contacted ion pairs: (a) 2+-Cl−, (b) 2+-BF4−, and (c) 2+-PF6−. Atom color code: yellow, green, orange, and green (spherical) refer to boron, fluorine, phosphorus, and chlorine, respectively.
Figure 5: Hirshfeld surface analysis mapped with dnorm of closely contacted ion pairs: (a) 2+-Cl−, (b) 2+-BF4−...
Theoretically performed energy decomposition analysis (EDA) based on FMO2-MP2 using the basis set of NOSeC-V-DZP with MCP [37-40] for the anion and closely located 2+, as suggested by Hirshfeld surface analysis, revealed that short counteranion–TATA+ plane distances resulted mainly from electrostatic interaction. Closely contacted ion pairs for 2+-Cl−, 2+-BF4−, and 2+-PF6− showed total energies (Etot) of −98.1, −102.2, and −101.9 kcal/mol, respectively, with the main contribution from electrostatic forces (Ees) of −84.6, −75.6, and −71.2 kcal/mol, and dispersion forces (Edisp) of −12.5, −29.5, and −34.2 kcal/mol, respectively (Figure 6a–c and Figures S26–28 in Supporting Information File 1). Other sets of the ion pairs in the crystal structures showed smaller interaction energies. For example, the pair of 2+ and BF4− located at the side of TATA+ showed the Etot, Ees, and Edisp values of −55.7, −48.6, and −6.9 kcal/mol, respectively, which are smaller than those of the ion pair contacting the TATA+ plane.
![[1860-5397-20-215-6]](/bjoc/content/figures/1860-5397-20-215-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: (i) Single-crystal X-ray structures and (ii) interaction energies for the pairs (a) 2+-Cl−, (b) 2+-BF4−, and (c) 2+-PF6−. In (i), the cation and the anion are represented in cyan and magenta colors, respectively.
Figure 6: (i) Single-crystal X-ray structures and (ii) interaction energies for the pairs (a) 2+-Cl−, (b) 2+-...
Ion pairs of larger counteranions formed ion-pairing assemblies in which the counteranions were displaced from the top of the TATA+ planes. In 2+-B(C6F5)4−, the distance between the boron of B(C6F5)4− and the central carbon of TATA+ was 7.32 Å, with an offset angle of 47.1° (Figure 3d, Figure 7a, and Figures S14 and S20 in Supporting Information File 1). On the other hand, in 2+-PCCp−, the PCCp− unit (centroid of the five-membered ring) was located 5.55 Å from the central carbon of TATA+ with an offset angle of 69.4° (Figure 3e, Figure 7b, and Figures S15 and S21 in Supporting Information File 1). The PCCp− unit was inclined at 29.9° to the TATA+ plane, with the closest distance of 3.31 Å between the cyano-N and TATA+ mean plane. Two proximally located PCCp− units formed a partially stacked dimer with a stacking distance of 3.43 Å. The B(C6F5)4− and PCCp− ion pairs with the nearest set of ion pairs showed the Etot values of −115.1 and −100.7 kcal/mol, respectively, with Ees/Edisp values of −48.6/−73.7 and −51.5/−54.9 kcal/mol, respectively (Figure 8a,b and Figures S29 and S30 in Supporting Information File 1). The energy values for 2+-PCCp− are larger than other sets of ion pairs located at the side of TATA+ showing Etot of −63.1 kcal/mol. On the other hand, another proximally located ion pair in 2+-B(C6F5)4− showed similar Etot, Ees, and Edisp values of −101.2, −46.4, and −62.2 kcal/mol, respectively, indicating that the bulkier counteranion interacted with several surrounding cations. Smaller Ees values for 2+-B(C6F5)4− and 2+-PCCp− compared to those for the ion pairs comprising smaller counteranions can be ascribed to the larger distances between 2+ and the counteranions owing to steric hindrance.
![[1860-5397-20-215-7]](/bjoc/content/figures/1860-5397-20-215-7.png?scale=2.2&max-width=1024&background=FFFFFF)
Figure 7: Hirshfeld surface analysis mapped with dnorm of closely contacted ion pairs: (a) 2+-B(C6F5)4− and (b) 2+-PCCp−. Atom color code: brown, yellow, blue, and green refer to carbon, boron, nitrogen, and fluorine, respectively.
Figure 7: Hirshfeld surface analysis mapped with dnorm of closely contacted ion pairs: (a) 2+-B(C6F5)4− and (...
![[1860-5397-20-215-8]](/bjoc/content/figures/1860-5397-20-215-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: (i) Single-crystal X-ray structures and (ii) interaction energies for the pairs for (a) 2+-B(C6F5)4− and (b) 2+-PCCp−. In (i), the cation and the anion are represented in cyan and magenta colors, respectively.
Figure 8: (i) Single-crystal X-ray structures and (ii) interaction energies for the pairs for (a) 2+-B(C6F5)4−...
Conclusion
In this study, N-(2,6-dimethylphenyl)-substituted TATA+ was used as a bulky cation to combine with anions of different sizes for forming ion pairs. In the solid state, the bulky TATA+ cation formed ion-pairing assemblies without stacking of the TATA+ planes owing to the steric hindrance of the bulky N-substituents, indicating that the arrangement of the TATA+ cation was largely dependent on the coexisting counteranions. In particular, the TATA+ cation in the BF4− and PF6− ion pairs formed herringbone structures by the aid of intermolecular C–H···π interactions. Such characteristic anion-driven assemblies of TATA+ are not known, and further studies on ion-pairing assemblies of modified TATA+ can lead to the development of functional materials, comprising multiple components associated via iπ–iπ interactions that are the interactions between charged (ionic) π-systems, with interesting electronic properties like electric conductivities.
Experimental
General procedures
Starting materials were purchased from FUJIFILM Wako Pure Chemical Corp., Nacalai Tesque Inc., and Sigma-Aldrich Co. and were used without further purification unless otherwise stated. According to the previous procedure [30], 4,8,12-tris(2,6-dimethylphenyl)-4,8,12-triazatriangulenium as a Cl− ion pair, 2+-Cl−, was prepared. NMR spectra used in the characterization of products were recorded on a JEOL ECA-600 600 MHz spectrometer. All NMR spectra were referenced to residual solvent. UV–visible absorption spectra were recorded on a Hitachi U-4500 spectrometer. Fluorescence spectra were recorded on a Hitachi F-4500 fluorescence spectrometer. MALDI–TOF MS was recorded on a Shimadzu Axima-CFRplus. TLC analyses were carried out on aluminum sheets coated with silica gel 60 (Merck 5554). Column chromatography was performed on Wakogel C-300.
