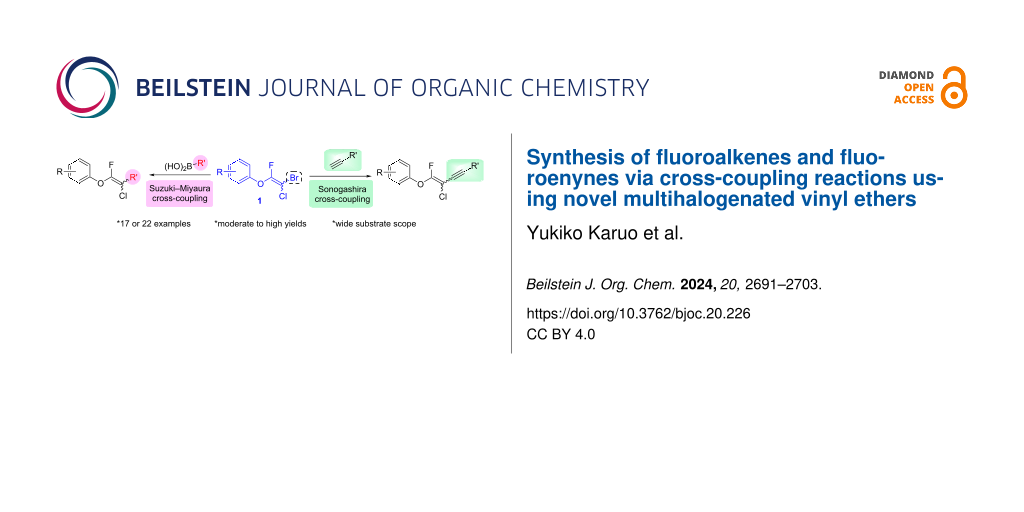
Abstract
In this study, we develop the synthesis methods of fluoroalkenes and fluoroenynes via Suzuki–Miyaura and Sonogashira cross-coupling reactions using novel multihalogenated fluorovinyl ethers, which are easily prepared from the reaction between phenols and 2-bromo-2-chloro-1,1,1-trifluoroethane (halothane). These reactions make use of the unique structure of multihalogenated fluorovinyl ethers, which contains a reactive bromine atom, to afford a series of fluoroalkenes and fluoroenynes in moderate to high yields.

Graphical Abstract
Introduction
Fluoroalkenes are one of the important frameworks for a wide range of industrial fields. For example, they are used as a bioisostere of amide bonds in medicines and agrochemicals, and contribute to the synthesis of peptide medicines that are stable to enzymatic metabolism and possess high lipophilicity [1]. In fact, several inhibitors of the β-site amyloyd β A4 precursor protein cleaving enzyme (BACE1), which is involved in the production of β-amyloid, and fluoroalkene analogs of dipeptidyl peptidase-4 inhibitors have previously been reported [2,3]. These inhibitors possess higher drug efficacies than their parent compounds. Furthermore, fluoroalkenes can be utilized as feedstock for fluoropolymers. Teflon, which is a well-known fluoropolymer with excellent water-repellent and oleophobic properties, is synthesized by polymerizing a monomer called tetrafluoroethylene. As a consequence, convenient and diverse synthetic methods for fluoroalkenes have attracted considerably and become increasingly necessary in pharmaceutical and industrial fields.
Fluoroalkenes have been constructed in a variety of methods [4-14], and one of the methods is to make use of fluorine-containing building blocks. When using them as nucleophilic reagents [15-20], the reaction between anion species, such as fluorine-containing Horner–Wadsworth–Emmons reagents, and carbonyl compounds led to E-selective olefination (Scheme 1A) [15]. On the other hand, some reactions with electrophilic fluorine-containing building blocks have been developed [21-25]. Jubault and Poisson et al. reported SN2’ reactions of hydride or alcohols to electrophilic fluorine-containing alkenes gave the corresponding fluoroalkenes (Scheme 1B) [21]. In recent years, many fluorine-containing coupling reagents have been developed. These reagents are easily being converted into multisubstituted fluoroalkenes through cross-coupling using palladium, nickel, copper, ruthenium, and manganese catalysts [26-41]. Hosoya and Niwa et al. published the development of a dual-reactive fluorine-containing C2-unit, which was prepared from trifluoroethanol in two steps in 63% yield, allowed the convergent synthesis of fluoroalkenes (Scheme 1C) [26]. We recently found multihalogenated vinyl ethers 1 could be obtained by the reaction of phenols with 2-bromo-2-chloro-1,1,1-trifluoroethane (halothane) in good yields (Scheme 1D) [42]. Compound 1 has a unique structure possessing three types of halogen atoms, namely bromine, chlorine, and fluorine, and it would be expected to afford multisubstituted fluoroalkenes by installing various substituents to bromine or chlorine atoms as reported by Hosoya and Niwa et al. In this study, we investigated the synthesis of fluoroalkenes 2 or fluoroenynes 3 by Suzuki–Miyaura or Sonogashira cross-couplings with a key building block 1 (Scheme 1D).
![[1860-5397-20-226-i1]](/bjoc/content/inline/1860-5397-20-226-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of monofluoroalkenes using fluorine-containing building blocks.
Scheme 1: Synthesis of monofluoroalkenes using fluorine-containing building blocks.
Results and Discussion
Optimization of the conditions of cross-coupling reactions
First, we optimized the conditions of the Suzuki–Miyaura cross-coupling in reference to the report by Yang et al. (Table 1) [43]. Upon the treatment of multihalogenated vinyl ether 1a with phenylboronic acid 4a (1.3 equiv) and palladium diacetate (10 mol %) as a catalyst at 40 °C, Suzuki–Miyaura cross-coupling proceeded to produce fluoroalkene 2a in 50% yield (Table 1, entry 1). Increasing the amount of 4a to 2.0 equiv and decreasing the amount of palladium diacetate to 5 mol % improved the reaction yield (Table 1, entry 2). When the reaction mixture was heated to 60 °C or reflux conditions, 2a could be synthesized in 84% yield under reflux conditions (Table 1, entries 3 and 4). Next, we examined an effective catalyst for the cross-coupling. Reactions using palladium dichloride or bis(2,4-pentanedionato)palladium significantly reduced the yields of 2a (Table 1, entries 5 and 6, respectively). When an allylpalladium chloride dimer or bis(triphenylphosphine)palladium dichloride were used as catalyst, the reaction proceeded with the same yield as that in Table 1, entry 4 (entries 7 and 8). Utilizing palladium catalyst such as bis(triphenylphosphine)palladium dichloride, all these reactions could convert 1a into 2a in good yields (Table 1, entries 9–11). Cross-coupling with palladium bis(trifluoroacetate), which is more reactive than palladium diacetate, gave the corresponding product in high yield of 96% (Table 1, entry 12). Without the addition of triphenylphosphine, the reaction proceeded in only 12% yield (Table 1, entry 13). Thus, it was concluded that triphenylphosphine is necessary for Suzuki–Miyaura cross-coupling of 1 with 4 and that it is involved in the production of palladium(0).
