Abstract
Nitrogen-containing heterocyclic compounds are widely used in pharmacology due to their pronounced biological activities and low toxicities. The introduction of a hydroxy function into uracil and pyridine molecules has led to compounds with antioxidant, anti-inflammatory, and immunomodulatory activity (3-hydroxy-6-methyl-2-ethylpyridine, 5-hydroxy-6-methyluracil, etc.). One of the successful methods for hydroxylation is peroxydisulfate oxidation. By modifying the Elbs reaction through catalysis and the introduction of additional oxidants, we have been able to significantly increase the yields of practically useful compounds.

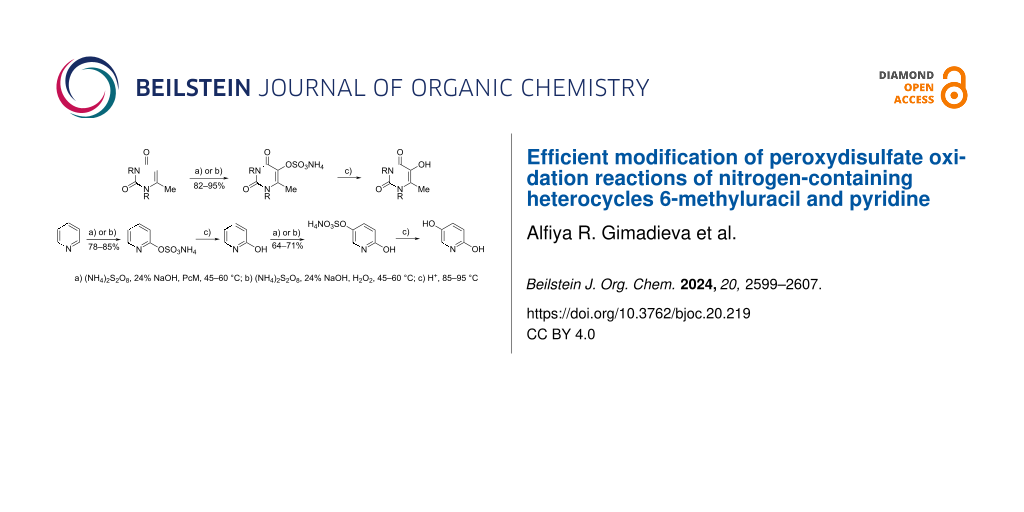
Graphical Abstract
Introduction
The Elbs and Boyland–Sims peroxydisulfate oxidation reactions offer a convenient means of introducing the hydroxy function into phenols and aromatic amines [1]. The oxidation of phenol using peroxydisulfate was first demonstrated by Karl Elbs in 1893 [2], with E. Boyland later expanding this reaction to include aromatic amines [3]. Concurrently, the successful oxidation of several pyrimidine derivatives was also reported [4]. Since then, the reaction has been extensively researched on various classes of compounds such as phenols, coumarins, pyridines, pyrimidines, quinolines, and others. This has resulted in the production of numerous valuable products. The reaction's appeal lies in its simplicity and the fact that there is no requirement to protect sensitive functional groups, thus making the introduction of hydroxy groups into various compound classes an attractive option. However, both reactions suffer from a significant downside – low yields of target products, rendering them unsuitable for industrial application.
Considering the practical value of the products, as well as the simplicity and convenience of the process, our objective is to enhance the efficiency of the Elbs and Boyland–Sims peroxydisulfate oxidation reactions.
Results and Discussion
We conducted research to enhance the product yield of peroxydisulfate oxidation reactions of specific nitrogen-containing heterocyclic compounds, such as 6-methyluracil (MU), 1,3,6-trimethyluracil (TMU), and pyridine (Py), and the results of the experiments are presented in this article. The hydroxy derivatives of MU and Py, obtained through oxidation followed by acid hydrolysis, possess compelling biological properties, rendering them practically useful. 5-Hydroxy-6-methyluracil (HMU) (Figure 1a) and 5-hydroxy-1,3,6-trimethyluracil (HTMU) (Figure 1b) have been identified as effective antioxidants and radical traps [5-7]. Additionally, 2-hydroxypyridine (HPy) has significant synthetic potential in the design of various bioactive compounds, including drugs and alkaloids [8]. Several drugs are known to contain the 2-pyridone structure, such as the cardiotonic drugs milrinone (Figure 1c) and amrinone (Figure 1d) [9,10], as well as the antibiotic pilicide (Figure 1e) [11,12].
![[1860-5397-20-219-1]](/bjoc/content/figures/1860-5397-20-219-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Derivatives of 6-methyluracil and 2-hydroxypyridine demonstrating pharmacological activity: 5-hydroxy-6-methyluracil (a), 5-hydroxy-1,3,6-trimethyluracil (b), milrinone (c), amrinone (d), and pilicide (e).
Figure 1: Derivatives of 6-methyluracil and 2-hydroxypyridine demonstrating pharmacological activity: 5-hydro...
As described in [13] during preliminary experiments, it was determined that room temperature is inadequate for fully oxidizing substrates. The optimal oxidation temperature for uracils 1 and 4 is between 60–65 °C, whereas for pyridines 7 and 9 it is 45 °C. Heating the reaction mixture at a temperature higher than the optimum level causes the substrates to be overoxidized and leads to destruction of the heterocyclic ring. As a result, the yield of the final products decreases. Furthermore, oxidation is incomplete at temperatures lower than the optimal temperature.
The oxidation of MU (1), TMU (4), and pyridine (7) using ammonium peroxydisulfate (APS) was conducted in two different ways: with metallophthalocyanine catalysts present and by including hydrogen peroxide as a co-oxidant (Scheme 1).
![[1860-5397-20-219-i1]](/bjoc/content/inline/1860-5397-20-219-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Peroxydisulfate oxidation of 6-methyluracil and 1,3,6-trimethyluracil. Сonditions: a) (NH4)2S2O8, 24% NaOH, 60 °C, PcM; b) (NH4)2S2O8, 24% NaOH, 60 °C, H2O2; c) H2SO4, 80 °C.
Scheme 1: Peroxydisulfate oxidation of 6-methyluracil and 1,3,6-trimethyluracil. Сonditions: a) (NH4)2S2O8, 2...
