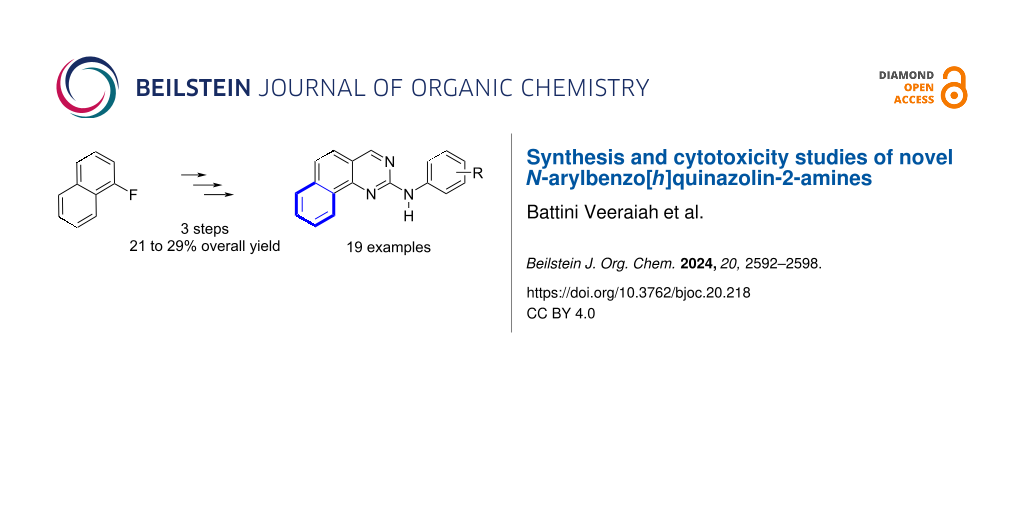
Abstract
In this paper, we report a short and efficient synthesis of novel N-arylbenzo[h]quinazoline-2-amines. We have prepared a focused library of nineteen representative examples which have been submitted to cytotoxicity assays against a representative panel of eight cancer cell lines and several molecules gave attractive results in this area.

Graphical Abstract
Introduction
Nitrogen-containing heterocyclic molecules are ubiquitous in living systems. Among them, quinazolines, and especially the 4-aminoquinazolines (type A molecules, Figure 1), are recognized as a “privileged scaffold” in bioorganic and medicinal chemistry [1]. On the other hand, the 2-anilinoquinazolines have been less studied but our groups have developed recently the synthesis of new molecules of this family and discovered a compound (DB18) with a potent inhibition (at 10–30 nM) of the CLK1, CLK2 and CLK4 kinases, and a remarkable selectivity since it does not show any action (at 100 μM) against the closely related DYRKs kinases [2,3]. Very recently, this skeleton proved to be also very useful in the research of antimicrobial agents [4], and the closely related dihydropteridinone derivatives were found as potent inhibitors of vaccinia-related kinase 1 and casein kinase 1δ/ε [5]. In addition, it is worth mentioning that closely related compounds like 2-(arylamino)benzo[h]quinazolin-4-ones [6-8] and N2-arylbenzo[h]quinazoline-2,4-diamines [9-11] are well identified scaffolds for bioactive molecules. Thus, it appeared interesting to explore novel molecules with extended aromatic units around this basic quinazoline core and our first choice was N-arylbenzo[h]quinazoline-2-amines with the general structure C as indicated in Figure 1.
![[1860-5397-20-218-1]](/bjoc/content/figures/1860-5397-20-218-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Aminoquinazolines and our new target molecules.
Figure 1: Aminoquinazolines and our new target molecules.
Very limited studies have been devoted to the synthesis of this core structure and, to the best of our knowledge, only one molecule of this type was obtained in low yield as a secondary product during studies on the use of the Friedländer annulation reaction starting from naphthylamines [12]. Therefore, the goal of this paper is to describe a short and efficient synthesis for this type of novel skeleton and to prepare a focused library for these targets C. Further, we will report results on their cytotoxic activities.