Table 1: Optimization of reaction conditions for Suzuki–Miyaura cross-coupling using multihalogenated vinyl ether 1a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-226-i2.svg?max-width=637&scale=1.0)
|
|||||
| Entry | 4a (equiv) | Pd cat. (mol %) | Temp. (°C) | Time (h) | 2a (%)a |
| 1 | 1.3 | Pd(OAc)2 (10) | 40 | 3.0 | 50 |
| 2 | 2.0 | Pd(OAc)2 (5) | 40 | 2.5 | 73 |
| 3 | 2.0 | Pd(OAc)2 (5) | 60 | 2.5 | 68 |
| 4 | 2.0 | Pd(OAc)2 (5) | reflux | 3.5 | 84 |
| 5 | 2.0 | PdCl2 (5) | reflux | 3.5 | 61 |
| 6 | 2.0 | Pd(acac)2 (5) | reflux | 3.5 | 12 |
| 7 | 2.0 | [Pd(allyl)Cl]2 (5) | reflux | 3.5 | 81 |
| 8 | 2.0 | Pd(PPh3)2Cl2 (5) | reflux | 3.5 | 84 |
| 9 | 2.0 | Pd[P(o-Tol)3]2Cl2 (5) | reflux | 3.5 | 89 |
| 10 | 2.0 | Pd(MeCN)2Cl2 (5) | reflux | 3.5 | 93 |
| 11 | 2.0 | Pd(PhCN)2Cl2 (5) | reflux | 3.5 | 92 |
| 12 | 2.0 | Pd(OCOCF3)2 (5) | reflux | 3.5 | 96 |
| 13b | 2.0 | Pd(OCOCF3)2 (5) | reflux | 3.5 | 12 |
aIsolated yields; bno PPh3.
Next, the reaction conditions for the Sonogashira cross-coupling were optimized (Table 2). On the basis of a previous study by Thorand [44], we performed the reaction between fluorine-containing vinyl ether 1a and 1.05 equiv of trimethylsilylacetylene (5a) to afford the corresponding enyne 3a in 55% yield (Table 2, entry 1). Cross-coupling utilizing a palladium(II) catalysts containing phosphine ligands produced low yields of 3a (Table 2, entries 2 and 3). In the case of palladium(II), which produced good yields of the Suzuki–Miyaura cross-coupling products, only a small amount of 3a was obtained (Table 2, entries 4–8). In particular, when the allylpalladium dichloride dimer was used, Sonogashira coupling hardly proceeded at all, and the starting ether 1a was recovered in an 83% yield (Table 2, entry 6). Zero-valent tetrakis(triphenylphosphine)palladium and tris(dibenzylideneacetone)dipalladium allowed the reaction to undergo in 37% or 23% yields, respectively (Table 2, entries 9 and 10). In entry 11, Table 2, we selected bis(triphenylphosphine)palladium as an effective catalyst, but increase of 5a to 1.5 equiv did not improve the reaction yield. Diluting the reaction concentration from 0.83 M to 0.2 M achieved to give 3a in a 63% yield (Table 2, entry 12). Increasing the amount of palladium catalyst to 4 mol % led to the conversion of 1a into 3a in 77% yield (Table 2, entry 13). In addition, using 2.0 equiv of 5a gave 3a in high 80% yield (Table 2, entry 14).
Table 2: Optimization of reaction conditions for Sonogashira cross-coupling using multihalogenated vinyl ether 1a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-226-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | 5a (equiv) | Pd cat. (mol %) | Time (h) | 3a (%)a |
| 1 | 1.05 | Pd(PPh3)2Cl2 (2) | 6.5 | 55 |
| 2 | 1.05 | Pd[P(o-Tol)3]2Cl2 (2) | 24 | 14 |
| 3 | 1.05 | Pd(PCy3)2Cl2 (2) | 24 | 0 |
| 4 | 1.05 | Pd(OAc)2 (2) | 20 | 6 |
| 5 | 1.05 | PdCl2 (2) | 24 | 13 |
| 6b | 1.05 | [Pd(allyl)Cl]2 (2) | 18.5 | 0 |
| 7 | 1.05 | Pd(MeCN)2Cl2 (2) | 25.5 | 13 |
| 8 | 1.05 | Pd(PhCN)2Cl2 (2) | 24 | 10 |
| 9 | 1.05 | Pd(PPh3)4 (2) | 21.5 | 37 |
| 10 | 1.05 | Pd2(dba)3 (2) | 24 | 23 |
| 11 | 1.5 | Pd(PPh3)2Cl2 (2) | 19 | 53 |
| 12c | 1.5 | Pd(PPh3)2Cl2 (2) | 17 | 63 |
| 13c | 1.5 | Pd(PPh3)2Cl2 (4) | 18 | 77 |
| 14c | 2.0 | Pd(PPh3)2Cl2 (4) | 19 | 80 |
aIsolated yields; bRecovery of 1a was 83% yield; cTHF (0.2 M) was used.
Based on these results, we determined entry 13 in Table 1 and entry 14 in Table 2 as the optimum reaction conditions. We used 1 as a mixture of diastereomers (diastereomer ratio = 1:1) for cross-coupling, and the corresponding compounds 2 and 3 were obtained as mixtures of diastereomers in a certain ratio as estimated by proton and fluorine NMR spectroscopy.
Substrate scope for cross-coupling reactions
The substrate scope was investigated using various boronic acids 4 and alkynes 5 in cross-coupling reactions using 1 (Table 3 and Table 4). p-Tolylboronic acid 4b provided 2b quantitatively, whereas m- and o-tolylboronic acids 4c and 4d produced 2c and 2d in low yields because the methyl group was positioned near the reaction site (Table 3, entries 1–3). Introduction of 3,4-methylenedioxyphenyl (4e) or p-fluorophenyl groups (4f) to 1a proceeded in high yields (Table 3, entries 4 and 5). Boronic acids with carbonyl groups such as acetyl, ester or formyl moieties in para position (4g–i) underwent the cross-coupling in 76, 96 or 77% yields (Table 3, entries 6–8). The reaction between 1a and 4j, which contains an electron-withdrawing nitro group, afforded 2j in 88% yield (Table 3, entry 9). Although p-hydroxyphenylboronic acid (4k) gave 2k in only 9% yield, m-aminophenylboronic acid (4l) provided 2l in high yield (Table 3, entries 10 and 11). We predicted that the product yield would decrease because 2k is labile in column chromatography. Utilizing a boronic acid bearing an n-butyl group as a primary alkyl group (4m), the cross-coupling did not proceed due to β-elimination (Table 3, entry 12). In contrast, the reaction with cyclopropylboronic acid (4n) achieved to give 2n in a 71% yield (Table 3, entry 13). When thiopheneboronic acid 4o was used as a coupling partner, the thiophene ring could be installed on 1a in a comparatively low yield of 31% (Table 3, entry 14). In addition, we investigated the substrate scope of 1 in the Suzuki–Miyaura cross-coupling. The reaction of 1b or 1c, which had a m-methoxy or p-nitro group on the benzene ring, with 4a proceeded smoothly to furnish 2p or 2q in good yieds (Table 3, entries 15 and 16). A phenyl group could be introduced into 1d possessing an ester moiety in moderate yield, whereas the cross-coupling between 1e, derived from m-aminophenol, and 4a proceeded in only 15% yield (Table 3, entries 17 and 18).
Table 3: Cross-coupling reactions between multihalogenated vinyl ethers 1 and various boronic acids 4.
