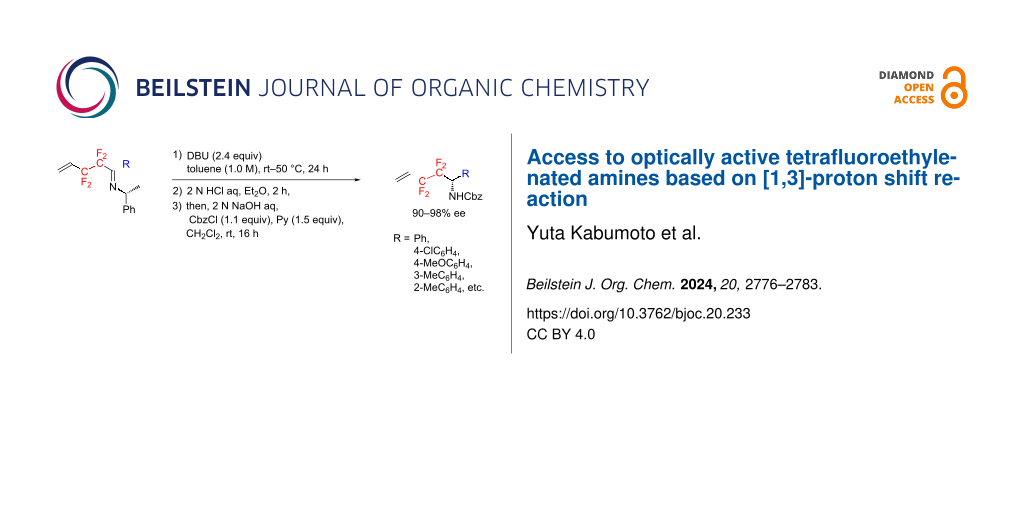
Abstract
Treatment of various (R)-N-(2,2,3,3-tetrafluoropent-4-en-1-ylidene)-1-phenylethylamine derivatives with 2.4 equiv of DBU in toluene at room temperature to 50 °C for 24 h led to a smooth [1,3]-proton shift reaction with a high chirality transfer, affording the corresponding rearranged products in acceptable yields. Without purification, these products were subjected to acid hydrolysis and the subsequent N-Cbz protection, providing the optically active tetrafluoroethylenated amides in moderate three-step yields.

Graphical Abstract
Introduction
A fluorine atom has quite peculiar chemical and physical properties compared to others, and hence changes in molecular properties resulting from the introduction of fluorine atom(s) into organic molecules are also significantly unique, and often extremely noticeable even when the number of the atom introduced is small [1-3]. By skillfully utilizing such characteristics, fluorine-containing organic molecules have established themselves as indispensable compounds in various frontlines, such as medicinal, agrochemical, and material fields [4-7].
In particular, tetrafluoroethylenated compounds possessing two fluorine atoms on each of two adjacent carbons, have been attracting an enormous attention these days. This stems from the fact that substances with a tetrafluoroethylene fragment exhibit significantly different molecular properties compared to monofluorinated, difluorinated, or trifluoromethylated molecules [8,9]. Therefore, more and more tetrafluoroethylenated molecules having a variety of applications, such as bioactive substances (Figure 1a, 1, 2) [10-12], liquid crystals (Figure 1b, 3, 4) [13-17], fluorescence materials (Figure 1c, 5) [18,19], and so on, have been developed in recent years.
![[1860-5397-20-233-1]](/bjoc/content/figures/1860-5397-20-233-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Various applications of tetrafluoroethylenated molecules.
Figure 1: Various applications of tetrafluoroethylenated molecules.
In sharp contrast to the major development of such non-chiral tetrafluoroethylenated compounds, there have been only few reports on the preparation of chiral molecules possessing a tetrafluoroethylene unit on an asymmetric carbon center in a high optical purity, and to the best of our knowledge, only the following have been published so far (Scheme 1).
![[1860-5397-20-233-i1]](/bjoc/content/inline/1860-5397-20-233-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Precedent synthetic approaches to optically active compounds possessing a tetrafluoroethylene group on an asymmetric carbon center.
Scheme 1: Precedent synthetic approaches to optically active compounds possessing a tetrafluoroethylene group...