Metal–phthalocyanine complexes (PcM) are recognized as catalysts for gentle, particular oxidation reactions under aerobic [14] and H2O2-based conditions [15-20]. The catalytic activity of metallophthalocyanines originates from their planar, cyclic structure with a developed π-conjugation system. This makes the fifth and sixth coordination sites of the central metal ion available for coordination with the reactant molecules of the catalytic reaction. Additionally, the π-conjugation system facilitates the redistribution of electron density within the reaction complex, thereby lowering the activation barrier of the reaction [21]. The study employed the following oxidation catalysts for phthalocyanines – PcCo, PcFe(II), PcFe(III), PcMn, PcNi, and PcZn.
The use of catalysts reduced the duration of the oxidation reaction and significantly increased the yield of sulfate derivatives.
In the presence of РсМ, the optimal duration for the oxidation reaction of MU (1) was found to be 4 hours. When the catalyst was not applied, the yield of MU-5-ammonium sulfate 2 was no more than 15%. Subsequently, upon acid hydrolysis of compound 2, HMU (3) was produced. The addition of PcM to the reaction mixture resulted in a significant increase in the yield of compound 2 [13,22]. The catalysts were added in quantities ranging from 0.00001–0.1 wt %, with the amount of catalyst increasing by a factor of 10 in each successive experiment.
As described in [13] PcFe(II), PcСo, and PcFe(III) exhibited the highest activity in oxidizing reactions of MU (1). Addition of these catalysts in the amount of 0.01–0.05 wt % increased the yield of MU-5-ammonium sulfate 2 to 82–95%. The maximum yield of compound 2, equal to 95%, was obtained when 0.05 wt % PcFe(II) was introduced into the reaction. However, on enhancing the catalyst's quantity to 0.1 wt %, the product yield decreased to 33–45%. Further increase in the quantity of catalyst led to a greater decline in the yield of MU-5-ammonium sulfate, probably due to the destruction of the pyrimidine ring. PcZn turned out to be the least active catalyst, resulting in a yield of MU-5-ammonium sulfate 2 of 72% (0.05 wt %). The catalysts’ activity decreased in the order of PcFe(II) > PcCo > PcFe(III) > PcMn > PcNi > PcZn.
The catalytic oxidation of TMU (4, Table 1) displayed a consistent pattern. When 0.01–0.05 wt % of PcСo and PcFe(II) were added to the reaction, a maximum yield of 83–85% of TMU-5-ammonium sulfate 5 was obtained. The catalysts exhibited varying degrees of activity in the following order: PcSo > PcFe(II) > PcFe(III) > PcMn > PcNi > PcZn.
Table 1: Yield dependence of 1,3,6-trimethyluracil-5-ammonium sulfate (5) on the amount and type of catalyst.a
| Amount of catalyst, wt % | Yield of 5, % | |||||
| PcСо | PcFe(II) | PcFe(III) | PcMn | PcNi | PcZn | |
| 0 | 19 | |||||
| 0.001 | 60 | 66 | 60 | 60 | 60 | 48 |
| 0.005 | 67 | 72 | 68 | 66 | 67 | 56 |
| 0.01 | 85 | 83 | 80 | 71 | 65 | 59 |
| 0.02 | 85 | 83 | 81 | 72 | 66 | 61 |
| 0.05 | 85 | 85 | 81 | 72 | 67 | 63 |
| 0.1 | 56 | 55 | 47 | 40 | 39 | 35 |
| 0.2 | 42 | 37 | 35 | 24 | 28 | 27 |
aMole ratio TMU/NaOH/APS 1:4:1.5; 55 °C; 8 h.
In previous studies, various pyridine derivatives such as 2-pyridone, its derivatives, and 4-pyridone [23-28], were subjected to the Elbs peroxydisulfate oxidation, resulting in yields of up to 38% for the corresponding 5-hydroxy derivatives (2,5-dihydroxy derivatives). Our study marks the first time pyridine has been involved in the peroxydisulfate oxidation reaction.
The oxidation of pyridine (7) using APS resulted in a single product – pyridin-2-yl ammonium sulfate (8). Upon acid hydrolysis, this product yielded HPy (9, Scheme 2). The physicochemical and spectral properties of HPy (9) are in agreement with those reported in the literature [29].
![[1860-5397-20-219-i2]](/bjoc/content/inline/1860-5397-20-219-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Peroxydisulfate oxidation of pyridine and 2-hydroxypyridine. Сonditions: a) (NH4)2S2O8, 24% NaOH, 45 °C, PcM; b) (NH4)2S2O8, 24% NaOH, 45 °C, H2O2; c) HCl, 85–95 °C.
Scheme 2: Peroxydisulfate oxidation of pyridine and 2-hydroxypyridine. Сonditions: a) (NH4)2S2O8, 24% NaOH, 4...
The most effective catalyst for the pyridine oxidation was found to be РсСо. Its addition to the reaction in the amount of 0.1–0.3 wt % increased the yield of pyridin-2-yl ammonium sulfate (8) up to 78–81% (Table 2) [30].
Table 2: Yield dependence of pyridine-2-ammonium sulfate (8)a and 2,5-dihydroxypyridine (11)b on the amount and type of catalyst.
| Amount of catalyst, wt % | Yield of 8/11, % | |||||
| PcСо | PcFe(II) | PcFe(III) | PcMn | PcNi | PcZn | |
| 0 | 19/19 | |||||
| 0.01 | 55/43 | 36/39 | 37/37 | 23/30 | 26/38 | 21/23 |
| 0.05 | 67/60 | 45/54 | 48/51 | 45/45 | 38/47 | 33/35 |
| 0.1 | 78/71 | 55/62 | 57/60 | 45/49 | 49/53 | 42/47 |
| 0.15 | –/72 | –/64 | –/62 | –/52 | –/55 | –/49 |
| 0.2 | 81/71 | 57/63 | 60/60 | 50/50 | 51/53 | 47/49 |
| 0.3 | 81/– | 57/– | 58/– | 48/– | 52/– | 46/– |
aMole ratio Py/NaOH/APS 1:4:1.5, 45 °C; 10 h; bmole ratio HPy/NaOH/APS 1:4:2; 45 °C; 8 h.