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-226-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry |
1
(diastereomeric ratio) |
4 |
2
(diastereomeric ratio) |
Time
(h) |
Yield
(%)a |
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-20-226-i5.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 5]](/bjoc/content/inline/1860-5397-20-226-i6.svg?max-width=637&scale=1.0)
4b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-20-226-i7.svg?max-width=637&scale=1.0)
2b (1:1) |
3.5 | 98 |
| 2b |
![[Graphic 7]](/bjoc/content/inline/1860-5397-20-226-i8.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 8]](/bjoc/content/inline/1860-5397-20-226-i9.svg?max-width=637&scale=1.0)
4c |
![[Graphic 9]](/bjoc/content/inline/1860-5397-20-226-i10.svg?max-width=637&scale=1.0)
2c (1:1) |
2.5 | 26 |
| 3 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-20-226-i11.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 11]](/bjoc/content/inline/1860-5397-20-226-i12.svg?max-width=637&scale=1.0)
4d |
![[Graphic 12]](/bjoc/content/inline/1860-5397-20-226-i13.svg?max-width=637&scale=1.0)
2d (1:1) |
3.5 | 16 |
| 4 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-20-226-i14.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 14]](/bjoc/content/inline/1860-5397-20-226-i15.svg?max-width=637&scale=1.0)
4e |
![[Graphic 15]](/bjoc/content/inline/1860-5397-20-226-i16.svg?max-width=637&scale=1.0)
2e (1:1) |
3.5 | 85 |
| 5 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-20-226-i17.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 17]](/bjoc/content/inline/1860-5397-20-226-i18.svg?max-width=637&scale=1.0)
4f |
![[Graphic 18]](/bjoc/content/inline/1860-5397-20-226-i19.svg?max-width=637&scale=1.0)
2f (1:1) |
3.5 | 94 |
| 6 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-20-226-i20.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 20]](/bjoc/content/inline/1860-5397-20-226-i21.svg?max-width=637&scale=1.0)
4g |
![[Graphic 21]](/bjoc/content/inline/1860-5397-20-226-i22.svg?max-width=637&scale=1.0)
2g (1:1) |
2.5 | 76 |
| 7 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-20-226-i23.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 23]](/bjoc/content/inline/1860-5397-20-226-i24.svg?max-width=637&scale=1.0)
4h |
![[Graphic 24]](/bjoc/content/inline/1860-5397-20-226-i25.svg?max-width=637&scale=1.0)
2h (1:1) |
1.5 | 96 |
| 8 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-20-226-i26.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 26]](/bjoc/content/inline/1860-5397-20-226-i27.svg?max-width=637&scale=1.0)
4i |
![[Graphic 27]](/bjoc/content/inline/1860-5397-20-226-i28.svg?max-width=637&scale=1.0)
2i (1:1) |
3.5 | 77 |
| 9 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-20-226-i29.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 29]](/bjoc/content/inline/1860-5397-20-226-i30.svg?max-width=637&scale=1.0)
4j |
![[Graphic 30]](/bjoc/content/inline/1860-5397-20-226-i31.svg?max-width=637&scale=1.0)
2j (1:1:1) |
3.0 | 88 |
| 10 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-20-226-i32.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 32]](/bjoc/content/inline/1860-5397-20-226-i33.svg?max-width=637&scale=1.0)
4k |
![[Graphic 33]](/bjoc/content/inline/1860-5397-20-226-i34.svg?max-width=637&scale=1.0)
2k (1:1.4) |
6.5 | 9 |
| 11 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-20-226-i35.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 35]](/bjoc/content/inline/1860-5397-20-226-i36.svg?max-width=637&scale=1.0)
4l |
![[Graphic 36]](/bjoc/content/inline/1860-5397-20-226-i37.svg?max-width=637&scale=1.0)
2l (1:1) |
4.0 | 92 |
| 12 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-20-226-i38.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 38]](/bjoc/content/inline/1860-5397-20-226-i39.svg?max-width=637&scale=1.0)
4m |
![[Graphic 39]](/bjoc/content/inline/1860-5397-20-226-i40.svg?max-width=637&scale=1.0)
2m (1:1) |
3.5 | trace |
| 13 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-20-226-i41.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 41]](/bjoc/content/inline/1860-5397-20-226-i42.svg?max-width=637&scale=1.0)
4n |
![[Graphic 42]](/bjoc/content/inline/1860-5397-20-226-i43.svg?max-width=637&scale=1.0)
2n (1:1) |
5.0 | 71 |
| 14 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-20-226-i44.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 44]](/bjoc/content/inline/1860-5397-20-226-i45.svg?max-width=637&scale=1.0)
4o |
![[Graphic 45]](/bjoc/content/inline/1860-5397-20-226-i46.svg?max-width=637&scale=1.0)
2o (1:1.1) |
4.0 | 31 |
| 15 |
![[Graphic 46]](/bjoc/content/inline/1860-5397-20-226-i47.svg?max-width=637&scale=1.0)
1b (1:1) |
![[Graphic 47]](/bjoc/content/inline/1860-5397-20-226-i48.svg?max-width=637&scale=1.0)
4a |
![[Graphic 48]](/bjoc/content/inline/1860-5397-20-226-i49.svg?max-width=637&scale=1.0)
2p (1:1) |
3.5 | 62 |
| 16 |
![[Graphic 49]](/bjoc/content/inline/1860-5397-20-226-i50.svg?max-width=637&scale=1.0)
1c (1:1) |
![[Graphic 50]](/bjoc/content/inline/1860-5397-20-226-i51.svg?max-width=637&scale=1.0)
4a |
![[Graphic 51]](/bjoc/content/inline/1860-5397-20-226-i52.svg?max-width=637&scale=1.0)
2q (1:1) |
5.5 | 85 |
| 17 |
![[Graphic 52]](/bjoc/content/inline/1860-5397-20-226-i53.svg?max-width=637&scale=1.0)
1d (1:1) |
![[Graphic 53]](/bjoc/content/inline/1860-5397-20-226-i54.svg?max-width=637&scale=1.0)
4a |
![[Graphic 54]](/bjoc/content/inline/1860-5397-20-226-i55.svg?max-width=637&scale=1.0)
2r (1:1.6) |
3.5 | 45 |
| 18c |
![[Graphic 55]](/bjoc/content/inline/1860-5397-20-226-i56.svg?max-width=637&scale=1.0)
1e (1:1) |
![[Graphic 56]](/bjoc/content/inline/1860-5397-20-226-i57.svg?max-width=637&scale=1.0)
4a |
![[Graphic 57]](/bjoc/content/inline/1860-5397-20-226-i58.svg?max-width=637&scale=1.0)
2s (1:1.2) |
21.5 | 15 |
aIsolated yields; b1 (1.5 equiv) and 2 (1.0 equiv) were used; cDME was used as a solvent.
We performed Sonogashira cross-couplings between 1 and a variety of alkynes 5 (Table 4). Arylacetylenes, which have electron-donating substituents on the aromatic ring (5b–f), and 2-naphthylacetylene (5g) provided the corresponding enynes (3b–g) in 43–92% yields (Table 4, entries 1–6). On the contrary, electron-withdrawing substituents such as chloro, trifluoromethyl and nitro groups resulted in low cross-coupling yields (3h–j) (Table 4, entries 7–9). p-Acetyl or p-formylphenylacetylene (5k or 5l) could be introduced into 1a in 76% or 52% yields, respectively (Table 4, entries 10 and 11). Reactions using acetylenes possessing a hydroxy group, amino group and thiophene proceeded well (3m–o) (Table 4, entries 12–14). Hexa-1-yne 5p and cyclopropylacetylene (5q) afforded 3p and 3q, respectively, in high yields without byproduct formation (Table 4, entries 15 and 16). Enyne compound 5r and 3-butyn-1-ol 5s also participated in cross-coupling reactions and products 3r and 3s could be obtained in moderate yields of 52 and 53% (Table 4, entries 17 and 18). Then, we attempted the cross-coupling between 1 derived from various phenols and 5a. Vinyl ethers 1b–d were converted into enynes 3t–v in 29–35% yields (Table 4, entries 19–21). The reaction using 1e, which bears an amino group on the benzene ring, did not complete despite requiring a long reaction time (Table 4, entry 22).
Table 4: Cross-coupling reactions between multihalogenated vinyl ethers 1 and various alkynes 5.