Results and Discussion
Chemical synthesis
For the preparation of our targets, we selected a flexible strategy which should allow us to introduce the molecular diversity on the anilino moiety in the last step through a palladium-catalyzed reaction (Scheme 1).
![[1860-5397-20-218-i1]](/bjoc/content/inline/1860-5397-20-218-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the desired targets 4.
Scheme 1: Synthesis of the desired targets 4.
Therefore, we prepared first the core structure 3 in two steps from the commercially available fluoronaphthalene 1. Metallation, followed by trapping with DMF afforded the known aldehyde 2 in 65% yield [13]. Then, reaction with guanidinium carbonate in DMA at high temperature [12], gave the desired intermediate 2-aminobenzo[h]quinazoline (3). In a final step, classical Buchwald–Hartwig coupling [14-17] with bromobenzene under the conditions described recently [18], gave the first target 4a (R = H) in 69% yield. In a similar way, we prepared a designed library with eighteen additional members 4b to 4s (Figure 2) having in the different ortho, meta and para positions either methyl groups, halogens (F, Cl) or methoxy groups. We have also prepared compounds with two substituents or with three fluorine atoms. All these compounds have been obtained in fair yields (51–70%) and they have spectral and analytical data in agreement with the indicated structures, as given in the experimental section and Supporting Information File 1.
![[1860-5397-20-218-2]](/bjoc/content/figures/1860-5397-20-218-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Target molecules 4 prepared with the yields for the last step.
Figure 2: Target molecules 4 prepared with the yields for the last step.
Cytotoxicity studies
Next, we performed a cytotoxicity screening of the nineteen molecules 4a–s, at 25 μM, investigating seven representative cancer cell lines: hepatocellular carcinoma HuH-7, colorectal adenocarcinoma CaCo-2, breast carcinoma MDA-MB-231 and MDA-MB-468, colorectal carcinoma HCT-116, prostate carcinoma PC3 and breast carcinoma MCF7. In parallel, human skin fibroblasts were used as reference for non-tumor cells and roscovitine (ROSCO), doxorubicin (DOXO) and Taxol as positive controls. For details see: ref. [2] and Supporting Information File 1, Table S1.
Compounds 4a–f, 4h, 4i, 4k, 4l, 4o and 4q that induced a more than 30% decrease of cell population for at least one cell line were further investigated.
The determination of IC50 confirmed that, although most molecules have low to no activity, a few of them exhibited significant cytotoxicity against several cell lines (Table 1). This is the case in particular for 4a which has demonstrated low micromolar toxicities (IC50 from 1.7 to 6 μM) against HuH-7, Caco-2, MDA-MB-468 and HCT-116 cells, with low to no action, on the other cells. Similarly, the meta-fluoro analogue 4i is active only on Caco-2, MDA-MB-468, HCT-116 and MCF7 (IC50 from 2 to 6 μM) while the meta-methoxy 4f and ortho-fluoro 4h show significant toxicity only on Caco-2 (IC50 4.3 and 4.6 μM, respectively). Further, the para-methyl compound 4d acts on HCT-116 only (IC50 5 μM). On the other hand, all molecules exhibited no effect on normal human fibroblasts at the highest concentration tested (25 μM). For these more potent molecules, further studies will be required to rationalize their activities and selectivities and explore their mechanism(s) of action.