Preparation of ion pairs
4,8,12-Tris(2,6-dimethylphenyl)-4,8,12-triazatriangulenium as a BF4− ion pair, 2+-BF4−. According to the literature procedure [26], to a Schlenk tube were added 2,6-dimethylaniline (7.0 mL, 59 mmol) and tris(2,6-dimethoxyphenyl)methylium as a Cl− ion pair [22] (504 mg, 1.09 mmol). The Schlenk tube was placed in an oil bath and was heated from rt to reflux over a period of 30 min. The reaction mixture was allowed to reach rt, and then the aniline was removed by short silica gel column chromatography (Wakogel C-300, eluent: MeOH/CH2Cl2 with an increment of MeOH). The residue was chromatographed over ion-exchanged resin (Amberlite IRA402BL, eluent: MeOH/CH2Cl2) and solvents were removed by evaporation. Then, to a MeOH solution (150 mL) of the solid was added AgBF4 (269 mg, 1.38 mmol), and the reaction mixture was stirred at rt for 10 min, followed by filtration, and evaporation to dryness. The residue was purified by silica gel column chromatography (Wakogel C-300; eluent: MeOH/AcOEt/CH2Cl2 1:2:8) and was recrystallized from CH2Cl2/n-hexane to afford 2+-BF4− (143 mg, 0.209 mmol, 19%) as a red solid. Rf 0.33 (MeOH/EtOAc/CH2Cl2 1:2:8); 1H NMR (600 MHz, CDCl3, 20 °C) δ (ppm) 7.68 (t, J = 8.4 Hz, 3H, TATA-H), 7.51 (t, J = 7.8 Hz, 3H, Ar-H), 7.44 (d, J = 7.8 Hz, 6H, Ar-H), 6.37 (d, J = 8.4 Hz, 6H, TATA-H), 2.10 (s, 18H, CH3); 13C NMR (151 MHz, CDCl3, 20 °C) δ (ppm) 142.15, 140.66, 138.90, 136.12, 134.80, 130.88, 130.80, 110.29, 106.31, 17.63; 19F NMR (564 MHz, CDCl3, 20 °C) δ (ppm) −157.91 (s, 10BF4−), −157.96 (s, 11BF4−); UV–vis (CH3CN) λmax, nm (ε, 105 M−1 cm−1): 272 (1.28), 337 (0.06), 349 (0.09), 524 (0.21); MALDI–TOF MS (m/z) (% intensity): (positive) 594.3 (100), 595.3 (40), 596.3 (10), [M − BF4]+ calcd for C43H36N3, 594.29; (negative) 86.0 (20), 87.0 (100); [M – C43H36N3]− calcd for BF4, 87.00. This compound was further characterized by single-crystal X-ray diffraction analysis.
4,8,12-Tris(2,6-dimethylphenyl)-4,8,12-triazatriangulenium as a PF6− ion pair, 2+-PF6−. To a MeOH solution (3.0 mL) of 2+-BF4− (4.98 mg, 7.3 μmol) was added NaPF6 (3.68 mg, 22 μmol), and the reaction mixture was stirred at rt for 15 min, followed by filtration and evaporation to dryness. The residue was purified by silica gel column chromatography (Wakogel C-300; eluent: 5% MeOH/CH2Cl2) and was recrystallized from CH2Cl2/n-hexane to afford 2+-PF6− (3.68 mg, 5.0 μmol, 68%) as a red solid. Rf 0.40 (5% MeOH/CH2Cl2); 1H NMR (600 MHz, CDCl3, 20 °C) δ (ppm) 7.63 (t, J = 8.4 Hz, 3H, TATA-H), 7.50 (t, J = 7.8 Hz, 3H, Ar-H), 7.43 (d, J = 7.8 Hz, 6H, Ar-H), 6.35 (d, J = 8.4 Hz, 6H, TATA-H), 2.11 (s, 18H, CH3); 13C NMR (151 MHz, CDCl3, 20 °C) δ (ppm) 142.40, 140.77, 138.59, 136.46, 135.03, 130.73, 130.69, 110.66, 106.15, 17.61; 19F NMR (564 MHz, CDCl3, 20 °C) δ (ppm) −77.26 (d, J = 712 Hz, 6F); UV–vis (CH3CN), λmax, nm (ε, 105 M−1 cm−1): 272 (1.30), 337 (0.07), 349 (0.09), 523 (0.21); MALDI–TOF MS (m/z) (% intensity): (positive) 594.3 (100), 595.3 (40), 596.3 (10), [M − F6P]+ calcd for C43H36N3, 594.29; (negative) 145.1 (100), [M − C49H30N3]− calcd for F6P, 144.96. This compound was further characterized by single-crystal X-ray diffraction analysis.
4,8,12-Tris(2,6-dimethylphenyl)-4,8,12-triazatriangulenium as a B(C6F5)4− ion pair, 2+-B(C6F5)4−. To a CH2Cl2 solution (3.0 mL) of 2+-BF4− (4.82 mg, 7.1 μmol) was added the Li+ salt of tetrakis(pentafluorophenyl)borate (LiB(C6F5)4, 12.6 mg, 18 μmol), and the reaction mixture was stirred at rt for 30 min, followed by filtration and evaporation to dryness. The residue was purified by silica gel column chromatography (Wakogel C-300; eluent: 5% MeOH/CH2Cl2) and was recrystallized from CH2Cl2/n-hexane to afford 2+-B(C6F5)4− (3.89 mg, 3.1 μmol, 44%) as a red solid. Rf 0.74 (5% MeOH/CH2Cl2); 1H NMR (600 MHz, CDCl3, 20 °C) δ (ppm) 7.68 (t, J = 8.4 Hz, 3H, TATA-H), 7.51 (t, J = 7.8 Hz, 3H, Ar-H), 7.44 (d, J = 7.2 Hz, 6H, Ar-H), 6.38 (d, J = 8.4 Hz, 6H, TATA-H), 2.11 (s, 18H, CH3); 13C NMR (151 MHz, CDCl3, 20 °C) δ (ppm) 148.27 (d, J13C–19F = 242 Hz), 142.18, 140.70, 138.92, 138.22 (d, J13C–19F = 241 Hz), 135.98, 136.31 (d, J13C–19F = 246 Hz), 134.76, 131.00, 130.84, 124.30, 110.24, 106.33, 17.37; 19F NMR (564 MHz, CDCl3, 20 °C) δ (ppm) −135.76 (s, 8F, Ar-F), −166.67 (t, J = 21.4 Hz, 4F, Ar-F), −170.25 (t, J = 18.0 Hz, 8F, Ar-F); UV–vis (CH3CN, λmax, nm (ε, 105 M−1 cm−1): 272 (1.26), 337 (0.07), 349 (0.10), 524 (0.20); MALDI–TOF MS (m/z) (% intensity): (positive) 594.3 (100), 595.3 (40), 596.3 (10), [M − C24BF20]+ calcd for C43H36N3, 594.29; (negative) 678.0 (20), 679.0 (100), 680.0 (20), [M – C43H36N3]– calcd for C24BF20, 678.98. This compound was further characterized by single-crystal X-ray diffraction analysis.