![[Graphic 58]](/bjoc/content/inline/1860-5397-20-226-i59.svg?max-width=637&scale=1.0)
|
|||||
| Entry |
1
(diastereomeric ratio) |
5 |
3
(diastereomeric ratio) |
Time
(h) |
Yield
(%)a |
| 1 |
![[Graphic 59]](/bjoc/content/inline/1860-5397-20-226-i60.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 60]](/bjoc/content/inline/1860-5397-20-226-i61.svg?max-width=637&scale=1.0)
5b |
![[Graphic 61]](/bjoc/content/inline/1860-5397-20-226-i62.svg?max-width=637&scale=1.0)
3b (1:1.1) |
18.5 | 92 |
| 2b |
![[Graphic 62]](/bjoc/content/inline/1860-5397-20-226-i63.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 63]](/bjoc/content/inline/1860-5397-20-226-i64.svg?max-width=637&scale=1.0)
5c |
![[Graphic 64]](/bjoc/content/inline/1860-5397-20-226-i65.svg?max-width=637&scale=1.0)
3c (1:1.4) |
3.5 | 74 |
| 3b |
![[Graphic 65]](/bjoc/content/inline/1860-5397-20-226-i66.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 66]](/bjoc/content/inline/1860-5397-20-226-i67.svg?max-width=637&scale=1.0)
5d |
![[Graphic 67]](/bjoc/content/inline/1860-5397-20-226-i68.svg?max-width=637&scale=1.0)
3d (1:1.4) |
2.5 | 49 |
| 4b |
![[Graphic 68]](/bjoc/content/inline/1860-5397-20-226-i69.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 69]](/bjoc/content/inline/1860-5397-20-226-i70.svg?max-width=637&scale=1.0)
5e |
![[Graphic 70]](/bjoc/content/inline/1860-5397-20-226-i71.svg?max-width=637&scale=1.0)
3e (1:1.4) |
4.5 | 70 |
| 5 |
![[Graphic 71]](/bjoc/content/inline/1860-5397-20-226-i72.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 72]](/bjoc/content/inline/1860-5397-20-226-i73.svg?max-width=637&scale=1.0)
5f |
![[Graphic 73]](/bjoc/content/inline/1860-5397-20-226-i74.svg?max-width=637&scale=1.0)
3f (1:1.1) |
4.5 | 43 |
| 6 |
![[Graphic 74]](/bjoc/content/inline/1860-5397-20-226-i75.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 75]](/bjoc/content/inline/1860-5397-20-226-i76.svg?max-width=637&scale=1.0)
5g |
![[Graphic 76]](/bjoc/content/inline/1860-5397-20-226-i77.svg?max-width=637&scale=1.0)
3g (1:1.2) |
17.5 | 59 |
| 7c |
![[Graphic 77]](/bjoc/content/inline/1860-5397-20-226-i78.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 78]](/bjoc/content/inline/1860-5397-20-226-i79.svg?max-width=637&scale=1.0)
5h |
![[Graphic 79]](/bjoc/content/inline/1860-5397-20-226-i80.svg?max-width=637&scale=1.0)
3h (1:1) |
16 | 39 |
| 8 |
![[Graphic 80]](/bjoc/content/inline/1860-5397-20-226-i81.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 81]](/bjoc/content/inline/1860-5397-20-226-i82.svg?max-width=637&scale=1.0)
5i |
![[Graphic 82]](/bjoc/content/inline/1860-5397-20-226-i83.svg?max-width=637&scale=1.0)
3i (1:1.2) |
14 | 49 |
| 9 |
![[Graphic 83]](/bjoc/content/inline/1860-5397-20-226-i84.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 84]](/bjoc/content/inline/1860-5397-20-226-i85.svg?max-width=637&scale=1.0)
5j |
![[Graphic 85]](/bjoc/content/inline/1860-5397-20-226-i86.svg?max-width=637&scale=1.0)
3j (1:1.3) |
17 | 42 |
| 10 |
![[Graphic 86]](/bjoc/content/inline/1860-5397-20-226-i87.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 87]](/bjoc/content/inline/1860-5397-20-226-i88.svg?max-width=637&scale=1.0)
5k |
![[Graphic 88]](/bjoc/content/inline/1860-5397-20-226-i89.svg?max-width=637&scale=1.0)
3k (1:1.1) |
16.5 | 76 |
| 11 |
![[Graphic 89]](/bjoc/content/inline/1860-5397-20-226-i90.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 90]](/bjoc/content/inline/1860-5397-20-226-i91.svg?max-width=637&scale=1.0)
5l |
![[Graphic 91]](/bjoc/content/inline/1860-5397-20-226-i92.svg?max-width=637&scale=1.0)
3l (1:1.1) |
28.5 | 52 |
| 12 |
![[Graphic 92]](/bjoc/content/inline/1860-5397-20-226-i93.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 93]](/bjoc/content/inline/1860-5397-20-226-i94.svg?max-width=637&scale=1.0)
5m |
![[Graphic 94]](/bjoc/content/inline/1860-5397-20-226-i95.svg?max-width=637&scale=1.0)
3m (1:1) |
15.5 | 38 |
| 13 |
![[Graphic 95]](/bjoc/content/inline/1860-5397-20-226-i96.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 96]](/bjoc/content/inline/1860-5397-20-226-i97.svg?max-width=637&scale=1.0)
5n |
![[Graphic 97]](/bjoc/content/inline/1860-5397-20-226-i98.svg?max-width=637&scale=1.0)
3n (1:1.1) |
15.5 | 87 |
| 14b |
![[Graphic 98]](/bjoc/content/inline/1860-5397-20-226-i99.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 99]](/bjoc/content/inline/1860-5397-20-226-i100.svg?max-width=637&scale=1.0)
5o |
![[Graphic 100]](/bjoc/content/inline/1860-5397-20-226-i101.svg?max-width=637&scale=1.0)
3o (1:1) |
15 | 37 |
| 15d |
![[Graphic 101]](/bjoc/content/inline/1860-5397-20-226-i102.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 102]](/bjoc/content/inline/1860-5397-20-226-i103.svg?max-width=637&scale=1.0)
5p |
![[Graphic 103]](/bjoc/content/inline/1860-5397-20-226-i104.svg?max-width=637&scale=1.0)
3p (1:1.1) |
15 | 89 |
| 16 |
![[Graphic 104]](/bjoc/content/inline/1860-5397-20-226-i105.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 105]](/bjoc/content/inline/1860-5397-20-226-i106.svg?max-width=637&scale=1.0)
5q |
![[Graphic 106]](/bjoc/content/inline/1860-5397-20-226-i107.svg?max-width=637&scale=1.0)
3q (1:1.4) |
14.5 | 86 |
| 17 |
![[Graphic 107]](/bjoc/content/inline/1860-5397-20-226-i108.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 108]](/bjoc/content/inline/1860-5397-20-226-i109.svg?max-width=637&scale=1.0)
5r |
![[Graphic 109]](/bjoc/content/inline/1860-5397-20-226-i110.svg?max-width=637&scale=1.0)
3r (1:1.4) |
15 | 52 |
| 18 |
![[Graphic 110]](/bjoc/content/inline/1860-5397-20-226-i111.svg?max-width=637&scale=1.0)
1a (1:1) |
![[Graphic 111]](/bjoc/content/inline/1860-5397-20-226-i112.svg?max-width=637&scale=1.0)
5s |
![[Graphic 112]](/bjoc/content/inline/1860-5397-20-226-i113.svg?max-width=637&scale=1.0)
3s (1:1) |
13.5 | 53 |
| 19 |
![[Graphic 113]](/bjoc/content/inline/1860-5397-20-226-i114.svg?max-width=637&scale=1.0)
1b (1:1) |
![[Graphic 114]](/bjoc/content/inline/1860-5397-20-226-i115.svg?max-width=637&scale=1.0)
5a |
![[Graphic 115]](/bjoc/content/inline/1860-5397-20-226-i116.svg?max-width=637&scale=1.0)
3t (1:1.1) |
12.5 | 35 |
| 20e |
![[Graphic 116]](/bjoc/content/inline/1860-5397-20-226-i117.svg?max-width=637&scale=1.0)
1c (1:1) |
![[Graphic 117]](/bjoc/content/inline/1860-5397-20-226-i118.svg?max-width=637&scale=1.0)
5a |
![[Graphic 118]](/bjoc/content/inline/1860-5397-20-226-i119.svg?max-width=637&scale=1.0)
3u (1:1.2) |
22 | 29 |
| 21 |
![[Graphic 119]](/bjoc/content/inline/1860-5397-20-226-i120.svg?max-width=637&scale=1.0)
1d (1:1.1) |
![[Graphic 120]](/bjoc/content/inline/1860-5397-20-226-i121.svg?max-width=637&scale=1.0)
5a |
![[Graphic 121]](/bjoc/content/inline/1860-5397-20-226-i122.svg?max-width=637&scale=1.0)
3v (1:1.6) |
20.5 | 35 |
| 22f |
![[Graphic 122]](/bjoc/content/inline/1860-5397-20-226-i123.svg?max-width=637&scale=1.0)
1e (1:1.1) |
![[Graphic 123]](/bjoc/content/inline/1860-5397-20-226-i124.svg?max-width=637&scale=1.0)
5a |
![[Graphic 124]](/bjoc/content/inline/1860-5397-20-226-i125.svg?max-width=637&scale=1.0)
3w (1:1.3) |
110 | 7 |
aIsolated yields; b1 (2.0 equiv) and 5 (1.0 equiv) were used; c1 (2.0 equiv) and 5 (1.0 equiv) were used; dreaction temperature was 50 °C; e5 (1.3 equiv) was used; freaction temperature was rt to 50 °C.