It is notable that even when a large excess of APS was used, only the product of pyridine monohydroxylation, HPy (9), was isolated from the reaction mass, probably due to the deactivating effect of the OSO3− group on the aromatic ring. Furthermore, 2,5-dihydroxypyridine (11) – the pyridine dihydroxylation product – was only obtained after the oxidation of the previously synthesized HPy (9, Scheme 2). The overall yield of the 2,5-dihydroxy derivative 11 with PcCo, PcFe(II), and PcFe(III) ranged from 37–72%, with the highest yield (72%) obtained at 0.15 wt % PcCo. Increasing the catalyst quantity did not increase the yield of pyridine 11 further (Table 2).
Despite the numerous works in the field of peroxydisulfate oxidation, there is still no unified view of the reaction mechanism. Consequently, in [31], the assumption of an electrophilic substitution mechanism for the Elbs and Boyland–Sims reactions was made. It has been suggested that a nucleophilic substitution of the peroxide oxygen atom occurs in peroxydisulfate [32]. Regarding phenols (Elbs reaction), there is also a nucleophilic substitution of the phenolate ion. For aromatic amines (Boyland–Sims reaction), a neutral nitrogen atom of the amino group is involved in the formation of an intermediate hydroxylamine derivative.
Scheme 3 demonstrates a possible reaction mechanism using the example of the peroxydisulfate oxidation of MU and TMU catalyzed by PcM. It is proposed that PcM provides the necessary polarization of peroxydi(mono)sulfate ions, thus enabling activated A or B particles to attack the substrate molecule, resulting in the intermediate σ-complex C. A like particle B [Pc–Me–Oδ−–Oδ+–SO3−] has been previously described in [33]. The formation of particle B is possible via the interaction of the catalyst PcM with the SO52− ion, which is formed by the non-radical decomposition of the peroxydisulfate ion in a strongly alkaline medium [34]:
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-219-i4.svg?max-width=637&scale=1.18182)
![[1860-5397-20-219-i3]](/bjoc/content/inline/1860-5397-20-219-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Potential mechanism of peroxydisulfate oxidation of 6-methyluracil and 1,3,6-trimethyluracil.
Scheme 3: Potential mechanism of peroxydisulfate oxidation of 6-methyluracil and 1,3,6-trimethyluracil.
The Elbs and Boyland–Sims reactions were also effectively modified by the use of H2O2 as a co-oxidant (binary oxidation mixture APS/H2O2). Adding 2.0–2.3 equiv of H2O2 (Table 3) [35] resulted in the highest yield of MU-5-ammonium sulfate 2. In this case, the most effective duration for oxidizing MU were 8 hours, with incomplete conversion observed at shorter reaction times and a slight decrease in the yield of MU-5-ammonium sulfate at durations longer than 8 hours, potentially due to pyrimidine ring destruction. The ideal oxidation of TMU (5) was achieved with the addition of 3.0–4.0 equiv H2O2 (Table 3).
Table 3: Yield dependence of 6-methyluracil-5-ammonium sulfate (2)a and 1,3,6-trimethyluracil-5-ammonium sulfate (5)b on the amount of H2O2.
| Ratio MU/H2O2, mol | Yield of 2, % | Ratio TMU/H2O2, mol | Yield of 5, % |
| 1:0.85 | 60 | 1:1 | 34 |
| 1:1.7 | 75 | 1:1.5 | 35 |
| 1:1.91 | 78 | 1:2 | 56 |
| 1:2.1 | 88 | 1:3 | 71 |
| 1:2.31 | 85 | 1:4 | 71 |
aMole ratio MU/NaOH/APS 1:4:1.5; 60 °C; 8 h; bmole ratio TMU/NaOH/APS 1:4:1.5; 55 °C; 8 h.
During the oxidation of pyridine (7) by the binary oxidation mixture APS/H2O2 at 45 °C, the yield of the reaction product – Py-sulfate 8 – increased gradually, reaching a maximum of 85% after 10 h of heating [36]. It is essential to include 2–3 equiv of H2O2 to obtain the best result (Table 4).
Table 4: Yield dependence of pyridine-2-ammonium sulfate (8) and 2-hydroxypyridine (9) on the amount of H2O2.a
| Ratio pyridine/H2O2, mol | Yield, % | |
| 8 | 9 | |
| 1:0 | 19 | 17 |
| 1:0,5 | 50 | 45 |
| 1:1 | 68 | 61 |
| 1:2 | 85 | 77 |
| 1:3 | 85 | 77 |
aMole ratio Py/NaOH/APS 1:4:1.5; 45 °С; 10 h.
The analysis of the results obtained from Table 3 and Table 4 indicates that the binary oxidation system of APS/H2O2 consumes APS more slowly and therefore the substrates (MU, TMU, and Py) are oxidized more completely due to a more efficient consumption of the oxidant (APS). Previously, the oxidation of orotic acid was studied and it was found that the presence of oxygen in the reaction medium affects the yield of the sulfate derivative [37]. Under anaerobic conditions, a low yield of the product is observed with a high rate of APS disappearance, possibly due to side reactions [38]. To create the necessary aerobic conditions, we hypothesize that the inclusion of H2O2 in the binary oxidation mixture is favorable. This phenomenon occurs as a consequence of the reaction between H2O2 and the peroxydisulfate ion, leading to the production of oxygen [39]:
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-219-i5.svg?max-width=637&scale=1.18182)
Oxygen accumulation is additionally achieved by hydrogen peroxide self-decay, which is known to be increased in alkaline conditions [40]:
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-219-i6.svg?max-width=637&scale=1.18182)
Additionally, it has been reported [41] that hydroxyl radicals, produced from the decomposition of H2O2, can generate sulfate anion radicals (SO4•−) [42] during their interaction with sulfate anions. This reaction occurs at a significant rate (k = 3.1∙108 min−1) [43] and leads to the recombination of peroxydisulfate in the reaction mixture.
![[Graphic 4]](/bjoc/content/inline/1860-5397-20-219-i7.svg?max-width=637&scale=1.18182)
![[Graphic 5]](/bjoc/content/inline/1860-5397-20-219-i8.svg?max-width=637&scale=1.18182)
![[Graphic 6]](/bjoc/content/inline/1860-5397-20-219-i9.svg?max-width=637&scale=1.18182)
It has been demonstrated that the efficiency of peroxydisulfate oxidation of nitrogen-containing heterocyclic compounds can be enhanced by the introduction of phthalocyanine catalysts or hydrogen peroxide into the reaction mixture. This approach has the potential to significantly increase the yield of the target compounds.