As a highly enantioselective synthesis, there has been a pioneering work by Linclau et al. They have reported that the asymmetric Sharpless dihydroxylation of readily available (E)-5-bromo-4,4,5,5-tetrafluoro-2-penten-1-ol derivative 6 led to the corresponding chiral diols 7 with an excellent enantiomeric excess, 96% ee (reaction 1 in Scheme 1) [20,21]. It has also been published that the asymmetric conjugate addition of 4-methylphenylboronic acid towards (E)-5-bromo-4,4,5,5-tetrafluoro-1-phenyl-2-penten-1-one (8) in the presence of a rhodium catalyst coordinated with (S)-BINAP gave the corresponding Michael adduct 9 in 94% enantiomeric excess (reaction 2, Scheme 1) [22].
As a diastereoselective synthesis, reductive coupling reactions of commercially available 4-bromo-3,3,4,4-tetrafluoro-1-butene and glyceraldehyde 10a, its imine derivative 11, or Garner's aldehyde 10b have been reported [23,24]. Although the diastereoselectivities were low in some cases, the diastereomers 12 and 13 are often easily separable, and each diastereomer of optically active alcohols or amines can be obtained with an excellent optical purity (reactions 3 and 4 in Scheme 1).
To the best of our knowledge, these are the only four works for the preparation of optically active substances having a tetrafluoroethylene group on an asymmetric carbon center. In order to overcome the current lack of synthetic methods for preparing such molecules, we came up with the idea of utilizing the [1,3]-proton shift reaction reported by Soloshonok et al.
In 1997, Soloshonok et al. reported that fluoroalkylated amines 15 with high optical purity could be easily prepared through [1,3]-proton shift reactions of optically active imines 14 which in turn were readily synthesized by condensation of various perfluoroalkyl ketones with optically active (R)-1-phenylethylamine (Scheme 2a) [25-32]. Therefore, we envisioned that optically active tetrafluoroethylenated amines 17 could be synthesized by applying the [1,3]-proton shift to optically active imines 16 derived from readily prepared tetrafluoroethylenated ketones (Scheme 2b).
![[1860-5397-20-233-i2]](/bjoc/content/inline/1860-5397-20-233-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic strategy for preparing fluorine-containing amines via [1,3]-proton shift reactions.
Scheme 2: Synthetic strategy for preparing fluorine-containing amines via [1,3]-proton shift reactions.
In this paper, we describe the details of the [1,3]-proton shift reaction of various tetrafluoroethylenated imines.
Results and Discussion
The preparation of substrates used in this study was as outlined in Scheme 3. Namely, the zinc reagent was prepared from commercially available 3,3,4,4-tetrafluoro-1-butene (18) [33] and reacted with various acid chlorides in the presence of a copper catalyst to afford the corresponding tetrafluoroethylenated ketones 19. The ketones were then condensed with (R)-1-phenylethylamine under the influence of TiCl4 [34,35] to prepare various optically active imines (R)-16 in high yields (Scheme 3). Based on the result of the NOESY spectrum of the imine (R)-16c, the stereochemistry of the imines (R)-16 was determined as E [36].
![[1860-5397-20-233-i3]](/bjoc/content/inline/1860-5397-20-233-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of the substrates used in this study.
Scheme 3: Preparation of the substrates used in this study.
Among the imines thus obtained, (R)-16b was used to investigate the optimum reaction conditions (Table 1). Treatment of (R)-16b with 1.2 equiv of DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) in THF at room temperature for 24 h gave the corresponding [1,3]-proton shift adduct (S)-20b in 31% yield (Table 1, entry 1). In this case, the HF-elimination product 21b was also obtained in 16% [37], and the starting material was recovered in 53%. As shown in entries 2–7 of Table 1, the reactions in various solvents were next examined. When CH3CN or CH2Cl2 was used, 17% or 36% of the target product (S)-20b were obtained and almost no HF-elimination product 21b was formed, while about 40% of the azocine derivative 22b was afforded as a byproduct [38], along with the recovery of (R)-16b. In the case of diethyl ether, toluene, hexane, and cyclohexane, (S)-20b was obtained in 30% to 40% yield and significant amounts of unreacted substrate were still observed, although formation of the byproduct 22b could be generally suppressed.