Therefore, Suzuki–Miyaura and Sonogashira cross-coupling with 1 has a broad substrate scope and can be used to synthesize various fluoroalkenes 2 and fluoroenyne 3. We speculate that these reaction mechanisms were similar to general cross-coupling mechanisms [45,46].
Conclusion
We used Suzuki–Miyaura and Sonogashira cross-coupling to exploit the unique structure of multihalogenated fluorovinyl ethers 1 for the synthesis of many kinds of fluoroalkenes 2 and fluoroenynes 3 in moderate to high yields. The synthesized alkenes 2 still possess reactive chlorine atoms and phenoxy groups. Thus, new multisubstituted fluoroalkenes could be synthesized by applying other cross-couplings to 2. In addition, enynes 3 could be converted into derivatives, such as fluorine-containing alkynylalcohols [47], allene compounds [48-50], and heterocycles [51,52]. However, further experiments are required to expand the abilities of 2 and 3 as new fluorine-containing building blocks.
Experimental
General information
1H NMR, 19F NMR, and 13C NMR spectra were recorded on JEOL ECZ 400S spectrometers. Chemical shifts of 1H NMR are reported in ppm from tetramethylsilane (TMS) as an internal standard. Chemical shifts of 13C NMR are reported in ppm from the center line of the triplet at 77.16 ppm for deuteriochloroform. Chemical shifts of 19F NMR are reported in ppm from CFCl3 as an internal standard. All data are reported as follows: chemical shifts, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, sep = septet, br = broad, brd = broad-doublet, m = multiplet), coupling constants (Hz), relative integration value. Mass spectra were obtained on a JEOL JMS-700T spectrometer (EI). Melting points were measured on a Yanaco MP-500V.
Materials
All commercially available materials were used as received without further purification. All experiments were carried out under argon atmosphere in flame-dried glassware using standard inert techniques for introducing reagents and solvents unless otherwise noted.
Suzuki–Miyaura cross-coupling with multihalogenated vinyl ethers 1
To a solution of 1 (1.0 equiv), triphenylphosphine (10 mol %), cesium carbonate (1.5 equiv), palladium bis(trifluoroacetate) (5 mol %) in THF (2.0 mL) was added the respective boronic acid derivative 4 (2.0 equiv). The reaction solution was refluxed for 3.5 h. The reaction mixture was quenched by the addition of water (40 mL) at 0 °C and extracted with EtOAc. The organic phase was washed with brine (40 mL), dried over Na2SO4 and filtered. Then, the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography and preparative TLC to afford 2.
2-Chloro-1-fluoro-2-phenylethenyl phenyl ether (2a): Compound 2a was purified by column chromatography and preparative TLC (hexane only), and obtained in 96% yield (122.0 mg) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.04–7.22 (m, 2H), 7.26–7.50 (m, 6H), 7.55–7.69 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 101.4 (d, J = 30.9 Hz), 102.5 (d, J = 48.0 Hz), 116.5 (d, J = 3.8 Hz), 124.8, 127.3, 127.4, 127.9 (d, J = 3.2 Hz), 128.2 (d, J = 5.5 Hz), 128.5 (d, J = 7.2 Hz), 128.6 (d, J = 11.8 Hz), 128.9, 130.1 (d, J = 4.2 Hz), 132.46 (d, J = 5.7 Hz), 132.52, 141.3, 151.2 (d, J = 286.1 Hz), 151.5 (d, J = 287.3 Hz), 154.3 (d, J = 3.4 Hz), 154.4 (d, J = 3.3 Hz); 19F NMR (376 MHz, CDCl3) δ −80.8 (s) and −87.6 (s) (1F, 1:1.1); EIMS (m/z): 248, 250 [M]+; HREIMS [M]+ (m/z): calcd. for C14H10ClFO, 248.0402; found, 248.0404.
Sonogashira cross-coupling with multihalogenated vinyl ethers 1
To a solution of 1 (1.0 equiv), copper iodide (4 mol %), bis(triphenylphosphine)palladium dichloride (4 mol %) and triethylamine (1.5 equiv) in THF (2.5 mL) was added the respective alkyne 5 (2.0 equiv). The reaction solution was stirred at room temperature until 1 was disappeared. The reaction mixture was evaporated and concentrated under reduced pressure. The residue was purified by column chromatography and preparative TLC to afford 3.
(3-Chloro-4-fluoro-4-phenoxybut-3-en-1-yn-1-yl)trimethylsilane (3a): Reaction time was 19 h. 3a was purified by column chromatography (pentane only), and obtained in 80% yield (107.2 mg) as a yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.13 (s) and 0.24 (s) (9H), 7.08 (d, J = 8.0 Hz, 2H), 7.07–7.15 (m, 2H), 7.16–7.23 (m, 1H) , 7.33–7.42 (m, 1H); 13C NMR (100 MHz, CDCl3) δ −0.36, −0.26, 85.2 (d, J = 44.8 Hz), 85.6 (d, J = 53.3 Hz), 94.5 (d, J = 47.8 Hz), 94.6 (d, J = 43.5 Hz), 103.7 (d, J = 63.4 Hz), 103.8 (d, J = 66.3 Hz), 117.2, 117.3, 125.2, 125.3, 130.0, 130.1, 154.00 (d, J = 75.1 Hz), 154.02 (d, J = 75.8 Hz), 158.2 (d, J = 292.4 Hz), 158.8 (d, J = 290.1 Hz); 19F NMR (376 MHz, CDCl3) δ −73.3 (s) and −78.4 (s) (1F, 1:1.2); EIMS m/z: 268 (M+); HREIMS [M]+ (m/z): calcd. for C13H14ClFOSi, 268.0486; found, 268.0490.