Table 1: Cytotoxic studies (IC50 determination) of selected quinazolines 4.a
| cells | HuH-7 | Caco-2 | MDA-MB-231 | MDA-MB-468 | HCT-116 | PC3 | MCF7 | Fibro |
| compounds | IC50 µM | IC50 µM | IC50 µM | IC50 µM | IC50 µM | IC50 µM | IC50 µM | IC50 µM |
| DMSO | NE | NE | NE | NE | NE | NE | NE | NE |
| roscovitine | 12 | 14 | 15 | 13 | 7 | 10 | 11 | 16 |
| doxorubicin | 0.03 | NE | 0.03 | 0.03 | 0.04 | 0.06 | 0.07 | NE |
| Taxol | 0.009 | 0.07 | 0.02 | 0.006 | 0.003 | 0.004 | 0.02 | NE |
| 4a |
1.7
(±0.4) |
4.7
(±1.3) |
29
(±5.5) |
6
(±2.0) |
4
(±0.4) |
34.5
(±4.5) |
13
(±3.6) |
NE |
| 4b | NE | NE | NE | NE | NE | NE | NE | NE |
| 4c | NE |
28
(±7.0) |
NE |
22
(±3.2) |
18
(±1.3) |
NE |
30
(±3.0) |
NE |
| 4d | NE | NE | NE |
31
(±6.0) |
5
(±1.9) |
30
(±8.5) |
NE | NE |
| 4e | NE |
20
(±4.1) |
25
(±2.3) |
25
(±4.1) |
26
(±11) |
NE |
22
(±4.6) |
NE |
| 4f |
23
(±3.9) |
4.3
(±1.7) |
NE |
33
(±6.9) |
10
(±1.0) |
26
(±6.0) |
NE | NE |
| 4h | NE |
4.6
(±1.6) |
NE | NE | NE | NE | NE | NE |
| 4i |
17
(±3.7) |
2
(±0.6) |
17
(±2.1) |
4
(±2.1) |
6
(±0.2) |
11
(±0.2) |
3
(±0.8) |
NE |
| 4k | NE | NE | NE | NE | NE | NE | NE | NE |
| 4l |
22
(±4.7) |
17
(±5.1) |
NE |
26
(±4.5) |
24
(±3.3) |
26
(±2.2) |
18
(±2.5) |
NE |
| 4o |
24
(±5.1) |
16
(±3.5) |
NE |
20
(±3.0) |
23
(±13) |
21
(±2.0) |
21
(±1.2) |
NE |
| 4q |
16
(±2.3) |
14
(±2.9) |
20
(±3.0) |
19
(±3.1) |
13
(±4.4) |
19
(±5.2) |
14
(±0.6) |
NE |
aIC50 determination of effects of quinazolines on seven representative tumor cell lines and normal human fibroblasts. IC50 (μM) were calculated from dose-response curves after 48 h exposure (mean of triplicates). NE: no effect.
Conclusion
In conclusion, we have designed a short and versatile strategy for the preparation of new N-arylbenzo[h]quinazoline-2-amines and it was exemplified through the preparation of a focused chemical library with 19 members. In addition, the cytotoxicity assays afforded interesting results demonstrating that the substitution on the anilino part can have significant effects on their bioactivity. Two molecules (4a and 4i) exhibited a relatively broad cytotoxicity on four cancer cell lines among the seven assayed. On the other hand, two others (4f and 4h) were active only on Caco-2, while another one (4d) only on HCT-116. Thus, the results obtained with these compounds indicate that such a novel heterocyclic scaffold, which has not been used earlier, should offer attractive potentialities in bioorganic and medicinal chemistry. Extension of these studies are ongoing in our laboratories and corresponding results will be reported in due course.
Experimental
Chemical synthesis
General information
All reactions were performed in heat gun-dried round-bottomed flasks under a dry argon or nitrogen atmosphere. Air and moisture-sensitive compounds were introduced via syringes or cannula, using standard inert atmosphere techniques. In addition, the gas stream was passed through glass cylinder filled with P2O5 to remove any traces of residual moisture. Reactions were monitored by thin-layer chromatography (TLC) using E. Merck silica gel plates and components were visualized by illumination with short wavelength UV light and/or staining (ninhydrin or basic KMnO4). All aldehydes were distilled right before use. All aryl bromides and other reagents were used as they were received from commercial suppliers, unless otherwise noted. THF and Et2O were dried over sodium-benzophenone and distilled prior to use.
1H NMR spectra were recorded at 300 and 400 MHz, and 13C NMR spectra at 75 and 100 MHz, in CDCl3 or DMSO-d6 using TMS (tetramethylsilane) as an internal standard. Multiplicity was tabulated using standard abbreviations: s for singlet, d for doublet, dd for doublet of doublets, t for triplet, q for quadruplet, ddd for doublet of doublets of doublets and m for multiplet (br means broad). When necessary, in particular in order to have better accuracy on small coupling constants, resolution in 1H NMR was enhanced using Traficante. All compounds were purified by flash column chromatography on silca gel unless otherwise noted.