Table 1: Investigation of the reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-233-i7.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Base/X equiv | Solvent | Yielda [%] of (S)-20b | Yielda [%] of 21b | Yielda [%] of 22b | Recoverya [%] of (R)-16b |
| 1 | DBU/1.2 | THF | 31 | 16 | 0 | 53 |
| 2 | DBU/1.2 | CH3CN | 17 | 0 | 39 | 44 |
| 3 | DBU/1.2 | Et2O | 38 | 14 | 17 | 31 |
| 4 | DBU/1.2 | toluene | 37 | 13 | 0 | 50 |
| 5 | DBU/1.2 | hexane | 26 | 12 | 0 | 62 |
| 6 | DBU/1.2 | CH2Cl2 | 36 | 3 | 38 | 23 |
| 7 | DBU/1.2 | cyclohexane | 32 | 17 | 0 | 51 |
| 8 | DBU/1.2 | toluene | 33 | 18 | 0 | 49 |
| 9 | Et3N/1.2 | toluene | 0 | 0 | 0 | 100 |
| 10 | DABCO/1.2 | toluene | 0 | 0 | 0 | 100 |
| 11 | DBU/2.4 | toluene | 50 | 2 | 43 | 5 |
| 12 | DBU/4.8 | toluene | 29 | 0 | 71 | 0 |
| 13 | DBU/6.8 | toluene | 31 | 0 | 69 | 0 |
| 14b | DBU/2.4 | toluene | 8 | 13 | 0 | 79 |
aDetermined by 19F NMR spectroscopy; bthe reaction was carried out at 0 °C.
We also examined the reaction using other bases instead of DBU. As shown in entries 9 and 10 of Table 1, the reaction did not proceed at all with triethylamine or DABCO, and (R)-16b was quantitatively recovered. The influence of the amount of DBU upon the reaction was also investigated (Table 1, entries 11–13). The results showed that when 2.4 equiv of DBU were used, the target compound (S)-20b was obtained in 50% yield, along with byproduct 22b in 43% yield (Table 1, entry 11), while increasing the number of equivalents of DBU decreased the yield of the target product (S)-20b and increased the yield of the byproduct 22b (Table 1, entries 12 and 13). Also carrying out the reaction at 0 °C gave unsatisfactory results and a large amount of (R)-16b was recovered (Table 1, entry 14).
Based on these results, the reaction conditions in entry 11 (Table 1) were determined as the optimum ones, which gave the highest yield, although the formation of the byproduct azocine derivative 22b could not be completely suppressed.
The thus obtained [1,3]-proton shift product (S)-20b was subjected to 2 N HCl aq in Et2O for 2 h, and subsequently 2 N NaOH aq, affording the corresponding free amine (S)-17b. Then, treatment of the amine with CbzCl and pyridine in CH2Cl2 gave the corresponding amide (S)-23b in 27% isolated yield over three-steps. The measurement of HPLC equipped with a chiral column, CHIRALPAK AD-H for (S)-23b, showed that the amide had an optical purity of 95% ee (Scheme 4).
![[1860-5397-20-233-i4]](/bjoc/content/inline/1860-5397-20-233-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Derivatization of (S)-20b to (S)-23b for determining the optical purity of (S)-20b.
Scheme 4: Derivatization of (S)-20b to (S)-23b for determining the optical purity of (S)-20b.
On the next stage, the substrate scope for the present reaction was explored by using various imines (R)-16 (Scheme 5). When the substituent R is an aromatic ring substituted by a halogen such as chlorine and bromine atoms, the amides (S)-23 were obtained in 22–27% yield and with very high optical purity ((S)-23b, (S)-23c), although as for (S)-23a, a satisfactory result (92% ee) could be obtained when the reaction was performed at 50 °C. The substrates with an aromatic ring substituted by not only an electron-withdrawing halogen atom but also an electron-donating group such as methoxy and methyl group also smoothly underwent the [1,3]-proton shift reaction, affording the desired products with high enantiomeric excess (90% ee for (S)-23d and (S)-23e). Furthermore, it was found that the substituent position on the aromatic ring did not significantly influence the reaction efficiency as well as optical purity and the reaction proceeded in a highly enantioselective manner (91% ee for (S)-23f and 94% ee for (S)-23g). Disappointingly, when R is an alkyl group, the desired rearrangement products were rarely obtained, resulting in unidentified products together with a large amount of starting material. This may be due to the following reasons. Namely, when the substituent R is an aryl group, the negative charge generated on the imine carbon can be delocalized, hence the transition state is stabilized and the reaction proceeds smoothly. On the other hand, when the substituent R is an alkyl group, the negative charge generated on the imine carbon is an unstable factor, and therefore the transition state is not stabilized, leading to the increase of the activation energy of the reaction. As a result, the reaction does not proceed smoothly.