Supporting Information
| Supporting Information File 1: Characterization data for 2b–s and 3b–w, and copies of 1H, 13C, and 19F NMR spectra. | ||
| Format: PDF | Size: 10.3 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Drouin, M.; Paquin, J.-F. Beilstein J. Org. Chem. 2017, 13, 2637–2658. doi:10.3762/bjoc.13.262
Return to citation in text: [1] -
Frohn, M.; Liu, L.; Siegmund, A. C.; Qian, W.; Amegadzie, A.; Chen, N.; Tan, H.; Hickman, D.; Wood, S.; Wen, P. H.; Bartberger, M. D.; Whittington, D. A.; Allen, J. R.; Bourbeau, M. P. Bioorg. Med. Chem. Lett. 2020, 30, 127240. doi:10.1016/j.bmcl.2020.127240
Return to citation in text: [1] -
Van der Veken, P.; Kertèsz, I.; Senten, K.; Haemers, A.; Augustyns, K. Tetrahedron Lett. 2003, 44, 6231–6234. doi:10.1016/s0040-4039(03)01542-9
Return to citation in text: [1] -
Lee, S. H.; Schwartz, J. J. Am. Chem. Soc. 1986, 108, 2445–2447. doi:10.1021/ja00269a052
Return to citation in text: [1] -
Suga, H.; Hamatani, T.; Guggisberg, Y.; Schlosser, M. Tetrahedron 1990, 46, 4255–4260. doi:10.1016/s0040-4020(01)86762-4
Return to citation in text: [1] -
Salim, S. S.; Bellingham, R. K.; Satcharoen, V.; Brown, R. C. D. Org. Lett. 2003, 5, 3403–3406. doi:10.1021/ol035065w
Return to citation in text: [1] -
Koh, M. J.; Nguyen, T. T.; Zhang, H.; Schrock, R. R.; Hoveyda, A. H. Nature 2016, 531, 459–465. doi:10.1038/nature17396
Return to citation in text: [1] -
Kojima, R.; Kubota, K.; Ito, H. Chem. Commun. 2017, 53, 10688–10691. doi:10.1039/c7cc05225a
Return to citation in text: [1] -
Hu, J.; Han, X.; Yuan, Y.; Shi, Z. Angew. Chem., Int. Ed. 2017, 56, 13342–13346. doi:10.1002/anie.201708224
Return to citation in text: [1] -
Vandamme, M.; Paquin, J.-F. Org. Lett. 2017, 19, 3604–3607. doi:10.1021/acs.orglett.7b01581
Return to citation in text: [1] -
Zhou, Y.; Fan, B.; Chen, J.; He, Z.; Fan, R.; Sun, W.; Li, K. Preparation method of fluorinated olefin and its application in preparation of medical material, pesticide special material and organic chemical material. Chin. Patent CN111170856, May 19, 2020.
Return to citation in text: [1] -
Du, H.-W.; Sun, J.; Gao, Q.-S.; Wang, J.-Y.; Wang, H.; Xu, Z.; Zhou, M.-D. Org. Lett. 2020, 22, 1542–1546. doi:10.1021/acs.orglett.0c00134
Return to citation in text: [1] -
Li, K.; Chen, J.; Yang, C.; Zhang, K.; Pan, C.; Fan, B. Org. Lett. 2020, 22, 4261–4265. doi:10.1021/acs.orglett.0c01294
Return to citation in text: [1] -
Yang, H.; Wang, J.; Jin, C.; Li, X.; Xu, X. J. Org. Chem. 2023, 88, 12074–12078. doi:10.1021/acs.joc.3c00594
Return to citation in text: [1] -
Volchkov, I.; Powell, B. V.; Zatolochnaya, O. V.; Leung, J. C.; Pennino, S.; Wu, L.; Gonnella, N. C.; Bhaskararao, B.; Kozlowski, M. C.; Reeves, J. T. J. Org. Chem. 2023, 88, 10881–10904. doi:10.1021/acs.joc.3c00917
Return to citation in text: [1] [2] -
Schlosser, M.; Zimmermann, M. Synthesis 1969, 75–76. doi:10.1055/s-1969-20377
Return to citation in text: [1] -
Tsai, H.-J. Tetrahedron Lett. 1996, 37, 629–632. doi:10.1016/0040-4039(95)02218-x
Return to citation in text: [1] -
Prakash, G. K. S.; Shakhmin, A.; Zibinsky, M.; Ledneczki, I.; Chacko, S.; Olah, G. A. J. Fluorine Chem. 2010, 131, 1192–1197. doi:10.1016/j.jfluchem.2010.06.009
Return to citation in text: [1] -
Liu, Q.; Shen, X.; Ni, C.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 619–623. doi:10.1002/anie.201610127
Return to citation in text: [1] -
Mandal, D.; Gupta, R.; Young, R. D. J. Am. Chem. Soc. 2018, 140, 10682–10686. doi:10.1021/jacs.8b06770
Return to citation in text: [1] -
Poutrel, P.; Pannecoucke, X.; Jubault, P.; Poisson, T. Org. Lett. 2020, 22, 4858–4863. doi:10.1021/acs.orglett.0c01701
Return to citation in text: [1] [2] -
Debien, L.; Quiclet-Sire, B.; Zard, S. S. Org. Lett. 2012, 14, 5118–5121. doi:10.1021/ol3023903
Return to citation in text: [1] -
Hamel, J.-D.; Cloutier, M.; Paquin, J.-F. Org. Lett. 2016, 18, 1852–1855. doi:10.1021/acs.orglett.6b00590
Return to citation in text: [1] -
Carreras, V.; Ollevier, T. J. Org. Chem. 2021, 86, 13160–13168. doi:10.1021/acs.joc.1c01724
Return to citation in text: [1] -
Li, H.; Zhu, C. J. Org. Chem. 2023, 88, 4134–4144. doi:10.1021/acs.joc.2c02568
Return to citation in text: [1] -
Isoda, M.; Uetake, Y.; Takimoto, T.; Tsuda, J.; Hosoya, T.; Niwa, T. J. Org. Chem. 2021, 86, 1622–1632. doi:10.1021/acs.joc.0c02474
Return to citation in text: [1] [2] -
Heinze, P. L.; Burton, D. J. J. Org. Chem. 1988, 53, 2714–2720. doi:10.1021/jo00247a010
Return to citation in text: [1] -
Matthews, D. P.; Gross, R. S.; McCarthy, J. R. Tetrahedron Lett. 1994, 35, 1027–1030. doi:10.1016/s0040-4039(00)79956-4
Return to citation in text: [1] -
Raghavanpillai, A.; Burton, D. J. J. Org. Chem. 2004, 69, 7083–7091. doi:10.1021/jo049179c
Return to citation in text: [1] -
Andrei, D.; Wnuk, S. F. J. Org. Chem. 2006, 71, 405–408. doi:10.1021/jo051980e
Return to citation in text: [1] -
Ohashi, M.; Kambara, T.; Hatanaka, T.; Saijo, H.; Doi, R.; Ogoshi, S. J. Am. Chem. Soc. 2011, 133, 3256–3259. doi:10.1021/ja109911p