Melting points were obtained using an Electrothermal IA9200 series digital apparatus with thin-wall glass tube used to hold mp samples for capillary mp determination. FTIR spectra were recorded on a Bruker Alpha apparatus with spectra further analysed with spectragryph 1.2.4 version. The absorption bands were reported in cm−1. High-resolution mass spectra were recorded in the Centre Régional de Mesures Physiques de l’Ouest, Rennes (CRMPO), on a Maxis 4G.
Representative synthesis: preparation of compound 4a
Step 1: Synthesis of 1-fluoro-2-naphthaldehyde (2): To a stirred solution of compound 1 (3.0 g, 0.02 mol) in THF (60 mL) was added n-butyllithium (1.6 M, 12.8 mL, 0.02 mol) at −78 °C. The mixture was stirred at −78 °C for 2 h, and then dry DMF (3.3 mL, 0.041 mol) was added. After stirring for 2 h at −78 °C, the mixture was quenched with water and the crude product was extracted with ethyl acetate (30 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford crude compound 2 as pale-yellow solid (65% yield). Its NMR data are in agreement with literature [5]. 1H NMR (400 MHz, DMSO-d6, δ ppm) 10.49 (s, 1H), 8.26 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.84–7.74 (m, 3H).
Step 2: Synthesis of benzo[h]quinazolin-2-amine (3): To a stirred solution of guanidinium carbonate (2.0 g, 0.011 mol) in DMA (15 mL) was added compound 2 (1.5 g, 0.008 mol). Then the reaction mixture was heated to stir at 150 °C for 16 h. After completion of the reaction (TLC), the mixture was poured into ice water, a solid was formed, filtered and dried to get compound 3 as a brown colored solid (63% yield). 1H NMR (400 MHz, DMSO-d6, δ ppm) 9.06 (s, 1H), 8.91 (d, J = 8.0 Hz, 1H), 7.93 (d, J = 7.6 Hz, 1H), 7.76–7.64 (m, 3H), 7.56 (d, J = 8.8 Hz, 1H), 6.99 (br s, 2H); 13C NMR (75 MHz, DMSO-d6, δ ppm) 162.18, 161.31, 152.03, 136.05, 130.07, 129.07, 128.36, 126.72, 124.51, 124.38, 122.67, 116.44; 1H-13C NMR ((300, 75) MHz, DMSO-d6, δ ppm) (9.09 161.23), (8.95 124.31), (7.95 128.17), (7.76 129.93), (7.69 126.77), (7.61 124.66), (7.58 122.55); FTIR (KBr 1%, cm−1) ν̃: 3440, 3316, 3192, 1625, 1611, 1598, 1571, 1496, 1471, 1460, 1410, 801, 763; HRESIMS (m/z): [M + H]+ calcd for C12H10N3,196.0869; found, 196.0872 (1 ppm); mp: 193–195 °C (lit. 194–196 °C [12]).
Step 3: Representative procedure for the preparation of compounds 4: To the stirred solution of benzo[h]quinazolin-2-amine (3a, 0.256 mmol, 1.0 equiv) in 1,4-dioxane (4 mL) was added bromobenzene (0.384 mmol, 1.5 equiv), Xanthphos (0.051 mmol, 0.2 equiv) and cesium carbonate (0.769 mmol, 3 equiv). This mixture was degassed for 15 minutes under N2 atmosphere. Pd2(dba)3 (0.0256 mmol, 0.1 equiv) was added and the reaction mixture was stirred for 16 h at 100 °C. After cooling to room temperature, the mixture was filtered through Celite and washed with EtOAc (2 × 10 mL). The filtrate was evaporated under reduced pressure and the crude product was purified by using 60–120 silica mesh column chromatography using 10–20% ethyl acetate in hexane as eluent afforded target compound 4a (69% yield).