![[1860-5397-20-233-i5]](/bjoc/content/inline/1860-5397-20-233-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Substrate scope for the present [1,3]-proton shift reaction. aYields are determined by 19F NMR spectroscopy. Values in parentheses show isolated yields. Enantiomeric excesses are deteremined by HPLC equipped with DAICEL CHIRALPAK AD-H. bCarried our at 50 °C in the [1,3]-proton shift reaction step.
Scheme 5: Substrate scope for the present [1,3]-proton shift reaction. aYields are determined by 19F NMR spec...
The absolute configurations of product (S)-23c, (S)-23d, and (S)-23e were determined on the basis of their X-ray crystallographic analyses. The validity of their absolute configurations was confirmed by the convergence of the Flack parameters of three compounds to values close to 0, i.e. −0.006(12), 0.1(4) and 0.2(4), respectively. As indicated in Scheme 5, therefore, the absolute configurations of all compounds were determined as S. Then, the [1,3]-proton shift reaction in this study is expected to proceed via the reaction mechanism reported by Soloshonok [25-32], as shown in Scheme 6.
![[1860-5397-20-233-i6]](/bjoc/content/inline/1860-5397-20-233-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
First, DBU interacts with the benzylic hydrogen of the imine (R)-16, and this hydrogen is about to be abstracted as a proton. This hydrogen which is being abstracted simultaneously interacts with the carbon possessing the tetrafluoroethylene fragment. At this time, transition states TS1 or TS2 are possible, but the reaction proceeds exclusively through the transition state TS1 to avoid significant steric repulsion between the substituent R and the phenyl group. Therefore, the product (S)-20 with S configuration is obtained preferentially.
Conclusion
In summary, we have succeeded in synthesizing optically active amines having a tetrafluoroethylene group on the asymmetric carbon center by applying the [1,3]-proton shift reaction using various optically active imines, which can be easily prepared starting from commercially available 4-bromo-3,3,4,4-tetrafluoro-1-butene. In this reaction, although the formation of azocine derivatives as byproducts could not be completely suppressed, the [1,3]-proton shift reaction proceeded in relatively good yield and a high asymmetric transfer was achieved. As a result, it was found that various optically active amine derivatives could be obtained with high optical purity.
Supporting Information
| Supporting Information File 1: Full experimental details, 1H, 13C, 19F NMR spectra of 16a–g and 23a–g, and HPLC charts of racemic as well as chiral compounds 23a–g. | ||
| Format: PDF | Size: 5.5 MB | Download |
| Supporting Information File 2: Crystallographic information files (CIF) for compounds 23c, 23d, and 23e. | ||
| Format: ZIP | Size: 297.4 KB | Download |
Data Availability Statement
All data that supports the findings of this study will be available in the published article and/or the supporting information of this article.