Return to citation in text: [1] -
Liu, J.; Ren, Q.; Zhang, X.; Gong, H. Angew. Chem., Int. Ed. 2016, 55, 15544–15548. doi:10.1002/anie.201607959
Return to citation in text: [1] -
Cai, S.-H.; Ye, L.; Wang, D.-X.; Wang, Y.-Q.; Lai, L.-J.; Zhu, C.; Feng, C.; Loh, T.-P. Chem. Commun. 2017, 53, 8731–8734. doi:10.1039/c7cc04131d
Return to citation in text: [1] -
Sakaguchi, H.; Uetake, Y.; Ohashi, M.; Niwa, T.; Ogoshi, S.; Hosoya, T. J. Am. Chem. Soc. 2017, 139, 12855–12862. doi:10.1021/jacs.7b08343
Return to citation in text: [1] -
Sakaguchi, H.; Ohashi, M.; Ogoshi, S. Angew. Chem., Int. Ed. 2018, 57, 328–332. doi:10.1002/anie.201710866
Return to citation in text: [1] -
Tian, H.; Yang, S.; Wang, X.; Xu, W.; Liu, Y.; Li, Y.; Wang, Q. J. Org. Chem. 2021, 86, 12772–12782. doi:10.1021/acs.joc.1c01363
Return to citation in text: [1] -
Wang, C.; Liu, Y.-C.; Xu, M.-Y.; Xiao, B. Org. Lett. 2021, 23, 4593–4597. doi:10.1021/acs.orglett.1c01289
Return to citation in text: [1] -
Wang, Y.; Ma, Q.; Tsui, G. C. Org. Lett. 2021, 23, 5241–5245. doi:10.1021/acs.orglett.1c01768
Return to citation in text: [1] -
Wu, X.; Zeng, Y.; Jiang, Z.-T.; Zhu, Y.; Xie, L.; Xia, Y. Org. Lett. 2022, 24, 8429–8434. doi:10.1021/acs.orglett.2c03544
Return to citation in text: [1] -
Liu, F.; Zhuang, Z.; Qian, Q.; Zhang, X.; Yang, C. J. Org. Chem. 2022, 87, 2730–2739. doi:10.1021/acs.joc.1c02662
Return to citation in text: [1] -
Wang, Y.; Tsui, G. C. Org. Lett. 2023, 25, 6217–6221. doi:10.1021/acs.orglett.3c02452
Return to citation in text: [1] -
Karuo, Y.; Tarui, A.; Sato, K.; Kawai, K.; Omote, M. Beilstein J. Org. Chem. 2022, 18, 1567–1574. doi:10.3762/bjoc.18.167
Return to citation in text: [1] -
Yang, Z.; Chen, X.; Kong, W.; Xia, S.; Zheng, R.; Luo, F.; Zhu, G. Org. Biomol. Chem. 2013, 11, 2175–2185. doi:10.1039/c3ob27307e
Return to citation in text: [1] -
Thorand, S.; Krause, N. J. Org. Chem. 1998, 63, 8551–8553. doi:10.1021/jo9808021
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/s0040-4039(00)91094-3
Return to citation in text: [1] -
Nguyen, K. D.; Herkommer, D.; Krische, M. J. J. Am. Chem. Soc. 2016, 138, 5238–5241. doi:10.1021/jacs.6b02279
Return to citation in text: [1] -
Mori, Y.; Onodera, G.; Kimura, M. Chem. Lett. 2014, 43, 97–99. doi:10.1246/cl.130865
Return to citation in text: [1] -
Huang, Y.; del Pozo, J.; Torker, S.; Hoveyda, A. H. J. Am. Chem. Soc. 2018, 140, 2643–2655. doi:10.1021/jacs.7b13296
Return to citation in text: [1] -
Xu, T.; Wu, S.; Zhang, Q.-N.; Wu, Y.; Hu, M.; Li, J.-H. Org. Lett. 2021, 23, 8455–8459. doi:10.1021/acs.orglett.1c03179
Return to citation in text: [1] -
Chun, Y. S.; Lee, J. H.; Kim, J. H.; Ko, Y. O.; Lee, S.-g. Org. Lett. 2011, 13, 6390–6393. doi:10.1021/ol202691b
Return to citation in text: [1] -
Wang, Y.-J.; Zhang, Y.; Qiang, Z.; Liang, J.-Y.; Chen, Z. Chem. Commun. 2021, 57, 12607–12610. doi:10.1039/d1cc05260h
Return to citation in text: [1]
| 1. | Drouin, M.; Paquin, J.-F. Beilstein J. Org. Chem. 2017, 13, 2637–2658. doi:10.3762/bjoc.13.262 |
| 15. | Volchkov, I.; Powell, B. V.; Zatolochnaya, O. V.; Leung, J. C.; Pennino, S.; Wu, L.; Gonnella, N. C.; Bhaskararao, B.; Kozlowski, M. C.; Reeves, J. T. J. Org. Chem. 2023, 88, 10881–10904. doi:10.1021/acs.joc.3c00917 |
| 48. | Mori, Y.; Onodera, G.; Kimura, M. Chem. Lett. 2014, 43, 97–99. doi:10.1246/cl.130865 |
| 49. | Huang, Y.; del Pozo, J.; Torker, S.; Hoveyda, A. H. J. Am. Chem. Soc. 2018, 140, 2643–2655. doi:10.1021/jacs.7b13296 |
| 50. | Xu, T.; Wu, S.; Zhang, Q.-N.; Wu, Y.; Hu, M.; Li, J.-H. Org. Lett. 2021, 23, 8455–8459. doi:10.1021/acs.orglett.1c03179 |
| 15. | Volchkov, I.; Powell, B. V.; Zatolochnaya, O. V.; Leung, J. C.; Pennino, S.; Wu, L.; Gonnella, N. C.; Bhaskararao, B.; Kozlowski, M. C.; Reeves, J. T. J. Org. Chem. 2023, 88, 10881–10904. doi:10.1021/acs.joc.3c00917 |
| 16. | Schlosser, M.; Zimmermann, M. Synthesis 1969, 75–76. doi:10.1055/s-1969-20377 |
| 17. | Tsai, H.-J. Tetrahedron Lett. 1996, 37, 629–632. doi:10.1016/0040-4039(95)02218-x |
| 18. | Prakash, G. K. S.; Shakhmin, A.; Zibinsky, M.; Ledneczki, I.; Chacko, S.; Olah, G. A. J. Fluorine Chem. 2010, 131, 1192–1197. doi:10.1016/j.jfluchem.2010.06.009 |
| 19. | Liu, Q.; Shen, X.; Ni, C.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 619–623. doi:10.1002/anie.201610127 |
| 20. | Mandal, D.; Gupta, R.; Young, R. D. J. Am. Chem. Soc. 2018, 140, 10682–10686. doi:10.1021/jacs.8b06770 |
| 51. | Chun, Y. S.; Lee, J. H.; Kim, J. H.; Ko, Y. O.; Lee, S.-g. Org. Lett. 2011, 13, 6390–6393. doi:10.1021/ol202691b |
| 52. | Wang, Y.-J.; Zhang, Y.; Qiang, Z.; Liang, J.-Y.; Chen, Z. Chem. Commun. 2021, 57, 12607–12610. doi:10.1039/d1cc05260h |
| 4. | Lee, S. H.; Schwartz, J. J. Am. Chem. Soc. 1986, 108, 2445–2447. doi:10.1021/ja00269a052 |
| 5. | Suga, H.; Hamatani, T.; Guggisberg, Y.; Schlosser, M. Tetrahedron 1990, 46, 4255–4260. doi:10.1016/s0040-4020(01)86762-4 |
| 6. | Salim, S. S.; Bellingham, R. K.; Satcharoen, V.; Brown, R. C. D. Org. Lett. 2003, 5, 3403–3406. doi:10.1021/ol035065w |