Conclusion
In conclusion, it is noted that we have identified effective modifications of peroxydisulfate-mediated oxidation reactions that allow us to obtain hydroxylated nitrogen-containing heterocycles in high yields. Our laboratory is currently investigating the application of these modifications to the oxidation of compounds of different classes.
Experimental
1H and 13C NMR spectra were recorded on a Bruker Avance III 500 MHz spectrometer at 500.13 MHz (1H) and 125.73 MHz (13C) with 5 mm QNP sensors at a constant sample temperature of 298 K. The solvents were DMSO-d6, D2O, CDCl3 and the internal standard was SiMe4. Chemical shifts in the 13C and 1H NMR spectra are given in parts per million (ppm). Elemental analyses were performed on a CHNS Euro-EA 3000 automatic analyzer. Melting points were determined on combinated Boetius tables. IR spectra were obtained on an IR Prestige-21 Shimadzu spectrophotometer in KBr pellets.
Freshly distilled water, 6-methyluracil (99%, Chemical Line LLC, Russia), pyridine (analytically pure grade, Reakhim LLC, Russia), (NH4)2S2O8 (analytically pure grade, Panreac), NaOH (analytically pure grade, Reakhim LLC, Russia), H2SO4 (reagent grade, LLC Sigma Tech, Russia), dimethyl sulfate (99%, Chemical Line LLC, Russia), chloroform (reagent grade, JSC Khimreaktivsnab, Russia) and ethyl alcohol (reagent grade, LLC TD Khimmed, Russia) were used in this work. The phthalocyanine catalysts (Co, FeII, FeIII, Mn, Ni, and Zn phthalocyanines) were synthesized according to the known procedure [44] from phthalonitrile (pure grade, Merck) and crystalline hydrates of the corresponding metal salts: CoCl2·6H2O (pure grade, Reakhim LLC, Russia), Ni(NO3)2·6H2O (analytically pure grade, Reakhim LLC, Russia), Fe(NO3)3·9H2O (pure grade, Reakhim LLC, Russia), FeCl2·4H2O (analytically pure grade, Panreac), MnSO4·5H2O (analytically pure grade, Reakhim LLC, Russia), and ZnSO4·7H2O (analytically pure grade, LLC Reachim, Russia); 1,3,6-trimethyluracil was synthesized according to the method [45] from 6-methyluracil, dimethyl sulfate, and NaOH.
General procedure for the peroxydisulfate oxidation of uracils 1 and 4
a) Catalysis by РсМ. As described in [13] to a three-necked flask of 100 mL capacity, equipped with a mechanical stirrer, a thermometer, and a reflux condenser, containing 10 mL of distilled water, was added 0.023 mol of the corresponding uracil 1 or 4, followed by the addition of 11 mL of a previously prepared 24% NaOH solution to the obtained suspension. To the obtained thick mass 0.034 mol of (NH4)2S2O8 was added in portions under stirring, after which the calculated amount of the corresponding PcM was added. The reaction mixture was stirred at 60 °C for 4 h (for uracil 4–8 h). After cooling the reaction mixture to room temperature, concentrated H2SO4 was slowly added until pH 6–7 according to litmus paper and left standing for 12 h. The precipitated white crystals were filtered off, washed with water, and dried in air.
Compound 2 yields are given in [13] and compound 5 yields are given in Table 1.
b) With addition of H2O2. To a three-necked flask equipped with a mechanical stirrer, a thermometer, and a reflux condenser containing 10 mL of distilled water was added 0.023 mol of powdered uracil 1 or 4, followed by the addition of 11 mL of a previously prepared 24% NaOH solution to the obtained suspension. To the obtained thick mass 0.034 mol of (NH4)2S2O8 was added in portions under stirring. After complete addition of (NH4)2S2O8, 30% H2O2 (0.048 mol for uracil 1, 0.069 mol for uracil 4) was added. The reaction mixture was stirred at 60 °C for 8 h. After cooling the reaction mixture to room temperature, concentrated H2SO4 was slowly added until pH 6–7 according to litmus paper and left standing overnight (12 h). The precipitated crystals were filtered off, washed with water, acetone, and dried in air. The crude product was recrystallized from water with activated carbon. The yield of compound 2 was 88% and for compound 5 70%.
Preparation of compounds
6-Methyluracil-5-ammonium sulfate (2). The spectral characteristics are given in [13].
1,3,6-Trimethyluracil-5-ammonium sulfate (5). The spectral characteristics are given in [13].
General procedure for the hydrolysis of uracils 2 and 5. As described in [13] in a three-necked flask of 100 mL capacity, equipped with a mechanical stirrer, a reflux condenser, and a dropping funnel, 0.022 mol of compound 2 or 5 was placed, 30 mL of distilled water were added and the mixture heated to 80 °C under constant stirring. Then, 0.022 mol of concentrated H2SO4 was added and the reaction mixture was stirred at the same temperature for 1 h and cooled. The precipitated crystals were filtered off, washed with cold distilled water to pH 6–7 (2 × 5 mL), and recrystallized from ethanol.
5-Hydroxy-6-methyluracil (3). Yield 98%. The spectral characteristics are given in [13].
5-Hydroxy-1,3,6-trimethyluracil (6). Yield 88%. The spectral characteristics are given in [13].