References
-
Szabó, K. J.; Selander, N., Eds. Organofluorine Chemistry: Synthesis, Modeling, and Applications; Wiley-VCH: Weinheim, Germany, 2021. doi:10.1002/9783527825158
Return to citation in text: [1] -
Haufe, G.; Leroux, F. R., Eds. Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals; Academic Press: London, UK, 2019. doi:10.1016/c2016-0-03808-1
Return to citation in text: [1] -
Tressaud, A. Fluorine: A Paradoxical Element; Elsevier: Amsterdam, Netherlands, 2019. doi:10.1016/c2016-0-02485-3
Return to citation in text: [1] -
Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830
Return to citation in text: [1] -
Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. iScience 2020, 23, 101467. doi:10.1016/j.isci.2020.101467
Return to citation in text: [1] -
Han, J.; Kiss, L.; Mei, H.; Remete, A. M.; Ponikvar-Svet, M.; Sedgwick, D. M.; Roman, R.; Fustero, S.; Moriwaki, H.; Soloshonok, V. A. Chem. Rev. 2021, 121, 4678–4742. doi:10.1021/acs.chemrev.0c01263
Return to citation in text: [1] -
Wang, Y.; Yang, X.; Meng, Y.; Wen, Z.; Han, R.; Hu, X.; Sun, B.; Kang, F.; Li, B.; Zhou, D.; Wang, C.; Wang, G. Chem. Rev. 2024, 124, 3494–3589. doi:10.1021/acs.chemrev.3c00826
Return to citation in text: [1] -
Václavík, J.; Klimánková, I.; Budinská, A.; Beier, P. Eur. J. Org. Chem. 2018, 3554–3593. doi:10.1002/ejoc.201701590
Return to citation in text: [1] -
Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910
Return to citation in text: [1] -
Bianchi, D.; Cesti, P.; Spezia, S.; Garavaglia, C.; Mirenna, L. J. Agric. Food Chem. 1991, 39, 197–201. doi:10.1021/jf00001a040
Return to citation in text: [1] -
N'Go, I.; Golten, S.; Ardá, A.; Cañada, J.; Jiménez‐Barbero, J.; Linclau, B.; Vincent, S. P. Chem. – Eur. J. 2014, 20, 106–112. doi:10.1002/chem.201303693
Return to citation in text: [1] -
Sari, O.; Bassit, L.; Gavegnano, C.; McBrayer, T. R.; McCormick, L.; Cox, B.; Coats, S. J.; Amblard, F.; Schinazi, R. F. Tetrahedron Lett. 2017, 58, 642–644. doi:10.1016/j.tetlet.2017.01.006
Return to citation in text: [1] -
Kirsch, P.; Bremer, M.; Huber, F.; Lannert, H.; Ruhl, A.; Lieb, M.; Wallmichrath, T. J. Am. Chem. Soc. 2001, 123, 5414–5417. doi:10.1021/ja010024l
Return to citation in text: [1] -
Yamada, S.; Tamamoto, K.; Kida, T.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 9442–9454. doi:10.1039/c7ob02399e
Return to citation in text: [1] -
Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 1495–1509. doi:10.1039/c6ob02431a
Return to citation in text: [1] -
Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013
Return to citation in text: [1] -
Kumon, T.; Hashishita, S.; Kida, T.; Yamada, S.; Ishihara, T.; Konno, T. Beilstein J. Org. Chem. 2018, 14, 148–154. doi:10.3762/bjoc.14.10
Return to citation in text: [1] -
Ohsato, H.; Morita, M.; Yamada, S.; Agou, T.; Fukumoto, H.; Konno, T. Mol. Syst. Des. Eng. 2022, 7, 1129–1137. doi:10.1039/d2me00055e
Return to citation in text: [1] -
Ohsato, H.; Kawauchi, K.; Yamada, S.; Konno, T. Chem. Rec. 2023, 23, e202300080. doi:10.1002/tcr.202300080
Return to citation in text: [1] -
Boydell, A. J.; Vinader, V.; Linclau, B. Angew. Chem., Int. Ed. 2004, 43, 5677–5679. doi:10.1002/anie.200460746
Return to citation in text: [1] -
Linclau, B.; Boydell, A. J.; Timofte, R. S.; Brown, K. J.; Vinader, V.; Weymouth-Wilson, A. C. Org. Biomol. Chem. 2009, 7, 803–814. doi:10.1039/b817260a