4,8,12-Tris(2,6-dimethylphenyl)-4,8,12-triazatriangulenium as a PCCp− ion pair, 2+-PCCp−. To a MeOH solution (3.0 mL) of 2+-BF4− (4.88 mg, 7.2 μmol) was added sodium pentacyanocyclopentadienide [18,19] (NaPCCp, 4.74 mg, 22 μmol), and the reaction mixture was stirred at rt for 10 min, followed by filtration and evaporation to dryness. The residue was purified by silica gel column chromatography (Wakogel C-300; eluent: 5% MeOH/CH2Cl2) and was recrystallized from CH2Cl2/n-hexane to afford 2+-PCCp− (3.21 mg, 4.1 μmol, 57%) as a red solid. Rf 0.56 (5% MeOH/CH2Cl2); 1H NMR (600 MHz, CDCl3, 20 °C) δ (ppm) 7.68 (t, J = 8.4 Hz, 3H, TATA-H), 7.53 (t, J = 7.8 Hz, 3H, Ar-H), 7.45 (d, J = 7.8 Hz, 6H, Ar-H), 6.39 (d, J = 8.4 Hz, 6H, TATA-H), 2.09 (s, 18H, CH3); 13C NMR (151 MHz, CDCl3, 20 °C) δ (ppm) 142.24, 140.72, 138.96, 136.04, 134.80, 131.00, 130.87, 113.57, 110.34, 106.37, 102.64, 17.65; UV–vis (CH3CN, λmax, nm (ε, 105 M−1 cm−1): 272 (1.33), 337 (0.07), 349 (0.10), 524 (0.20); MALDI–TOF MS (m/z) (% intensity): (positive) 594.3 (100), 595.3 (40), 596.3 (10), [M − C10N5]+ calcd for C43H36N3, 594.29; (negative) 190.1 (100), 191.1 (10), [M − C43H36N3]− calcd for C10N5, 190.02. This compound was further characterized by single-crystal X-ray diffraction analysis.
Method for single-crystal X-ray analysis
Crystallographic data are summarized in Table 1. A single crystal of 2+-Cl− was obtained by vapor diffusion of n-heptane into a CHCl3 solution. The data crystal was a red prism of approximate dimensions 0.20 mm × 0.02 mm × 0.01 mm. A single crystal of 2+-BF4− was obtained by vapor diffusion of n-heptane into a CHCl3 solution. The data crystal was a red prism of approximate dimensions 0.30 mm × 0.10 mm × 0.05 mm. A single crystal of 2+-PF6− was obtained by vapor diffusion of n-hexane into a CH2Cl2 with small amounts of MeOH and toluene. The data crystal was a red prism of approximate dimensions 0.20 mm × 0.02 mm × 0.01 mm. A single crystal of 2+-PCCp− was obtained by vapor diffusion of n-heptane into a CHCl3 solution. The data crystal was a red prism of approximate dimensions 0.050 mm × 0.030 mm × 0.010 mm. A single crystal of 2+-B(C6F5)4− was obtained by vapor diffusion of n-hexane into a toluene solution. The data crystal was a red prism of approximate dimensions 0.05 mm × 0.01 mm × 0.01 mm. The data of 2+-Cl− and 2+-BF4− were collected at 90 K on a Bruker D8 Venture diffractometer with Mo Kα radiation (λ = 0.71073 Å) focused by a multilayer confocal mirror, whereas the data of 2+-PF6− and 2+-B(C6F5)4− were collected at 100 K on a DECTRIS EIGER X 1M diffractometer with Si(111) monochromated synchrotron radiation (λ = 0.8125 and 0.8120 Å, respectively) at BL40XU (SPring-8) [41,42]. The data of 2+-PCCp− was collected at 90 K on a Rigaku Pliatus 3 CdTe 1M with Si(311) monochromated synchrotron radiation (λ = 0.4144 Å) at BL02B1 (SPring-8) [43]. All the structures were solved by dual-space method. The structures were refined by a full-matrix least-squares method by using a SHELXL 2014 [44] (Yadokari-XG) [45,46]. In each structure, the non-hydrogen atoms were refined anisotropically. CIF files (CCDC-2364292–2364296) can be obtained free of charge from the Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif.
Table 1: Crystallographic details.