2-Pyridinyl sulfate (8): a) In a 150 mL three-necked flask equipped with a reflux condenser and a mechanical stirrer, to a solution of 0.06 mol of pyridine in 20 mL of azeotropic solution of water and acetone (1:1) was slowly added to 20 mL of 24% NaOH solution. The reaction temperature was raised to 45 °C and a solution of 0.09 mol of (NH4)2S2O8 in 30 mL of water was added. After complete addition of (NH4)2S2O8, 0.01 wt % of the catalyst, PcCo, was added. The reaction mixture was stirred at 45 °C for 10 h, cooled to room temperature, evaporated at reduced pressure to 1/3 volume, extracted with ethyl acetate (2 × 20 mL) to remove unreacted pyridine, followd by butanol (3 × 50 mL). The butanol fractions were combined and evaporated to dryness. The residue was washed with hot ethanol, kept for 12 h at 0 °C, the precipitate was decanted, and the filtrate was evaporated to afford 6.33 g (55%) of 2-pyridinyl sulfate as a thick mass of dark brown color. By the same method 2-pyridinyl sulfate (8) was prepared in the presence of other catalysts – PcFe(II), PcFe(III), PcMn, PcNi, PcZn. The yields of compound 8 are given in Table 2.
b) In a 150 mL three-neck flask equipped with a reflux condenser, a mechanical stirrer, a solution of 0.06 mol of pyridine in 20 mL of azeotropic solution of water and acetone (1:1) was slowly added to 20 mL of 24% NaOH solution, the reaction temperature was raised to 45 °C and a solution of 0.09 mol of (NH4)2S2O8 in 30 mL of water was added. After complete addition of (NH4)2S2O8, 0.012 mol of a 30% H2O2 solution was added. The reaction mixture was stirred at 45 °C for 10 h, cooled to room temperature, evaporated at reduced pressure to 1/3 volume, extracted with ethyl acetate (2 × 20 mL) to remove unreacted pyridine, followed by butanol (3 × 50 mL). The butanol fractions were combined and evaporated to dryness. The residue was washed with hot ethanol, kept for 12 h at 0 °C, the precipitate was decanted, and the filtrate was evaporated to give 9.78 g (85%) of 2-pyridinyl sulfate as a thick mass of dark brown color.
Pyridine-2(1H)-one (9). 0.0146 mol of 2-pyridinyl sulfate (8) was dissolved in 30 mL of distilled water at 90 °C under stirring and after complete dissolution 0.0146 mol of HCl was added. The reaction mixture was heated for 3 h, monitored by TLC (eluent ethanol/ammonia aqueous 4:1), cooled to room temperature, alkalinized with NaHCO3 solution to pH 7–8, extracted with chloroform (3 × 10 mL), and the organic layer was dried with MgSO4. After removal of the solvent 1.17 g (95%) of pyridin-2(1H)-one (9) was obtained as a thick mass of light brown color. After recrystallization from ethanol, 1.11 g (90%) of pyridine 9 were obtained as a light yellow powder. Mp 108–110 °С; 1H NMR (CDCl3) δ 6.16 (dd, 1H, С5H), 6.38 (d, 1H, С3H), 7.38 (d, 1H, С6H), 7.40 (dd, 1H, С4H), 11.5 (s, 1H, C1-OH); 13С NMR (CDCl3) δ 104.8 (С4), 119.7 (С3), 135.2 (С6), 140.8 (С4), 162.3 (С5). The spectral data is in accordance with [29].
2,5-Dihydroxypyridine (11). 0.9 g (0.01 mol) of 2-hydroxypyridine (9) was dissolved in a solution of 1.56 g (0.028 mol) of NaOH in 25 mL of distilled water in a flat-bottomed flask fitted with a magnetic stirrer. The flask was placed in an ice bath for cooling and when the temperature of the reaction mixture reached 5 °C, 2.28 g (0.01 mol) of (NH4)2S2O8 in 10 mL of distilled water were added. When the reaction temperature reached 23–25 °C, 0.1 wt % PcCo was added. The reaction was kept at room temperature for 20 h. Then, 1.96 g (0.02 mol) of concentrated H2SO4 were added to the reaction mixture while stirring and the solution was gently boiled for 2 h. After cooling, 40% NaOH solution was added to the reaction mixture until pH 6 and extracted with chloroform (2 × 20 mL). The aqueous layer was evaporated to dryness and treated with hot ethanol. 0.9 g (50%) to obtain a reddish brown glassy solid. After recrystallization in ethanol, light beige crystals of 2,5-dihydroxypyridine (11) were obtained. Mp 107–109 °С; 1H NMR (CDCl3) δ 6.96 (1H, С3H), 7.16 (1H, С6H), 7.60 (1H, С4H), 9.76 (C5-OH), 10.78 (C2-OH); 13С NMR (CDCl3) δ106.2 (С3), 118.2 (С4), 128.4 (С6), 136.5 (С5), 141.4 (С2); Anal. calcd for C5H5NO2: C, 53.98; H, 4.67; N, 12.71; found: C, 54.06; H, 4.51; N, 12.62.
Supporting Information
| Supporting Information File 1: NMR spectra of compounds 2, 3, and 6. | ||
| Format: PDF | Size: 233.5 KB | Download |
Funding
This work was financially supported by the Ministry of Science and Higher Education of the Russian Federation (State Assignment No. 122031400278-2). Spectral experiments were carried out using the equipment of the Center for Collective Use "Chemistry" at the Ufa Institute of Chemistry at the Ufa Federal Research Centre of the Russian Academy of Sciences.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Behrman, E. J. Mini-Rev. Org. Chem. 2021, 18, 621–625. doi:10.2174/1570193x17999200813153655
Return to citation in text: [1] -
Elbs, K. J. Prakt. Chem. 1893, 48, 179–185. doi:10.1002/prac.18930480123
Return to citation in text: [1] -
Boyland, E.; Manson, D.; Sims, P. J. Chem. Soc. 1953, 3623. doi:10.1039/jr9530003623
Return to citation in text: [1] -