Return to citation in text: [1] -
Yamashika, K.; Morishitabara, S.; Yamada, S.; Kubota, T.; Konno, T. J. Fluorine Chem. 2018, 207, 24–37. doi:10.1016/j.jfluchem.2017.12.013
Return to citation in text: [1] -
Fontenelle, C. Q.; Tizzard, G. J.; Linclau, B. J. Fluorine Chem. 2015, 174, 95–101. doi:10.1016/j.jfluchem.2014.07.015
Return to citation in text: [1] -
Konno, T.; Hoshino, T.; Kida, T.; Takano, S.; Ishihara, T. J. Fluorine Chem. 2013, 152, 106–113. doi:10.1016/j.jfluchem.2013.02.013
Return to citation in text: [1] -
Soloshonok, V. A.; Catt, H. T.; Ono, T. J. Fluorine Chem. 2010, 131, 261–265. doi:10.1016/j.jfluchem.2009.10.013
Return to citation in text: [1] [2] -
Nagy, P.; Ueki, H.; Berbasov, D. O.; Soloshonok, V. A. J. Fluorine Chem. 2008, 129, 409–415. doi:10.1016/j.jfluchem.2008.02.001
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Soloshonok, I. V.; Kukhar, V. P.; Svedas, V. K. J. Org. Chem. 1998, 63, 1878–1884. doi:10.1021/jo971777m
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ono, T. J. Org. Chem. 1997, 62, 3030–3031. doi:10.1021/jo970425c
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ono, T.; Soloshonok, I. V. J. Org. Chem. 1997, 62, 7538–7539. doi:10.1021/jo9710238
Return to citation in text: [1] [2] -
Ono, T.; Kukhar, V. P.; Soloshonok, V. A. J. Org. Chem. 1996, 61, 6563–6569. doi:10.1021/jo960503g
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Kukhar, V. P. Tetrahedron 1996, 52, 6953–6964. doi:10.1016/0040-4020(96)00300-6
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ono, T. Tetrahedron 1996, 52, 14701–14712. doi:10.1016/0040-4020(96)00920-9
Return to citation in text: [1] [2] -
Tamamoto, K.; Yamada, S.; Konno, T. Beilstein J. Org. Chem. 2018, 14, 2375–2383. doi:10.3762/bjoc.14.213
Return to citation in text: [1] -
Braconi, E.; Cramer, N. Angew. Chem., Int. Ed. 2022, 61, e202112148. doi:10.1002/anie.202112148
Return to citation in text: [1] -
Hou, W.; Tang, C.; Liu, G.; Huang, Z. Organometallics 2022, 41, 3115–3121. doi:10.1021/acs.organomet.2c00321
Return to citation in text: [1] -
In the NOESY spectrum of (R)-16c, a cross-peak was observed between the methine or methyl protons and the ortho proton of the 4-chlorophenyl group. This indicates that the 4-chlorophenyl and 1-phenylethyl groups are in close proximity, i.e., the stereochemistry of the imine is E.
Return to citation in text: [1] -
The HF elimination product, 21b, was too unstable in silica gel column chromatography to be isolated in a pure form, and hence its identification by 1H NMR analysis could not be carried out. A similar compound, however, N-benzylidene-(1-phenyl)-2,3,3-trifluoro-1,4-pentdienylamine, could be easily isolated, and the structure could be completely identified (see Supporting Information File 1). Accordingly, it was determined unambiguously that 21b also has an azatriene structure, on the basis of the comparison of the 19F NMR chemical shifts for 21b and N-benzylidene-(1-phenyl)-2,3,3-trifluoro-1,4-pentdienylamine.
Return to citation in text: [1] -
Electrocyclization of 5,6-difluoro-3-azaocta-1,3,5,7-tetraene derivative which resulted from two HF eliminations of the imine (R)-16b under the influence of DBU might take place, giving the corresponding azocine derivative 22b. The details are currently under investigation. See Supporting Information File 1.