| 7. | Koh, M. J.; Nguyen, T. T.; Zhang, H.; Schrock, R. R.; Hoveyda, A. H. Nature 2016, 531, 459–465. doi:10.1038/nature17396 |
| 8. | Kojima, R.; Kubota, K.; Ito, H. Chem. Commun. 2017, 53, 10688–10691. doi:10.1039/c7cc05225a |
| 9. | Hu, J.; Han, X.; Yuan, Y.; Shi, Z. Angew. Chem., Int. Ed. 2017, 56, 13342–13346. doi:10.1002/anie.201708224 |
| 10. | Vandamme, M.; Paquin, J.-F. Org. Lett. 2017, 19, 3604–3607. doi:10.1021/acs.orglett.7b01581 |
| 11. | Zhou, Y.; Fan, B.; Chen, J.; He, Z.; Fan, R.; Sun, W.; Li, K. Preparation method of fluorinated olefin and its application in preparation of medical material, pesticide special material and organic chemical material. Chin. Patent CN111170856, May 19, 2020. |
| 12. | Du, H.-W.; Sun, J.; Gao, Q.-S.; Wang, J.-Y.; Wang, H.; Xu, Z.; Zhou, M.-D. Org. Lett. 2020, 22, 1542–1546. doi:10.1021/acs.orglett.0c00134 |
| 13. | Li, K.; Chen, J.; Yang, C.; Zhang, K.; Pan, C.; Fan, B. Org. Lett. 2020, 22, 4261–4265. doi:10.1021/acs.orglett.0c01294 |
| 14. | Yang, H.; Wang, J.; Jin, C.; Li, X.; Xu, X. J. Org. Chem. 2023, 88, 12074–12078. doi:10.1021/acs.joc.3c00594 |
| 45. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 46. | Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/s0040-4039(00)91094-3 |
| 2. | Frohn, M.; Liu, L.; Siegmund, A. C.; Qian, W.; Amegadzie, A.; Chen, N.; Tan, H.; Hickman, D.; Wood, S.; Wen, P. H.; Bartberger, M. D.; Whittington, D. A.; Allen, J. R.; Bourbeau, M. P. Bioorg. Med. Chem. Lett. 2020, 30, 127240. doi:10.1016/j.bmcl.2020.127240 |
| 3. | Van der Veken, P.; Kertèsz, I.; Senten, K.; Haemers, A.; Augustyns, K. Tetrahedron Lett. 2003, 44, 6231–6234. doi:10.1016/s0040-4039(03)01542-9 |
| 47. | Nguyen, K. D.; Herkommer, D.; Krische, M. J. J. Am. Chem. Soc. 2016, 138, 5238–5241. doi:10.1021/jacs.6b02279 |
| 26. | Isoda, M.; Uetake, Y.; Takimoto, T.; Tsuda, J.; Hosoya, T.; Niwa, T. J. Org. Chem. 2021, 86, 1622–1632. doi:10.1021/acs.joc.0c02474 |
| 43. | Yang, Z.; Chen, X.; Kong, W.; Xia, S.; Zheng, R.; Luo, F.; Zhu, G. Org. Biomol. Chem. 2013, 11, 2175–2185. doi:10.1039/c3ob27307e |
| 26. | Isoda, M.; Uetake, Y.; Takimoto, T.; Tsuda, J.; Hosoya, T.; Niwa, T. J. Org. Chem. 2021, 86, 1622–1632. doi:10.1021/acs.joc.0c02474 |
| 27. | Heinze, P. L.; Burton, D. J. J. Org. Chem. 1988, 53, 2714–2720. doi:10.1021/jo00247a010 |
| 28. | Matthews, D. P.; Gross, R. S.; McCarthy, J. R. Tetrahedron Lett. 1994, 35, 1027–1030. doi:10.1016/s0040-4039(00)79956-4 |
| 29. | Raghavanpillai, A.; Burton, D. J. J. Org. Chem. 2004, 69, 7083–7091. doi:10.1021/jo049179c |
| 30. | Andrei, D.; Wnuk, S. F. J. Org. Chem. 2006, 71, 405–408. doi:10.1021/jo051980e |
| 31. | Ohashi, M.; Kambara, T.; Hatanaka, T.; Saijo, H.; Doi, R.; Ogoshi, S. J. Am. Chem. Soc. 2011, 133, 3256–3259. doi:10.1021/ja109911p |
| 32. | Liu, J.; Ren, Q.; Zhang, X.; Gong, H. Angew. Chem., Int. Ed. 2016, 55, 15544–15548. doi:10.1002/anie.201607959 |
| 33. | Cai, S.-H.; Ye, L.; Wang, D.-X.; Wang, Y.-Q.; Lai, L.-J.; Zhu, C.; Feng, C.; Loh, T.-P. Chem. Commun. 2017, 53, 8731–8734. doi:10.1039/c7cc04131d |
| 34. | Sakaguchi, H.; Uetake, Y.; Ohashi, M.; Niwa, T.; Ogoshi, S.; Hosoya, T. J. Am. Chem. Soc. 2017, 139, 12855–12862. doi:10.1021/jacs.7b08343 |
| 35. | Sakaguchi, H.; Ohashi, M.; Ogoshi, S. Angew. Chem., Int. Ed. 2018, 57, 328–332. doi:10.1002/anie.201710866 |
| 36. | Tian, H.; Yang, S.; Wang, X.; Xu, W.; Liu, Y.; Li, Y.; Wang, Q. J. Org. Chem. 2021, 86, 12772–12782. doi:10.1021/acs.joc.1c01363 |
| 37. | Wang, C.; Liu, Y.-C.; Xu, M.-Y.; Xiao, B. Org. Lett. 2021, 23, 4593–4597. doi:10.1021/acs.orglett.1c01289 |
| 38. | Wang, Y.; Ma, Q.; Tsui, G. C. Org. Lett. 2021, 23, 5241–5245. doi:10.1021/acs.orglett.1c01768 |
| 39. | Wu, X.; Zeng, Y.; Jiang, Z.-T.; Zhu, Y.; Xie, L.; Xia, Y. Org. Lett. 2022, 24, 8429–8434. doi:10.1021/acs.orglett.2c03544 |
| 40. | Liu, F.; Zhuang, Z.; Qian, Q.; Zhang, X.; Yang, C. J. Org. Chem. 2022, 87, 2730–2739. doi:10.1021/acs.joc.1c02662 |
| 41. | Wang, Y.; Tsui, G. C. Org. Lett. 2023, 25, 6217–6221. doi:10.1021/acs.orglett.3c02452 |
| 44. | Thorand, S.; Krause, N. J. Org. Chem. 1998, 63, 8551–8553. doi:10.1021/jo9808021 |
| 21. | Poutrel, P.; Pannecoucke, X.; Jubault, P.; Poisson, T. Org. Lett. 2020, 22, 4858–4863. doi:10.1021/acs.orglett.0c01701 |
| 21. | Poutrel, P.; Pannecoucke, X.; Jubault, P.; Poisson, T. Org. Lett. 2020, 22, 4858–4863. doi:10.1021/acs.orglett.0c01701 |
| 22. | Debien, L.; Quiclet-Sire, B.; Zard, S. S. Org. Lett. 2012, 14, 5118–5121. doi:10.1021/ol3023903 |
| 23. | Hamel, J.-D.; Cloutier, M.; Paquin, J.-F. Org. Lett. 2016, 18, 1852–1855. doi:10.1021/acs.orglett.6b00590 |
| 24. | Carreras, V.; Ollevier, T. J. Org. Chem. 2021, 86, 13160–13168. doi:10.1021/acs.joc.1c01724 |
| 25. | Li, H.; Zhu, C. J. Org. Chem. 2023, 88, 4134–4144. doi:10.1021/acs.joc.2c02568 |
| 42. | Karuo, Y.; Tarui, A.; Sato, K.; Kawai, K.; Omote, M. Beilstein J. Org. Chem. 2022, 18, 1567–1574. doi:10.3762/bjoc.18.167 |
© 2024 Karuo et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.