Hull, R. J. Chem. Soc. 1956, 2033. doi:10.1039/jr9560002033
Return to citation in text: [1] -
Krivonogov, V. P.; Tolstikov, G. A.; Murinov, Y. I.; Zarudii, F. S.; Lazareva, D. N.; Ismagilova, A. F.; Volkova, S. S.; Sakhautdinova, G. M.; Spirikhin, L. V.; Abdrakhmanov, I. B.; Krivonogova, I. I. Pharm. Chem. J. 1993, 27, 112–120. doi:10.1007/bf00781072
Return to citation in text: [1] -
Grabovskiy, S. A.; Abdrakhmanova, A. R.; Murinov, Y. I.; Kabal'nova, N. N. Curr. Org. Chem. 2009, 13, 1733–1736. doi:10.2174/138527209789578081
Return to citation in text: [1] -
Grabovskiy, S. A.; Murinov, Y. I.; Kabal’nova, N. N. Tetrahedron Lett. 2012, 53, 6025–6028. doi:10.1016/j.tetlet.2012.08.133
Return to citation in text: [1] -
Amer, M. M. K.; Aziz, M. A.; Shehab, W. S.; Abdellattif, M. H.; Mouneir, S. M. J. Saudi Chem. Soc. 2021, 25, 101259. doi:10.1016/j.jscs.2021.101259
Return to citation in text: [1] -
Pastelin, G.; Mendez, R.; Kabela, E.; Farah, A. Life Sci. 1983, 33, 1787–1796. doi:10.1016/0024-3205(83)90686-0
Return to citation in text: [1] -
Mirković, J. M.; Mijin, D. Ž.; Petrović, S. D. Hem. Ind. 2013, 67, 17–25. doi:10.2298/hemind120410057m
Return to citation in text: [1] -
Åberg, V.; Almqvist, F. Org. Biomol. Chem. 2007, 5, 1827–1834. doi:10.1039/b702397a
Return to citation in text: [1] -
Cegelski, L.; Pinkner, J. S.; Hammer, N. D.; Cusumano, C. K.; Hung, C. S.; Chorell, E.; Åberg, V.; Walker, J. N.; Seed, P. C.; Almqvist, F.; Chapman, M. R.; Hultgren, S. J. Nat. Chem. Biol. 2009, 5, 913–919. doi:10.1038/nchembio.242
Return to citation in text: [1] -
Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Pereira Monteiro, C. J.; Ferreira Faustino, M. A.; Pinho Morgado Silva Neves, M. d. G.; Quialheiro Simões, M. M.; Sanjust, E. Catalysts 2021, 11, 122. doi:10.3390/catal11010122
Return to citation in text: [1] -
Kluson, P.; Drobek, M.; Zsigmond, A.; Baranyi, J.; Bata, P.; Zarubova, S.; Kalaji, A. Appl. Catal., B 2009, 91, 605–609. doi:10.1016/j.apcatb.2009.06.033
Return to citation in text: [1] -
Sorokin, A.; Meunier, B.; Séris, J.-L. Science 1995, 268, 1163–1166. doi:10.1126/science.268.5214.1163
Return to citation in text: [1] -
Sorokin, A.; De Suzzoni-Dezard, S.; Poullain, D.; Noël, J.-P.; Meunier, B. J. Am. Chem. Soc. 1996, 118, 7410–7411. doi:10.1021/ja960177m
Return to citation in text: [1] -
Sorokin, A.; Fraisse, L.; Rabion, A.; Meunier, B. J. Mol. Catal. A: Chem. 1997, 117, 103–114. doi:10.1016/s1381-1169(96)00415-3
Return to citation in text: [1] -
Kruid, J.; Fogel, R.; Limson, J. Environ. Sci. Pollut. Res. 2018, 25, 32346–32357. doi:10.1007/s11356-018-3215-4
Return to citation in text: [1] -
Platonova, Y. B.; Morozov, A. S.; Burtsev, I. D.; Korostei, Y. S.; Ionidi, V. Y.; Romanovsky, B. V.; Tomilova, L. G. Mendeleev Commun. 2018, 28, 198–199. doi:10.1016/j.mencom.2018.03.030
Return to citation in text: [1] -
Enikolopyan, N. S.; Bogdanova, K. A.; Askarov, K. A. Russ. Chem. Rev. 1983, 52, 13–26. doi:10.1070/rc1983v052n01abeh002794
Return to citation in text: [1] -
Mustafin, A. G.; Gimadieva, A. R.; Khazimullina, Yu. Z.; Abdrakhmanov, I. B.; Bajmetov, B. Z. Catalytic method of producing 6-methyluracil-5-ammonium sulfate. Russ. Pat. RU2700422, Sept 17, 2019.
Return to citation in text: [1] -
íçLí, S. Tetrahedron 1990, 46, 2891–2902. doi:10.1016/s0040-4020(01)88381-2
Return to citation in text: [1] -
Hunt, A. H.; Mynderse, J. S.; Samlaska, S. K.; Fukuda, D. S.; Maciak, G. M.; Kirst, H. A.; Occolowitz, J. L.; Swartzendruber, J. K.; Jones, N. D. J. Antibiot. 1988, 41, 771–779. doi:10.7164/antibiotics.41.771
Return to citation in text: [1] -
Behrman, E. J. Chem. Cent. J. 2009, 3, 1. doi:10.1186/1752-153x-3-1
Return to citation in text: [1] -
Behrman, E. J. J. Chem. Res. 2014, 38, 121–122. doi:10.3184/174751914x13896383516701
Return to citation in text: [1] -
Nantka-Namirski, P.; Rykowski, A. Acta Pol. Pharm. 1972, 29, 225–229.
Return to citation in text: [1] -
Lorenz, P.; Hradecky, M.; Berger, M.; Bertrams, J.; Meyer, U.; Stintzing, F. C. Phytochem. Anal. 2010, 21, 234–245. doi:10.1002/pca.1190
Return to citation in text: [1] -
Forlani, L.; Cristoni, G.; Boga, C.; Todesco, P. E.; Del Vecchio, E.; Selva, S.; Monari, M. ARKIVOC 2002, No. xi, 198–215. doi:10.3998/ark.5550190.0003.b18
Return to citation in text: [1] [2] -
Gimadieva, A. R.; Khazimullina, Yu. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Method of producing pyridine-2(1H)-one. Russ. Pat. RU2784429, Nov 24, 2022.
Return to citation in text: [1] -
Behrman, E. J. Org. React. 1988, 35, 421–511. doi:10.1002/0471264180.or035.02
Return to citation in text: [1] -
Behrman, E. J. Beilstein J. Org. Chem. 2006, 2, 22. doi:10.1186/1860-5397-2-22
Return to citation in text: [1] -
Bressan, M.; d’Alessandro, N.; Liberatore, L.; Morvillo, A. Coord. Chem. Rev. 1999, 185–186, 385–402. doi:10.1016/s0010-8545(99)00024-7
Return to citation in text: [1] -
Singh, U. C.; Venkatarao, K. J. Inorg. Nucl. Chem. 1976, 38, 541–543. doi:10.1016/0022-1902(76)80300-4
Return to citation in text: [1] -
Mustafin, A. G.; Gimadieva, A. R.; Khazimullina, Yu. Z.; Abdrakhmanov, I. B.; Safiullin, R. L.; Bajmetov, B. Z. Method of producing 5-hydroxy-6-methyluracil. Russ. Pat. RU2700687, Sept 19, 2019.