Return to citation in text: [1]
| 1. | Szabó, K. J.; Selander, N., Eds. Organofluorine Chemistry: Synthesis, Modeling, and Applications; Wiley-VCH: Weinheim, Germany, 2021. doi:10.1002/9783527825158 |
| 2. | Haufe, G.; Leroux, F. R., Eds. Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals; Academic Press: London, UK, 2019. doi:10.1016/c2016-0-03808-1 |
| 3. | Tressaud, A. Fluorine: A Paradoxical Element; Elsevier: Amsterdam, Netherlands, 2019. doi:10.1016/c2016-0-02485-3 |
| 13. | Kirsch, P.; Bremer, M.; Huber, F.; Lannert, H.; Ruhl, A.; Lieb, M.; Wallmichrath, T. J. Am. Chem. Soc. 2001, 123, 5414–5417. doi:10.1021/ja010024l |
| 14. | Yamada, S.; Tamamoto, K.; Kida, T.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 9442–9454. doi:10.1039/c7ob02399e |
| 15. | Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 1495–1509. doi:10.1039/c6ob02431a |
| 16. | Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013 |
| 17. | Kumon, T.; Hashishita, S.; Kida, T.; Yamada, S.; Ishihara, T.; Konno, T. Beilstein J. Org. Chem. 2018, 14, 148–154. doi:10.3762/bjoc.14.10 |
| 38. | Electrocyclization of 5,6-difluoro-3-azaocta-1,3,5,7-tetraene derivative which resulted from two HF eliminations of the imine (R)-16b under the influence of DBU might take place, giving the corresponding azocine derivative 22b. The details are currently under investigation. See Supporting Information File 1. |
| 10. | Bianchi, D.; Cesti, P.; Spezia, S.; Garavaglia, C.; Mirenna, L. J. Agric. Food Chem. 1991, 39, 197–201. doi:10.1021/jf00001a040 |
| 11. | N'Go, I.; Golten, S.; Ardá, A.; Cañada, J.; Jiménez‐Barbero, J.; Linclau, B.; Vincent, S. P. Chem. – Eur. J. 2014, 20, 106–112. doi:10.1002/chem.201303693 |
| 12. | Sari, O.; Bassit, L.; Gavegnano, C.; McBrayer, T. R.; McCormick, L.; Cox, B.; Coats, S. J.; Amblard, F.; Schinazi, R. F. Tetrahedron Lett. 2017, 58, 642–644. doi:10.1016/j.tetlet.2017.01.006 |
| 25. | Soloshonok, V. A.; Catt, H. T.; Ono, T. J. Fluorine Chem. 2010, 131, 261–265. doi:10.1016/j.jfluchem.2009.10.013 |
| 26. | Nagy, P.; Ueki, H.; Berbasov, D. O.; Soloshonok, V. A. J. Fluorine Chem. 2008, 129, 409–415. doi:10.1016/j.jfluchem.2008.02.001 |
| 27. | Soloshonok, V. A.; Soloshonok, I. V.; Kukhar, V. P.; Svedas, V. K. J. Org. Chem. 1998, 63, 1878–1884. doi:10.1021/jo971777m |
| 28. | Soloshonok, V. A.; Ono, T. J. Org. Chem. 1997, 62, 3030–3031. doi:10.1021/jo970425c |
| 29. | Soloshonok, V. A.; Ono, T.; Soloshonok, I. V. J. Org. Chem. 1997, 62, 7538–7539. doi:10.1021/jo9710238 |
| 30. | Ono, T.; Kukhar, V. P.; Soloshonok, V. A. J. Org. Chem. 1996, 61, 6563–6569. doi:10.1021/jo960503g |
| 31. | Soloshonok, V. A.; Kukhar, V. P. Tetrahedron 1996, 52, 6953–6964. doi:10.1016/0040-4020(96)00300-6 |
| 32. | Soloshonok, V. A.; Ono, T. Tetrahedron 1996, 52, 14701–14712. doi:10.1016/0040-4020(96)00920-9 |
| 8. | Václavík, J.; Klimánková, I.; Budinská, A.; Beier, P. Eur. J. Org. Chem. 2018, 3554–3593. doi:10.1002/ejoc.201701590 |
| 9. | Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910 |
| 36. | In the NOESY spectrum of (R)-16c, a cross-peak was observed between the methine or methyl protons and the ortho proton of the 4-chlorophenyl group. This indicates that the 4-chlorophenyl and 1-phenylethyl groups are in close proximity, i.e., the stereochemistry of the imine is E. |
| 4. | Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830 |