Compound 4a: N-phenylbenzo[h]quinazolin-2-amine: 69% yield (off white solid). 1H NMR (400 MHz, DMSO-d6, δ ppm) 10.01 (br s, 1H, NH), 9.29 (s, 1H), 9.03 (dd, J = 7.2, 0.8 Hz, 1H), 8.07–8.01 (m, 3H), 7.84–7.72 (m, 3H), 7.43 (d, J = 2.0 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.05 (t, J = 6.8 Hz, 1H); 13C NMR (75 MHz, DMSO-d6, δ ppm) δ 161.2, 158.1, 151.2, 141.0, 136.1, 130.5, 129.2, 129.1, 128.6, 127.4, 124.5, 124.4, 124.3, 122.1, 119.3, 117.6; FTIR (KBr 1%, cm−1) ν̃: 3423, 3274, 1643, 1601, 1591, 1542, 1504, 1450, 1393, 1025, 993, 804, 750; HRESIMS (m/z): [M + H]+ calcd for C18H14N3, 272.1182; found, 272.1181 (0 ppm); mp: 195–197 °C.
Protocole ImPACell for cytotoxicity studies
Cell culture. Skin normal fibroblastic cells are purchased from Lonza (Basel, Switzerland), HuH-7, Caco-2, MDA-MB-231, HCT-116, PC3, MCF7 and NCI-H727 cancer cell lines were obtained from ATCC (American Type Culture Collection). Cells are grown at 37 °C, 5% CO2 in ATCC recommended media: DMEM for HuH-7, MDA-MB-231, MDA-MB-468 and fibroblast, EMEM for MCF7 and CaCo-2, McCoy’s for HCT-116 and RPMI for PC3 and NCI-H727. All culture media are supplemented by 10% of FBS, 1% of penicillin-streptomycin and 2 mM glutamine.
Cytotoxic assays: primary screen (unique concentration) and secondary screen (IC50)
Both studies have been performed following the protocols described in a previous paper, see ref. [2].
Supporting Information
| Supporting Information File 1: Supplementary Table S1; spectral and analytical data for compounds 4b–s as well as all copies of 1H and 13C NMR spectra of compounds 4. | ||
| Format: PDF | Size: 7.4 MB | Download |
Acknowledgements
We thank Chemveda Life Sciences for providing laboratory facility for carrying out these research experiments. This work has been performed also as part of the Indo-French “Joint Laboratory for Natural Products and Synthesis towards Affordable Health”. We thank CSIR, CNRS, IICT and University of Rennes for their support. We thank CRMPO (University of Rennes) for the mass spectra analysis.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Das, D.; Hong, J. Eur. J. Med. Chem. 2019, 170, 55–72. doi:10.1016/j.ejmech.2019.03.004
See for a recent review.
Return to citation in text: [1] -
Brahmaiah, D.; Kanaka Durga Bhavani, A.; Aparna, P.; Sampath Kumar, N.; Solhi, H.; Le Guevel, R.; Baratte, B.; Ruchaud, S.; Bach, S.; Singh Jadav, S.; Raji Reddy, C.; Roisnel, T.; Mosset, P.; Levoin, N.; Grée, R. Bioorg. Med. Chem. 2021, 31, 115962. doi:10.1016/j.bmc.2020.115962
Return to citation in text: [1] [2] [3] -