Return to citation in text: [1] -
Gimadieva, A. R.; Khazimullina, Yu. Z.; Gilimkhanova, A. A.; Abdrakhmanov, I. B.; Mustafin, A. G. Method of producing pyridine-2(1H)-one. Russ. Pat. RU2818919, May 7, 2024.
Return to citation in text: [1] -
Behrman, E. J. J. Chem. Res., Synop. 2003, 702–703. doi:10.3184/030823403322862996
Return to citation in text: [1] -
Krivonogov, V. P.; Tolstikov, G. A.; Akhunov, I. R.; Kazakov, V. P.; Komissarov, V. D.; Murinov, Y. I. Pharm. Chem. J. 1997, 31, 663–666. doi:10.1007/bf02464252
Return to citation in text: [1] -
House, D. A. Chem. Rev. 1962, 62, 185–203. doi:10.1021/cr60217a001
Return to citation in text: [1] -
Yan, N.; Liu, F.; Liu, B.; Brusseau, M. L. Environ. Sci. Pollut. Res. 2018, 25, 32088–32095. doi:10.1007/s11356-018-3153-1
Return to citation in text: [1] -
Batoeva, A. A.; Sizykh, M. R.; Aseev, D. G. J. Sib. Fed. Univ., Chem. 2022, 15, 69–80. doi:10.17516/1998-2836-0272
Return to citation in text: [1] -
Antoniou, M. G.; de la Cruz, A. A.; Dionysiou, D. D. Appl. Catal., B 2010, 96, 290–298. doi:10.1016/j.apcatb.2010.02.013
Return to citation in text: [1] -
Neta, P.; Huie, R. E. J. Phys. Chem. 1986, 90, 4644–4648. doi:10.1021/j100410a035
Return to citation in text: [1] -
Aleksanyan, K. G.; Stokolos, O. A.; Zaitseva, Y. N.; Solodova, E. V.; Belysheva, D. A.; Botin, A. A. NefteGazoKhimia 2018, 3, 44.
Return to citation in text: [1] -
Zajac, M. A.; Zakrzewski, A. G.; Kowal, M. G.; Narayan, S. Synth. Commun. 2003, 33, 3291–3297. doi:10.1081/scc-120023986
Return to citation in text: [1]
| 41. | Batoeva, A. A.; Sizykh, M. R.; Aseev, D. G. J. Sib. Fed. Univ., Chem. 2022, 15, 69–80. doi:10.17516/1998-2836-0272 |
| 42. | Antoniou, M. G.; de la Cruz, A. A.; Dionysiou, D. D. Appl. Catal., B 2010, 96, 290–298. doi:10.1016/j.apcatb.2010.02.013 |
| 43. | Neta, P.; Huie, R. E. J. Phys. Chem. 1986, 90, 4644–4648. doi:10.1021/j100410a035 |
| 1. | Behrman, E. J. Mini-Rev. Org. Chem. 2021, 18, 621–625. doi:10.2174/1570193x17999200813153655 |
| 5. | Krivonogov, V. P.; Tolstikov, G. A.; Murinov, Y. I.; Zarudii, F. S.; Lazareva, D. N.; Ismagilova, A. F.; Volkova, S. S.; Sakhautdinova, G. M.; Spirikhin, L. V.; Abdrakhmanov, I. B.; Krivonogova, I. I. Pharm. Chem. J. 1993, 27, 112–120. doi:10.1007/bf00781072 |
| 6. | Grabovskiy, S. A.; Abdrakhmanova, A. R.; Murinov, Y. I.; Kabal'nova, N. N. Curr. Org. Chem. 2009, 13, 1733–1736. doi:10.2174/138527209789578081 |
| 7. | Grabovskiy, S. A.; Murinov, Y. I.; Kabal’nova, N. N. Tetrahedron Lett. 2012, 53, 6025–6028. doi:10.1016/j.tetlet.2012.08.133 |
| 23. | íçLí, S. Tetrahedron 1990, 46, 2891–2902. doi:10.1016/s0040-4020(01)88381-2 |
| 24. | Hunt, A. H.; Mynderse, J. S.; Samlaska, S. K.; Fukuda, D. S.; Maciak, G. M.; Kirst, H. A.; Occolowitz, J. L.; Swartzendruber, J. K.; Jones, N. D. J. Antibiot. 1988, 41, 771–779. doi:10.7164/antibiotics.41.771 |
| 25. | Behrman, E. J. Chem. Cent. J. 2009, 3, 1. doi:10.1186/1752-153x-3-1 |
| 26. | Behrman, E. J. J. Chem. Res. 2014, 38, 121–122. doi:10.3184/174751914x13896383516701 |
| 27. | Nantka-Namirski, P.; Rykowski, A. Acta Pol. Pharm. 1972, 29, 225–229. |
| 28. | Lorenz, P.; Hradecky, M.; Berger, M.; Bertrams, J.; Meyer, U.; Stintzing, F. C. Phytochem. Anal. 2010, 21, 234–245. doi:10.1002/pca.1190 |
| 13. | Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144 |
| 29. | Forlani, L.; Cristoni, G.; Boga, C.; Todesco, P. E.; Del Vecchio, E.; Selva, S.; Monari, M. ARKIVOC 2002, No. xi, 198–215. doi:10.3998/ark.5550190.0003.b18 |
| 13. | Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144 |
| 3. | Boyland, E.; Manson, D.; Sims, P. J. Chem. Soc. 1953, 3623. doi:10.1039/jr9530003623 |
| 13. | Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144 |
| 22. | Mustafin, A. G.; Gimadieva, A. R.; Khazimullina, Yu. Z.; Abdrakhmanov, I. B.; Bajmetov, B. Z. Catalytic method of producing 6-methyluracil-5-ammonium sulfate. Russ. Pat. RU2700422, Sept 17, 2019. |
| 13. | Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144 |
| 13. | Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144 |
| 13. | Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144 |
| 13. | Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144 |