| 5. | Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. iScience 2020, 23, 101467. doi:10.1016/j.isci.2020.101467 |
| 6. | Han, J.; Kiss, L.; Mei, H.; Remete, A. M.; Ponikvar-Svet, M.; Sedgwick, D. M.; Roman, R.; Fustero, S.; Moriwaki, H.; Soloshonok, V. A. Chem. Rev. 2021, 121, 4678–4742. doi:10.1021/acs.chemrev.0c01263 |
| 7. | Wang, Y.; Yang, X.; Meng, Y.; Wen, Z.; Han, R.; Hu, X.; Sun, B.; Kang, F.; Li, B.; Zhou, D.; Wang, C.; Wang, G. Chem. Rev. 2024, 124, 3494–3589. doi:10.1021/acs.chemrev.3c00826 |
| 37. | The HF elimination product, 21b, was too unstable in silica gel column chromatography to be isolated in a pure form, and hence its identification by 1H NMR analysis could not be carried out. A similar compound, however, N-benzylidene-(1-phenyl)-2,3,3-trifluoro-1,4-pentdienylamine, could be easily isolated, and the structure could be completely identified (see Supporting Information File 1). Accordingly, it was determined unambiguously that 21b also has an azatriene structure, on the basis of the comparison of the 19F NMR chemical shifts for 21b and N-benzylidene-(1-phenyl)-2,3,3-trifluoro-1,4-pentdienylamine. |
| 23. | Fontenelle, C. Q.; Tizzard, G. J.; Linclau, B. J. Fluorine Chem. 2015, 174, 95–101. doi:10.1016/j.jfluchem.2014.07.015 |
| 24. | Konno, T.; Hoshino, T.; Kida, T.; Takano, S.; Ishihara, T. J. Fluorine Chem. 2013, 152, 106–113. doi:10.1016/j.jfluchem.2013.02.013 |
| 33. | Tamamoto, K.; Yamada, S.; Konno, T. Beilstein J. Org. Chem. 2018, 14, 2375–2383. doi:10.3762/bjoc.14.213 |
| 22. | Yamashika, K.; Morishitabara, S.; Yamada, S.; Kubota, T.; Konno, T. J. Fluorine Chem. 2018, 207, 24–37. doi:10.1016/j.jfluchem.2017.12.013 |
| 34. | Braconi, E.; Cramer, N. Angew. Chem., Int. Ed. 2022, 61, e202112148. doi:10.1002/anie.202112148 |
| 35. | Hou, W.; Tang, C.; Liu, G.; Huang, Z. Organometallics 2022, 41, 3115–3121. doi:10.1021/acs.organomet.2c00321 |
| 20. | Boydell, A. J.; Vinader, V.; Linclau, B. Angew. Chem., Int. Ed. 2004, 43, 5677–5679. doi:10.1002/anie.200460746 |
| 21. | Linclau, B.; Boydell, A. J.; Timofte, R. S.; Brown, K. J.; Vinader, V.; Weymouth-Wilson, A. C. Org. Biomol. Chem. 2009, 7, 803–814. doi:10.1039/b817260a |
| 18. | Ohsato, H.; Morita, M.; Yamada, S.; Agou, T.; Fukumoto, H.; Konno, T. Mol. Syst. Des. Eng. 2022, 7, 1129–1137. doi:10.1039/d2me00055e |
| 19. | Ohsato, H.; Kawauchi, K.; Yamada, S.; Konno, T. Chem. Rec. 2023, 23, e202300080. doi:10.1002/tcr.202300080 |
| 25. | Soloshonok, V. A.; Catt, H. T.; Ono, T. J. Fluorine Chem. 2010, 131, 261–265. doi:10.1016/j.jfluchem.2009.10.013 |
| 26. | Nagy, P.; Ueki, H.; Berbasov, D. O.; Soloshonok, V. A. J. Fluorine Chem. 2008, 129, 409–415. doi:10.1016/j.jfluchem.2008.02.001 |
| 27. | Soloshonok, V. A.; Soloshonok, I. V.; Kukhar, V. P.; Svedas, V. K. J. Org. Chem. 1998, 63, 1878–1884. doi:10.1021/jo971777m |
| 28. | Soloshonok, V. A.; Ono, T. J. Org. Chem. 1997, 62, 3030–3031. doi:10.1021/jo970425c |
| 29. | Soloshonok, V. A.; Ono, T.; Soloshonok, I. V. J. Org. Chem. 1997, 62, 7538–7539. doi:10.1021/jo9710238 |
| 30. | Ono, T.; Kukhar, V. P.; Soloshonok, V. A. J. Org. Chem. 1996, 61, 6563–6569. doi:10.1021/jo960503g |
| 31. | Soloshonok, V. A.; Kukhar, V. P. Tetrahedron 1996, 52, 6953–6964. doi:10.1016/0040-4020(96)00300-6 |
| 32. | Soloshonok, V. A.; Ono, T. Tetrahedron 1996, 52, 14701–14712. doi:10.1016/0040-4020(96)00920-9 |
© 2024 Kabumoto et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.