Brahmaiah, D.; Bhavani, A. K. D.; Aparna, P.; Kumar, N. S.; Solhi, H.; Le Guevel, R.; Baratte, B.; Robert, T.; Ruchaud, S.; Bach, S.; Jadav, S. S.; Reddy, C. R.; Mosset, P.; Gouault, N.; Levoin, N.; Grée, R. Molecules 2022, 27, 6149. doi:10.3390/molecules27196149
Return to citation in text: [1] -
Das, N.; Roy, J.; Patra, B.; Saunders, E.; Sarkar, D.; Goon, S.; Sinha, B. P.; Roy, S.; Roy, S.; Sarif, J.; Bandopadhyay, P.; Barik, S.; Mukherjee, S.; McNamara, N.; Varghese, S.; Simpson, K.; Baell, J.; McConville, M.; Ganguly, D.; Talukdar, A. Eur. J. Med. Chem. 2024, 269, 116256. doi:10.1016/j.ejmech.2024.116256
Return to citation in text: [1] -
de Souza Gama, F. H.; Dutra, L. A.; Hawgood, M.; Vinícius dos Reis, C.; Serafim, R. A. M.; Ferreira, M. A., Jr.; Teodoro, B. V. M.; Takarada, J. E.; Santiago, A. S.; Balourdas, D.-I.; Nilsson, A.-S.; Urien, B.; Almeida, V. M.; Gileadi, C.; Ramos, P. Z.; Salmazo, A.; Vasconcelos, S. N. S.; Cunha, M. R.; Mueller, S.; Knapp, S.; Massirer, K. B.; Elkins, J. M.; Gileadi, O.; Mascarello, A.; Lemmens, B. B. L. G.; Guimarães, C. R. W.; Azevedo, H.; Counago, R. M. J. Med. Chem. 2024, 67, 8609–8629. doi:10.1021/acs.jmedchem.3c02250
Return to citation in text: [1] [2] -
Alagarsamy, V.; Chitra, K.; Saravanan, G.; Solomon, V. R.; Sulthana, M. T.; Narendhar, B. Eur. J. Med. Chem. 2018, 151, 628–685. doi:10.1016/j.ejmech.2018.03.076
Return to citation in text: [1] -
Collet, J. W.; van der Nol, E. A.; Roose, T. R.; Maes, B. U. W.; Ruijter, E.; Orru, R. V. A. J. Org. Chem. 2020, 85, 7378–7385. doi:10.1021/acs.joc.0c00771
Return to citation in text: [1] -
Dong, Y.; Zhang, J.; Yang, J.; Yan, C.; Wu, Y. New J. Chem. 2021, 45, 15344–15349. doi:10.1039/d1nj03179a
Return to citation in text: [1] -
Sengupta, S. K.; Chatterjee, S.; Protopapa, H. K.; Modest, E. J. J. Org. Chem. 1972, 37, 1323–1328. doi:10.1021/jo00974a010
Return to citation in text: [1] -
Rosowsky, A.; Papathanasopoulos, N. J. Org. Chem. 1974, 39, 3293–3295. doi:10.1021/jo00936a033
Return to citation in text: [1] -
Khan, I.; Ibrar, A.; Abbas, N.; Saeed, A. Eur. J. Med. Chem. 2014, 76, 193–244. doi:10.1016/j.ejmech.2014.02.005
Return to citation in text: [1] -
Malinowski, Z.; Fornal, E.; Warpas, A.; Nowak, M. Monatsh. Chem. 2018, 149, 1999–2011. doi:10.1007/s00706-018-2268-x
Return to citation in text: [1] [2] [3] -
Leroux, F.; Mangano, G.; Schlosser, M. Eur. J. Org. Chem. 2005, 5049–5054. doi:10.1002/ejoc.200500514
Return to citation in text: [1] -
Paul, F.; Patt, J.; Hartwig, J. F. J. Am. Chem. Soc. 1994, 116, 5969–5970. doi:10.1021/ja00092a058
Return to citation in text: [1] -
Guram, A. S.; Buchwald, S. L. J. Am. Chem. Soc. 1994, 116, 7901–7902. doi:10.1021/ja00096a059
Return to citation in text: [1] -