| 2+-Cl− | 2+-BF4− | 2+-PF6− | 2+-B(C6F5)4− | 2+-PCCp− | |
| formula |
C43H36N3Cl·
3CHCl3 |
C43H36N3BF4·
2CHCl3 |
C43H36N3·F6P |
C43H36N3·C24BF20·
C7H8·0.5C6H14 |
C43H36N3·C10N5·
CHCl3 |
| fw | 988.30 | 920.29 | 739.72 | 1409.02 | 904.26 |
| crystal size, mm | 0.20 × 0.02 × 0.01 | 0.30 × 0.10 × 0.05 | 0.20 × 0.02 × 0.01 | 0.05 × 0.01 × 0.01 | 0.050 × 0.030 × 0.010 |
| crystal system | triclinic | monoclinic | monoclinic | triclinic | triclinic |
| space group |
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-215-i1.svg?max-width=637&scale=1.2) (no. 2) (no. 2)
|
P21/n (no. 14) | P21/n (no. 14) |
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-215-i2.svg?max-width=637&scale=1.2) (no. 2) (no. 2)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-215-i3.svg?max-width=637&scale=1.2) (no. 2) (no. 2)
|
| a, Å | 10.3193(6) | 19.3043(12) | 15.1182(2) | 14.6519(5) | 11.079(10) |
| b, Å | 12.8955(8) | 11.7445(6) | 11.95190(10) | 15.4923(6) | 14.418(12) |
| c, Å | 18.2429(12) | 19.3182(12) | 19.4163(2) | 15.9933(5) | 16.284(15) |
| α, ° | 78.520(2) | 90 | 90 | 99.408(3) | 90.562(7) |
| β, ° | 78.621(2) | 103.358(2) | 91.4290(10) | 99.229(3) | 105.230(8) |
| γ, ° | 76.892(2) | 90 | 90 | 108.879(3) | 102.795(7) |
| V, Å3 | 2287.4(2) | 4261.3(4) | 3507.26(7) | 3299.2(2) | 2441(4) |
| ρcalcd, g cm–3 | 1.435 | 1.434 | 1.401 | 1.418 | 1.230 |
| Z | 2 | 4 | 4 | 2 | 2 |
| T, K | 90(2) | 90(2) | 100(2) | 100(2) | 90(2) |
| μ, mm–1 | 0.646a | 0.458a | 0.209b | 0.167b | 0.065b |
| no. of reflns | 39674 | 81192 | 41635 | 35844 | 20901 |
| no. of unique reflns | 7912 | 8668 | 8216 | 14804 | 9959 |
| variables | 559 | 566 | 485 | 908 | 592 |
| λ, Å | 0.71073a | 0.71073a | 0.8125b | 0.8120b | 0.4144b |
| R1 (I > 2σ(I)) | 0.0708 | 0.0470 | 0.0395 | 0.0737 | 0.0577 |
| wR2 (I > 2σ(I)) | 0.1633 | 0.1218 | 0.1142 | 0.2198 | 0.1738 |
| GOF | 1.061 | 1.016 | 1.050 | 1.058 | 0.963 |
aMo Kα. bSynchrotron radiation.
DFT calculations. DFT calculations for the geometrical optimizations were carried out by using the Gaussian 16 program [31].
Supporting Information
| Supporting Information File 1: 1H, 13C, and 19F NMR spectra of new ion pairs, details for crystal structures, and theoretical calculations. | ||
| Format: PDF | Size: 4.1 MB | Download |
Acknowledgements
Theoretical calculations were partially performed at the Research Center for Computational Science, Okazaki, Japan (Projects: 22-IMS-C077, 23-IMS-C069, and 24-IMS-C067). The synchrotron radiation experiments were performed at the BL02B1 (2023A1645) and BL40XU (2023B1390) of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI). We thank Mr. Makoto Kobayashi, Ritsumeikan University, for the initial examinations of ion-pairing assemblies of TATA+, Dr. Yuiga Nakamura and Dr. Kouhei Ichiyanagi, JASRI, for single-crystal synchrotron X-ray analysis, Prof. Osamu Tsutsumi, Ritsumeikan University, for single-crystal X-ray analysis, and Prof. Hitoshi Tamiaki, Ritsumeikan University, for various measurements.
Funding
This work was supported by JSPS KAKENHI Grant Numbers JP22H02067 and JP23K23335 for Scientific Research (B), JP24K08389 for Scientific Research (C), JP23K17951 for Challenging Research (Exploratory), and JP20H05863 for Transformative Research Areas (A) “Condensed Conjugation” and Ritsumeikan Global Innovation Research Organization (R-GIRO) project (2017–22 and 2022–27).
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Bosson, J.; Gouin, J.; Lacour, J. Chem. Soc. Rev. 2014, 43, 2824–2840. doi:10.1039/c3cs60461f
Return to citation in text: [1] -
Winter, A.; Schubert, U. S. Mater. Chem. Front. 2019, 3, 2308–2325. doi:10.1039/c9qm00532c
Return to citation in text: [1] -
Thyrhaug, E.; Sørensen, T. J.; Gryczynski, I.; Gryczynski, Z.; Laursen, B. W. J. Phys. Chem. A 2013, 117, 2160–2168. doi:10.1021/jp312376k
Return to citation in text: [1] -
Shivalingam, A.; Vyšniauskas, A.; Albrecht, T.; White, A. J. P.; Kuimova, M. K.; Vilar, R. Chem. – Eur. J. 2016, 22, 4129–4139. doi:10.1002/chem.201504099
Return to citation in text: [1] -
Rosenberg, M.; Rostgaard, K. R.; Liao, Z.; Madsen, A. Ø.; Martinez, K. L.; Vosch, T.; Laursen, B. W. Chem. Sci. 2018, 9, 3122–3130. doi:10.1039/c8sc00089a
Return to citation in text: [1] -
Gouin, J.; Bürgi, T.; Guénée, L.; Lacour, J. Org. Lett. 2014, 16, 3800–3803. doi:10.1021/ol501692r
Return to citation in text: [1] -
Delgado, I. H.; Pascal, S.; Wallabregue, A.; Duwald, R.; Besnard, C.; Guénée, L.; Nançoz, C.; Vauthey, E.; Tovar, R. C.; Lunkley, J. L.; Muller, G.; Lacour, J. Chem. Sci. 2016, 7, 4685–4693. doi:10.1039/c6sc00614k