| 15. | Kluson, P.; Drobek, M.; Zsigmond, A.; Baranyi, J.; Bata, P.; Zarubova, S.; Kalaji, A. Appl. Catal., B 2009, 91, 605–609. doi:10.1016/j.apcatb.2009.06.033 |
| 16. | Sorokin, A.; Meunier, B.; Séris, J.-L. Science 1995, 268, 1163–1166. doi:10.1126/science.268.5214.1163 |
| 17. | Sorokin, A.; De Suzzoni-Dezard, S.; Poullain, D.; Noël, J.-P.; Meunier, B. J. Am. Chem. Soc. 1996, 118, 7410–7411. doi:10.1021/ja960177m |
| 18. | Sorokin, A.; Fraisse, L.; Rabion, A.; Meunier, B. J. Mol. Catal. A: Chem. 1997, 117, 103–114. doi:10.1016/s1381-1169(96)00415-3 |
| 19. | Kruid, J.; Fogel, R.; Limson, J. Environ. Sci. Pollut. Res. 2018, 25, 32346–32357. doi:10.1007/s11356-018-3215-4 |
| 20. | Platonova, Y. B.; Morozov, A. S.; Burtsev, I. D.; Korostei, Y. S.; Ionidi, V. Y.; Romanovsky, B. V.; Tomilova, L. G. Mendeleev Commun. 2018, 28, 198–199. doi:10.1016/j.mencom.2018.03.030 |
| 13. | Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144 |
| 11. | Åberg, V.; Almqvist, F. Org. Biomol. Chem. 2007, 5, 1827–1834. doi:10.1039/b702397a |
| 12. | Cegelski, L.; Pinkner, J. S.; Hammer, N. D.; Cusumano, C. K.; Hung, C. S.; Chorell, E.; Åberg, V.; Walker, J. N.; Seed, P. C.; Almqvist, F.; Chapman, M. R.; Hultgren, S. J. Nat. Chem. Biol. 2009, 5, 913–919. doi:10.1038/nchembio.242 |
| 21. | Enikolopyan, N. S.; Bogdanova, K. A.; Askarov, K. A. Russ. Chem. Rev. 1983, 52, 13–26. doi:10.1070/rc1983v052n01abeh002794 |
| 13. | Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144 |
| 9. | Pastelin, G.; Mendez, R.; Kabela, E.; Farah, A. Life Sci. 1983, 33, 1787–1796. doi:10.1016/0024-3205(83)90686-0 |
| 10. | Mirković, J. M.; Mijin, D. Ž.; Petrović, S. D. Hem. Ind. 2013, 67, 17–25. doi:10.2298/hemind120410057m |
| 44. | Aleksanyan, K. G.; Stokolos, O. A.; Zaitseva, Y. N.; Solodova, E. V.; Belysheva, D. A.; Botin, A. A. NefteGazoKhimia 2018, 3, 44. |
| 8. | Amer, M. M. K.; Aziz, M. A.; Shehab, W. S.; Abdellattif, M. H.; Mouneir, S. M. J. Saudi Chem. Soc. 2021, 25, 101259. doi:10.1016/j.jscs.2021.101259 |
| 14. | Pereira Monteiro, C. J.; Ferreira Faustino, M. A.; Pinho Morgado Silva Neves, M. d. G.; Quialheiro Simões, M. M.; Sanjust, E. Catalysts 2021, 11, 122. doi:10.3390/catal11010122 |
| 45. | Zajac, M. A.; Zakrzewski, A. G.; Kowal, M. G.; Narayan, S. Synth. Commun. 2003, 33, 3291–3297. doi:10.1081/scc-120023986 |
| 32. | Behrman, E. J. Beilstein J. Org. Chem. 2006, 2, 22. doi:10.1186/1860-5397-2-22 |
| 30. | Gimadieva, A. R.; Khazimullina, Yu. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Method of producing pyridine-2(1H)-one. Russ. Pat. RU2784429, Nov 24, 2022. |
| 13. | Gimadieva, A. R.; Khazimullina, Y. Z.; Abdrakhmanov, I. B.; Mustafin, A. G. Russ. J. Appl. Chem. 2022, 95, 436–441. doi:10.1134/s1070427222030144 |
| 31. | Behrman, E. J. Org. React. 1988, 35, 421–511. doi:10.1002/0471264180.or035.02 |
| 29. | Forlani, L.; Cristoni, G.; Boga, C.; Todesco, P. E.; Del Vecchio, E.; Selva, S.; Monari, M. ARKIVOC 2002, No. xi, 198–215. doi:10.3998/ark.5550190.0003.b18 |
| 40. | Yan, N.; Liu, F.; Liu, B.; Brusseau, M. L. Environ. Sci. Pollut. Res. 2018, 25, 32088–32095. doi:10.1007/s11356-018-3153-1 |
| 37. | Behrman, E. J. J. Chem. Res., Synop. 2003, 702–703. doi:10.3184/030823403322862996 |
| 38. | Krivonogov, V. P.; Tolstikov, G. A.; Akhunov, I. R.; Kazakov, V. P.; Komissarov, V. D.; Murinov, Y. I. Pharm. Chem. J. 1997, 31, 663–666. doi:10.1007/bf02464252 |
| 35. | Mustafin, A. G.; Gimadieva, A. R.; Khazimullina, Yu. Z.; Abdrakhmanov, I. B.; Safiullin, R. L.; Bajmetov, B. Z. Method of producing 5-hydroxy-6-methyluracil. Russ. Pat. RU2700687, Sept 19, 2019. |
| 36. | Gimadieva, A. R.; Khazimullina, Yu. Z.; Gilimkhanova, A. A.; Abdrakhmanov, I. B.; Mustafin, A. G. Method of producing pyridine-2(1H)-one. Russ. Pat. RU2818919, May 7, 2024. |
| 33. | Bressan, M.; d’Alessandro, N.; Liberatore, L.; Morvillo, A. Coord. Chem. Rev. 1999, 185–186, 385–402. doi:10.1016/s0010-8545(99)00024-7 |
| 34. | Singh, U. C.; Venkatarao, K. J. Inorg. Nucl. Chem. 1976, 38, 541–543. doi:10.1016/0022-1902(76)80300-4 |
© 2024 Gimadieva et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.