Guram, A. S.; Rennels, R. A.; Buchwald, S. L. Angew. Chem., Int. Ed. Engl. 1995, 34, 1348–1350. doi:10.1002/anie.199513481
Return to citation in text: [1] -
Louie, J.; Hartwig, J. F. Tetrahedron Lett. 1995, 36, 3609–3612. doi:10.1016/0040-4039(95)00605-c
Return to citation in text: [1] -
Xiang, J.; Wang, Y.; Wang, W.; Yu, J.; Zheng, L.; Hong, Y.; Shi, L.; Zhang, C.; Chen, N.; Xu, J.; Gong, X.; Zhang, Z.; Cui, H.; Zhou, Q.; Zhang, D.; Liu, Y.; Ke, Y.; Shen, J.; Xia, G.; Bai, X. Bioorg. Chem. 2023, 140, 106765. doi:10.1016/j.bioorg.2023.106765
Return to citation in text: [1]
| 1. |
Das, D.; Hong, J. Eur. J. Med. Chem. 2019, 170, 55–72. doi:10.1016/j.ejmech.2019.03.004
See for a recent review. |
| 6. | Alagarsamy, V.; Chitra, K.; Saravanan, G.; Solomon, V. R.; Sulthana, M. T.; Narendhar, B. Eur. J. Med. Chem. 2018, 151, 628–685. doi:10.1016/j.ejmech.2018.03.076 |
| 7. | Collet, J. W.; van der Nol, E. A.; Roose, T. R.; Maes, B. U. W.; Ruijter, E.; Orru, R. V. A. J. Org. Chem. 2020, 85, 7378–7385. doi:10.1021/acs.joc.0c00771 |
| 8. | Dong, Y.; Zhang, J.; Yang, J.; Yan, C.; Wu, Y. New J. Chem. 2021, 45, 15344–15349. doi:10.1039/d1nj03179a |
| 2. | Brahmaiah, D.; Kanaka Durga Bhavani, A.; Aparna, P.; Sampath Kumar, N.; Solhi, H.; Le Guevel, R.; Baratte, B.; Ruchaud, S.; Bach, S.; Singh Jadav, S.; Raji Reddy, C.; Roisnel, T.; Mosset, P.; Levoin, N.; Grée, R. Bioorg. Med. Chem. 2021, 31, 115962. doi:10.1016/j.bmc.2020.115962 |
| 5. | de Souza Gama, F. H.; Dutra, L. A.; Hawgood, M.; Vinícius dos Reis, C.; Serafim, R. A. M.; Ferreira, M. A., Jr.; Teodoro, B. V. M.; Takarada, J. E.; Santiago, A. S.; Balourdas, D.-I.; Nilsson, A.-S.; Urien, B.; Almeida, V. M.; Gileadi, C.; Ramos, P. Z.; Salmazo, A.; Vasconcelos, S. N. S.; Cunha, M. R.; Mueller, S.; Knapp, S.; Massirer, K. B.; Elkins, J. M.; Gileadi, O.; Mascarello, A.; Lemmens, B. B. L. G.; Guimarães, C. R. W.; Azevedo, H.; Counago, R. M. J. Med. Chem. 2024, 67, 8609–8629. doi:10.1021/acs.jmedchem.3c02250 |
| 4. | Das, N.; Roy, J.; Patra, B.; Saunders, E.; Sarkar, D.; Goon, S.; Sinha, B. P.; Roy, S.; Roy, S.; Sarif, J.; Bandopadhyay, P.; Barik, S.; Mukherjee, S.; McNamara, N.; Varghese, S.; Simpson, K.; Baell, J.; McConville, M.; Ganguly, D.; Talukdar, A. Eur. J. Med. Chem. 2024, 269, 116256. doi:10.1016/j.ejmech.2024.116256 |
| 5. | de Souza Gama, F. H.; Dutra, L. A.; Hawgood, M.; Vinícius dos Reis, C.; Serafim, R. A. M.; Ferreira, M. A., Jr.; Teodoro, B. V. M.; Takarada, J. E.; Santiago, A. S.; Balourdas, D.-I.; Nilsson, A.-S.; Urien, B.; Almeida, V. M.; Gileadi, C.; Ramos, P. Z.; Salmazo, A.; Vasconcelos, S. N. S.; Cunha, M. R.; Mueller, S.; Knapp, S.; Massirer, K. B.; Elkins, J. M.; Gileadi, O.; Mascarello, A.; Lemmens, B. B. L. G.; Guimarães, C. R. W.; Azevedo, H.; Counago, R. M. J. Med. Chem. 2024, 67, 8609–8629. doi:10.1021/acs.jmedchem.3c02250 |