Return to citation in text: [1] -
Fabri, B.; Funaioli, T.; Frédéric, L.; Elsner, C.; Bordignon, E.; Zinna, F.; Di Bari, L.; Pescitelli, G.; Lacour, J. J. Am. Chem. Soc. 2024, 146, 8308–8319. doi:10.1021/jacs.3c13487
Return to citation in text: [1] -
Gueret, R.; Poulard, L.; Oshinowo, M.; Chauvin, J.; Dahmane, M.; Dupeyre, G.; Lainé, P. P.; Fortage, J.; Collomb, M.-N. ACS Catal. 2018, 8, 3792–3802. doi:10.1021/acscatal.7b04000
Return to citation in text: [1] -
Ho, P.-Y.; Cheng, S.-C.; Yu, F.; Yeung, Y.-Y.; Ni, W.-X.; Ko, C.-C.; Leung, C.-F.; Lau, T.-C.; Robert, M. ACS Catal. 2023, 13, 5979–5985. doi:10.1021/acscatal.3c00036
Return to citation in text: [1] -
Sørensen, T. J.; Hildebrandt, C. B.; Elm, J.; Andreasen, J. W.; Madsen, A. Ø.; Westerlund, F.; Laursen, B. W. J. Mater. Chem. 2012, 22, 4797–4805. doi:10.1039/c2jm15954f
Return to citation in text: [1] [2] [3] -
Sørensen, T. J.; Hildebrandt, C. B.; Glyvradal, M.; Laursen, B. W. Dyes Pigm. 2013, 98, 297–303. doi:10.1016/j.dyepig.2013.03.006
Return to citation in text: [1] [2] [3] -
Westerlund, F.; Lemke, H. T.; Hassenkam, T.; Simonsen, J. B.; Laursen, B. W. Langmuir 2013, 29, 6728–6736. doi:10.1021/la400797b
Return to citation in text: [1] -
Shi, D.; Schwall, C.; Sfintes, G.; Thyrhaug, E.; Hammershøj, P.; Cárdenas, M.; Simonsen, J. B.; Laursen, B. W. Chem. – Eur. J. 2014, 20, 6853–6856. doi:10.1002/chem.201402523
Return to citation in text: [1] -
Laursen, B. W.; Krebs, F. C. Angew. Chem., Int. Ed. 2000, 39, 3432–3434. doi:10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s
Return to citation in text: [1] [2] [3] [4] -
Laursen, B. W.; Krebs, F. C. Chem. – Eur. J. 2001, 7, 1773–1783. doi:10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f
Return to citation in text: [1] [2] [3] -
Haketa, Y.; Yamasumi, K.; Maeda, H. Chem. Soc. Rev. 2023, 52, 7170–7196. doi:10.1039/d3cs00581j
Return to citation in text: [1] -
Webster, O. W. J. Am. Chem. Soc. 1965, 87, 1820–1821. doi:10.1021/ja01086a055
Return to citation in text: [1] [2] -
Sakai, T.; Seo, S.; Matsuoka, J.; Mori, Y. J. Org. Chem. 2013, 78, 10978–10985. doi:10.1021/jo401946j
Return to citation in text: [1] [2] -
Bando, Y.; Haketa, Y.; Sakurai, T.; Matsuda, W.; Seki, S.; Takaya, H.; Maeda, H. Chem. – Eur. J. 2016, 22, 7843–7850. doi:10.1002/chem.201600686
Return to citation in text: [1] -
Sasano, Y.; Tanaka, H.; Haketa, Y.; Kobayashi, Y.; Ishibashi, Y.; Morimoto, T.; Sato, R.; Shigeta, Y.; Yasuda, N.; Asahi, T.; Maeda, H. Chem. Sci. 2021, 12, 9645–9657. doi:10.1039/d1sc02260a
Return to citation in text: [1] [2] -
Haketa, Y.; Sasaki, S.; Ohta, N.; Masunaga, H.; Ogawa, H.; Mizuno, N.; Araoka, F.; Takezoe, H.; Maeda, H. Angew. Chem., Int. Ed. 2010, 49, 10079–10083. doi:10.1002/anie.201006356
Return to citation in text: [1] [2] [3] [4] -
Haketa, Y.; Honsho, Y.; Seki, S.; Maeda, H. Chem. – Eur. J. 2012, 18, 7016–7020. doi:10.1002/chem.201200951
Return to citation in text: [1] -
Maeda, H.; Nishimura, T.; Haketa, Y.; Tanaka, H.; Fujita, M.; Yasuda, N. J. Org. Chem. 2022, 87, 7818–7825. doi:10.1021/acs.joc.2c00463
Return to citation in text: [1] -
Kugizaki, R.; Haketa, Y.; Kamada, K.; Maeda, H. Chem. – Eur. J. 2024, 30, e202401932. doi:10.1002/chem.202401932
Return to citation in text: [1] -
Hammershøj, P.; Sørensen, T. J.; Han, B.-H.; Laursen, B. W. J. Org. Chem. 2012, 77, 5606–5612. doi:10.1021/jo3007732
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Hu, X.-M.; Chen, Q.; Sui, Z.-Y.; Zhao, Z.-Q.; Bovet, N.; Laursen, B. W.; Han, B.-H. RSC Adv. 2015, 5, 90135–90143. doi:10.1039/c5ra18047c
Return to citation in text: [1] [2] -
Plajer, A. J.; Percástegui, E. G.; Santella, M.; Rizzuto, F. J.; Gan, Q.; Laursen, B. W.; Nitschke, J. R. Angew. Chem., Int. Ed. 2019, 58, 4200–4204. doi:10.1002/anie.201814149
Return to citation in text: [1] [2] -
Zhang, D.; Gan, Q.; Plajer, A. J.; Lavendomme, R.; Ronson, T. K.; Lu, Z.; Jensen, J. D.; Laursen, B. W.; Nitschke, J. R. J. Am. Chem. Soc. 2022, 144, 1106–1112. doi:10.1021/jacs.1c11536
Return to citation in text: [1] [2] -
Li, D.; Silveira, O. J.; Matsuda, T.; Hayashi, H.; Maeda, H.; Foster, A. S.; Kawai, S. Angew. Chem., Int. Ed. 2024, 63, e202411893. doi:10.1002/anie.202411893
Return to citation in text: [1] [2] -
Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2016.
Return to citation in text: [1] [2] -
The pseudo-polymorph of 2+-BF4– in the absence of included solvent molecules was observed from a low-quality data and will be reported somewhere after updating.