| 2. | Brahmaiah, D.; Kanaka Durga Bhavani, A.; Aparna, P.; Sampath Kumar, N.; Solhi, H.; Le Guevel, R.; Baratte, B.; Ruchaud, S.; Bach, S.; Singh Jadav, S.; Raji Reddy, C.; Roisnel, T.; Mosset, P.; Levoin, N.; Grée, R. Bioorg. Med. Chem. 2021, 31, 115962. doi:10.1016/j.bmc.2020.115962 |
| 3. | Brahmaiah, D.; Bhavani, A. K. D.; Aparna, P.; Kumar, N. S.; Solhi, H.; Le Guevel, R.; Baratte, B.; Robert, T.; Ruchaud, S.; Bach, S.; Jadav, S. S.; Reddy, C. R.; Mosset, P.; Gouault, N.; Levoin, N.; Grée, R. Molecules 2022, 27, 6149. doi:10.3390/molecules27196149 |
| 12. | Malinowski, Z.; Fornal, E.; Warpas, A.; Nowak, M. Monatsh. Chem. 2018, 149, 1999–2011. doi:10.1007/s00706-018-2268-x |
| 12. | Malinowski, Z.; Fornal, E.; Warpas, A.; Nowak, M. Monatsh. Chem. 2018, 149, 1999–2011. doi:10.1007/s00706-018-2268-x |
| 18. | Xiang, J.; Wang, Y.; Wang, W.; Yu, J.; Zheng, L.; Hong, Y.; Shi, L.; Zhang, C.; Chen, N.; Xu, J.; Gong, X.; Zhang, Z.; Cui, H.; Zhou, Q.; Zhang, D.; Liu, Y.; Ke, Y.; Shen, J.; Xia, G.; Bai, X. Bioorg. Chem. 2023, 140, 106765. doi:10.1016/j.bioorg.2023.106765 |
| 13. | Leroux, F.; Mangano, G.; Schlosser, M. Eur. J. Org. Chem. 2005, 5049–5054. doi:10.1002/ejoc.200500514 |
| 2. | Brahmaiah, D.; Kanaka Durga Bhavani, A.; Aparna, P.; Sampath Kumar, N.; Solhi, H.; Le Guevel, R.; Baratte, B.; Ruchaud, S.; Bach, S.; Singh Jadav, S.; Raji Reddy, C.; Roisnel, T.; Mosset, P.; Levoin, N.; Grée, R. Bioorg. Med. Chem. 2021, 31, 115962. doi:10.1016/j.bmc.2020.115962 |
| 12. | Malinowski, Z.; Fornal, E.; Warpas, A.; Nowak, M. Monatsh. Chem. 2018, 149, 1999–2011. doi:10.1007/s00706-018-2268-x |
| 9. | Sengupta, S. K.; Chatterjee, S.; Protopapa, H. K.; Modest, E. J. J. Org. Chem. 1972, 37, 1323–1328. doi:10.1021/jo00974a010 |
| 10. | Rosowsky, A.; Papathanasopoulos, N. J. Org. Chem. 1974, 39, 3293–3295. doi:10.1021/jo00936a033 |
| 11. | Khan, I.; Ibrar, A.; Abbas, N.; Saeed, A. Eur. J. Med. Chem. 2014, 76, 193–244. doi:10.1016/j.ejmech.2014.02.005 |
| 14. | Paul, F.; Patt, J.; Hartwig, J. F. J. Am. Chem. Soc. 1994, 116, 5969–5970. doi:10.1021/ja00092a058 |
| 15. | Guram, A. S.; Buchwald, S. L. J. Am. Chem. Soc. 1994, 116, 7901–7902. doi:10.1021/ja00096a059 |
| 16. | Guram, A. S.; Rennels, R. A.; Buchwald, S. L. Angew. Chem., Int. Ed. Engl. 1995, 34, 1348–1350. doi:10.1002/anie.199513481 |
| 17. | Louie, J.; Hartwig, J. F. Tetrahedron Lett. 1995, 36, 3609–3612. doi:10.1016/0040-4039(95)00605-c |
© 2024 Veeraiah et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.