Return to citation in text: [1] -
Spackman, P. R.; Turner, M. J.; McKinnon, J. J.; Wolff, S. K.; Grimwood, D. J.; Jayatilaka, D.; Spackman, M. A. J. Appl. Crystallogr. 2021, 54, 1006–1011. doi:10.1107/s1600576721002910
Return to citation in text: [1] -
Phipps, M. J. S.; Fox, T.; Tautermann, C. S.; Skylaris, C.-K. Chem. Soc. Rev. 2015, 44, 3177–3211. doi:10.1039/c4cs00375f
Return to citation in text: [1] -
Spackman, M. A.; Jayatilaka, D. CrystEngComm 2009, 11, 19–32. doi:10.1039/b818330a
Return to citation in text: [1] -
McKinnon, J. J.; Spackman, M. A.; Mitchell, A. S. Acta Crystallogr., Sect. B: Struct. Sci. 2004, 60, 627–668. doi:10.1107/s0108768104020300
Return to citation in text: [1] -
Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.; Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, N.; Nguyen, K. A.; Su, S.; Windus, T. L.; Dupuis, M.; Montgomery, J. A., Jr. J. Comput. Chem. 1993, 14, 1347–1363. doi:10.1002/jcc.540141112
Return to citation in text: [1] -
Gordon, M. S.; Schmidt, M. W. Advances in electronic structure theory: GAMESS a decade later. In Theory and Applications of Computational Chemistry; Dykstra, C. E.; Frenking, G.; Kim, K. S.; Scuseria, G. E., Eds.; Elsevier, 2005; pp 1167–1189. doi:10.1016/b978-044451719-7/50084-6
Return to citation in text: [1] -
Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M. Chem. Phys. Lett. 1999, 313, 701–706. doi:10.1016/s0009-2614(99)00874-x
Return to citation in text: [1] -
Fedorov, D. G.; Kitaura, K. J. Comput. Chem. 2007, 28, 222–237. doi:10.1002/jcc.20496
Return to citation in text: [1] -
Yasuda, N.; Murayama, H.; Fukuyama, Y.; Kim, J.; Kimura, S.; Toriumi, K.; Tanaka, Y.; Moritomo, Y.; Kuroiwa, Y.; Kato, K.; Tanaka, H.; Takata, M. J. Synchrotron Radiat. 2009, 16, 352–357. doi:10.1107/s090904950900675x
Return to citation in text: [1] -
Yasuda, N.; Fukuyama, Y.; Toriumi, K.; Kimura, S.; Takata, M. AIP Conf. Proc. 2010, 1234, 147–150. doi:10.1063/1.3463161
Return to citation in text: [1] -
Sugimoto, K.; Ohsumi, H.; Aoyagi, S.; Nishibori, E.; Moriyoshi, C.; Kuroiwa, Y.; Sawa, H.; Takata, M. AIP Conf. Proc. 2010, 1234, 887–890. doi:10.1063/1.3463359
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930
Return to citation in text: [1] -
Yadokari-XG, Software for Crystal Structure Analyses; 2001.
Return to citation in text: [1] -
Kabuto, C.; Akine, S.; Nemoto, T.; Kwon, E. J. Crystallogr. Soc. Jpn. 2009, 51, 218–224. doi:10.5940/jcrsj.51.218
Return to citation in text: [1]
| 26. | Hammershøj, P.; Sørensen, T. J.; Han, B.-H.; Laursen, B. W. J. Org. Chem. 2012, 77, 5606–5612. doi:10.1021/jo3007732 |
| 22. | Haketa, Y.; Sasaki, S.; Ohta, N.; Masunaga, H.; Ogawa, H.; Mizuno, N.; Araoka, F.; Takezoe, H.; Maeda, H. Angew. Chem., Int. Ed. 2010, 49, 10079–10083. doi:10.1002/anie.201006356 |
| 18. | Webster, O. W. J. Am. Chem. Soc. 1965, 87, 1820–1821. doi:10.1021/ja01086a055 |
| 19. | Sakai, T.; Seo, S.; Matsuoka, J.; Mori, Y. J. Org. Chem. 2013, 78, 10978–10985. doi:10.1021/jo401946j |
| 1. | Bosson, J.; Gouin, J.; Lacour, J. Chem. Soc. Rev. 2014, 43, 2824–2840. doi:10.1039/c3cs60461f |
| 2. | Winter, A.; Schubert, U. S. Mater. Chem. Front. 2019, 3, 2308–2325. doi:10.1039/c9qm00532c |
| 11. | Sørensen, T. J.; Hildebrandt, C. B.; Elm, J.; Andreasen, J. W.; Madsen, A. Ø.; Westerlund, F.; Laursen, B. W. J. Mater. Chem. 2012, 22, 4797–4805. doi:10.1039/c2jm15954f |
| 12. | Sørensen, T. J.; Hildebrandt, C. B.; Glyvradal, M.; Laursen, B. W. Dyes Pigm. 2013, 98, 297–303. doi:10.1016/j.dyepig.2013.03.006 |
| 13. | Westerlund, F.; Lemke, H. T.; Hassenkam, T.; Simonsen, J. B.; Laursen, B. W. Langmuir 2013, 29, 6728–6736. doi:10.1021/la400797b |
| 14. | Shi, D.; Schwall, C.; Sfintes, G.; Thyrhaug, E.; Hammershøj, P.; Cárdenas, M.; Simonsen, J. B.; Laursen, B. W. Chem. – Eur. J. 2014, 20, 6853–6856. doi:10.1002/chem.201402523 |
| 28. | Plajer, A. J.; Percástegui, E. G.; Santella, M.; Rizzuto, F. J.; Gan, Q.; Laursen, B. W.; Nitschke, J. R. Angew. Chem., Int. Ed. 2019, 58, 4200–4204. doi:10.1002/anie.201814149 |
| 29. | Zhang, D.; Gan, Q.; Plajer, A. J.; Lavendomme, R.; Ronson, T. K.; Lu, Z.; Jensen, J. D.; Laursen, B. W.; Nitschke, J. R. J. Am. Chem. Soc. 2022, 144, 1106–1112. doi:10.1021/jacs.1c11536 |
| 9. | Gueret, R.; Poulard, L.; Oshinowo, M.; Chauvin, J.; Dahmane, M.; Dupeyre, G.; Lainé, P. P.; Fortage, J.; Collomb, M.-N. ACS Catal. 2018, 8, 3792–3802. doi:10.1021/acscatal.7b04000 |
| 10. | Ho, P.-Y.; Cheng, S.-C.; Yu, F.; Yeung, Y.-Y.; Ni, W.-X.; Ko, C.-C.; Leung, C.-F.; Lau, T.-C.; Robert, M. ACS Catal. 2023, 13, 5979–5985. doi:10.1021/acscatal.3c00036 |
| 30. | Li, D.; Silveira, O. J.; Matsuda, T.; Hayashi, H.; Maeda, H.; Foster, A. S.; Kawai, S. Angew. Chem., Int. Ed. 2024, 63, e202411893. doi:10.1002/anie.202411893 |
| 6. | Gouin, J.; Bürgi, T.; Guénée, L.; Lacour, J. Org. Lett. 2014, 16, 3800–3803. doi:10.1021/ol501692r |
| 7. | Delgado, I. H.; Pascal, S.; Wallabregue, A.; Duwald, R.; Besnard, C.; Guénée, L.; Nançoz, C.; Vauthey, E.; Tovar, R. C.; Lunkley, J. L.; Muller, G.; Lacour, J. Chem. Sci. 2016, 7, 4685–4693. doi:10.1039/c6sc00614k |
| 8. | Fabri, B.; Funaioli, T.; Frédéric, L.; Elsner, C.; Bordignon, E.; Zinna, F.; Di Bari, L.; Pescitelli, G.; Lacour, J. J. Am. Chem. Soc. 2024, 146, 8308–8319. doi:10.1021/jacs.3c13487 |
| 26. | Hammershøj, P.; Sørensen, T. J.; Han, B.-H.; Laursen, B. W. J. Org. Chem. 2012, 77, 5606–5612. doi:10.1021/jo3007732 |
| 3. | Thyrhaug, E.; Sørensen, T. J.; Gryczynski, I.; Gryczynski, Z.; Laursen, B. W. J. Phys. Chem. A 2013, 117, 2160–2168. doi:10.1021/jp312376k |
| 4. | Shivalingam, A.; Vyšniauskas, A.; Albrecht, T.; White, A. J. P.; Kuimova, M. K.; Vilar, R. Chem. – Eur. J. 2016, 22, 4129–4139. doi:10.1002/chem.201504099 |
| 5. | Rosenberg, M.; Rostgaard, K. R.; Liao, Z.; Madsen, A. Ø.; Martinez, K. L.; Vosch, T.; Laursen, B. W. Chem. Sci. 2018, 9, 3122–3130. doi:10.1039/c8sc00089a |
| 27. | Hu, X.-M.; Chen, Q.; Sui, Z.-Y.; Zhao, Z.-Q.; Bovet, N.; Laursen, B. W.; Han, B.-H. RSC Adv. 2015, 5, 90135–90143. doi:10.1039/c5ra18047c |
| 20. | Bando, Y.; Haketa, Y.; Sakurai, T.; Matsuda, W.; Seki, S.; Takaya, H.; Maeda, H. Chem. – Eur. J. 2016, 22, 7843–7850. doi:10.1002/chem.201600686 |
| 21. | Sasano, Y.; Tanaka, H.; Haketa, Y.; Kobayashi, Y.; Ishibashi, Y.; Morimoto, T.; Sato, R.; Shigeta, Y.; Yasuda, N.; Asahi, T.; Maeda, H. Chem. Sci. 2021, 12, 9645–9657. doi:10.1039/d1sc02260a |
| 22. | Haketa, Y.; Sasaki, S.; Ohta, N.; Masunaga, H.; Ogawa, H.; Mizuno, N.; Araoka, F.; Takezoe, H.; Maeda, H. Angew. Chem., Int. Ed. 2010, 49, 10079–10083. doi:10.1002/anie.201006356 |
| 23. | Haketa, Y.; Honsho, Y.; Seki, S.; Maeda, H. Chem. – Eur. J. 2012, 18, 7016–7020. doi:10.1002/chem.201200951 |
| 24. | Maeda, H.; Nishimura, T.; Haketa, Y.; Tanaka, H.; Fujita, M.; Yasuda, N. J. Org. Chem. 2022, 87, 7818–7825. doi:10.1021/acs.joc.2c00463 |
| 25. | Kugizaki, R.; Haketa, Y.; Kamada, K.; Maeda, H. Chem. – Eur. J. 2024, 30, e202401932. doi:10.1002/chem.202401932 |
| 44. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930 |
| 18. | Webster, O. W. J. Am. Chem. Soc. 1965, 87, 1820–1821. doi:10.1021/ja01086a055 |
| 19. | Sakai, T.; Seo, S.; Matsuoka, J.; Mori, Y. J. Org. Chem. 2013, 78, 10978–10985. doi:10.1021/jo401946j |
| 26. | Hammershøj, P.; Sørensen, T. J.; Han, B.-H.; Laursen, B. W. J. Org. Chem. 2012, 77, 5606–5612. doi:10.1021/jo3007732 |
| 27. | Hu, X.-M.; Chen, Q.; Sui, Z.-Y.; Zhao, Z.-Q.; Bovet, N.; Laursen, B. W.; Han, B.-H. RSC Adv. 2015, 5, 90135–90143. doi:10.1039/c5ra18047c |
| 28. | Plajer, A. J.; Percástegui, E. G.; Santella, M.; Rizzuto, F. J.; Gan, Q.; Laursen, B. W.; Nitschke, J. R. Angew. Chem., Int. Ed. 2019, 58, 4200–4204. doi:10.1002/anie.201814149 |
| 29. | Zhang, D.; Gan, Q.; Plajer, A. J.; Lavendomme, R.; Ronson, T. K.; Lu, Z.; Jensen, J. D.; Laursen, B. W.; Nitschke, J. R. J. Am. Chem. Soc. 2022, 144, 1106–1112. doi:10.1021/jacs.1c11536 |
| 45. | Yadokari-XG, Software for Crystal Structure Analyses; 2001. |
| 46. | Kabuto, C.; Akine, S.; Nemoto, T.; Kwon, E. J. Crystallogr. Soc. Jpn. 2009, 51, 218–224. doi:10.5940/jcrsj.51.218 |
| 17. | Haketa, Y.; Yamasumi, K.; Maeda, H. Chem. Soc. Rev. 2023, 52, 7170–7196. doi:10.1039/d3cs00581j |
| 41. | Yasuda, N.; Murayama, H.; Fukuyama, Y.; Kim, J.; Kimura, S.; Toriumi, K.; Tanaka, Y.; Moritomo, Y.; Kuroiwa, Y.; Kato, K.; Tanaka, H.; Takata, M. J. Synchrotron Radiat. 2009, 16, 352–357. doi:10.1107/s090904950900675x |
| 42. | Yasuda, N.; Fukuyama, Y.; Toriumi, K.; Kimura, S.; Takata, M. AIP Conf. Proc. 2010, 1234, 147–150. doi:10.1063/1.3463161 |
| 15. | Laursen, B. W.; Krebs, F. C. Angew. Chem., Int. Ed. 2000, 39, 3432–3434. doi:10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s |
| 16. | Laursen, B. W.; Krebs, F. C. Chem. – Eur. J. 2001, 7, 1773–1783. doi:10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f |
| 21. | Sasano, Y.; Tanaka, H.; Haketa, Y.; Kobayashi, Y.; Ishibashi, Y.; Morimoto, T.; Sato, R.; Shigeta, Y.; Yasuda, N.; Asahi, T.; Maeda, H. Chem. Sci. 2021, 12, 9645–9657. doi:10.1039/d1sc02260a |
| 43. | Sugimoto, K.; Ohsumi, H.; Aoyagi, S.; Nishibori, E.; Moriyoshi, C.; Kuroiwa, Y.; Sawa, H.; Takata, M. AIP Conf. Proc. 2010, 1234, 887–890. doi:10.1063/1.3463359 |
| 15. | Laursen, B. W.; Krebs, F. C. Angew. Chem., Int. Ed. 2000, 39, 3432–3434. doi:10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s |
| 26. | Hammershøj, P.; Sørensen, T. J.; Han, B.-H.; Laursen, B. W. J. Org. Chem. 2012, 77, 5606–5612. doi:10.1021/jo3007732 |
| 37. | Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.; Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, N.; Nguyen, K. A.; Su, S.; Windus, T. L.; Dupuis, M.; Montgomery, J. A., Jr. J. Comput. Chem. 1993, 14, 1347–1363. doi:10.1002/jcc.540141112 |
| 38. | Gordon, M. S.; Schmidt, M. W. Advances in electronic structure theory: GAMESS a decade later. In Theory and Applications of Computational Chemistry; Dykstra, C. E.; Frenking, G.; Kim, K. S.; Scuseria, G. E., Eds.; Elsevier, 2005; pp 1167–1189. doi:10.1016/b978-044451719-7/50084-6 |
| 39. | Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M. Chem. Phys. Lett. 1999, 313, 701–706. doi:10.1016/s0009-2614(99)00874-x |
| 40. | Fedorov, D. G.; Kitaura, K. J. Comput. Chem. 2007, 28, 222–237. doi:10.1002/jcc.20496 |
| 30. | Li, D.; Silveira, O. J.; Matsuda, T.; Hayashi, H.; Maeda, H.; Foster, A. S.; Kawai, S. Angew. Chem., Int. Ed. 2024, 63, e202411893. doi:10.1002/anie.202411893 |
| 11. | Sørensen, T. J.; Hildebrandt, C. B.; Elm, J.; Andreasen, J. W.; Madsen, A. Ø.; Westerlund, F.; Laursen, B. W. J. Mater. Chem. 2012, 22, 4797–4805. doi:10.1039/c2jm15954f |
| 12. | Sørensen, T. J.; Hildebrandt, C. B.; Glyvradal, M.; Laursen, B. W. Dyes Pigm. 2013, 98, 297–303. doi:10.1016/j.dyepig.2013.03.006 |
| 15. | Laursen, B. W.; Krebs, F. C. Angew. Chem., Int. Ed. 2000, 39, 3432–3434. doi:10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s |
| 16. | Laursen, B. W.; Krebs, F. C. Chem. – Eur. J. 2001, 7, 1773–1783. doi:10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f |
| 22. | Haketa, Y.; Sasaki, S.; Ohta, N.; Masunaga, H.; Ogawa, H.; Mizuno, N.; Araoka, F.; Takezoe, H.; Maeda, H. Angew. Chem., Int. Ed. 2010, 49, 10079–10083. doi:10.1002/anie.201006356 |
| 33. | Spackman, P. R.; Turner, M. J.; McKinnon, J. J.; Wolff, S. K.; Grimwood, D. J.; Jayatilaka, D.; Spackman, M. A. J. Appl. Crystallogr. 2021, 54, 1006–1011. doi:10.1107/s1600576721002910 |
| 34. | Phipps, M. J. S.; Fox, T.; Tautermann, C. S.; Skylaris, C.-K. Chem. Soc. Rev. 2015, 44, 3177–3211. doi:10.1039/c4cs00375f |
| 35. | Spackman, M. A.; Jayatilaka, D. CrystEngComm 2009, 11, 19–32. doi:10.1039/b818330a |
| 36. | McKinnon, J. J.; Spackman, M. A.; Mitchell, A. S. Acta Crystallogr., Sect. B: Struct. Sci. 2004, 60, 627–668. doi:10.1107/s0108768104020300 |
| 11. | Sørensen, T. J.; Hildebrandt, C. B.; Elm, J.; Andreasen, J. W.; Madsen, A. Ø.; Westerlund, F.; Laursen, B. W. J. Mater. Chem. 2012, 22, 4797–4805. doi:10.1039/c2jm15954f |
| 12. | Sørensen, T. J.; Hildebrandt, C. B.; Glyvradal, M.; Laursen, B. W. Dyes Pigm. 2013, 98, 297–303. doi:10.1016/j.dyepig.2013.03.006 |
| 15. | Laursen, B. W.; Krebs, F. C. Angew. Chem., Int. Ed. 2000, 39, 3432–3434. doi:10.1002/1521-3773(20001002)39:19<3432::aid-anie3432>3.0.co;2-s |
| 16. | Laursen, B. W.; Krebs, F. C. Chem. – Eur. J. 2001, 7, 1773–1783. doi:10.1002/1521-3765(20010417)7:8<1773::aid-chem17730>3.0.co;2-f |
| 22. | Haketa, Y.; Sasaki, S.; Ohta, N.; Masunaga, H.; Ogawa, H.; Mizuno, N.; Araoka, F.; Takezoe, H.; Maeda, H. Angew. Chem., Int. Ed. 2010, 49, 10079–10083. doi:10.1002/anie.201006356 |
| 26. | Hammershøj, P.; Sørensen, T. J.; Han, B.-H.; Laursen, B. W. J. Org. Chem. 2012, 77, 5606–5612. doi:10.1021/jo3007732 |
| 32. | The pseudo-polymorph of 2+-BF4– in the absence of included solvent molecules was observed from a low-quality data and will be reported somewhere after updating. |
| 26. | Hammershøj, P.; Sørensen, T. J.; Han, B.-H.; Laursen, B. W. J. Org. Chem. 2012, 77, 5606–5612. doi:10.1021/jo3007732 |
© 2024 Haketa et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.