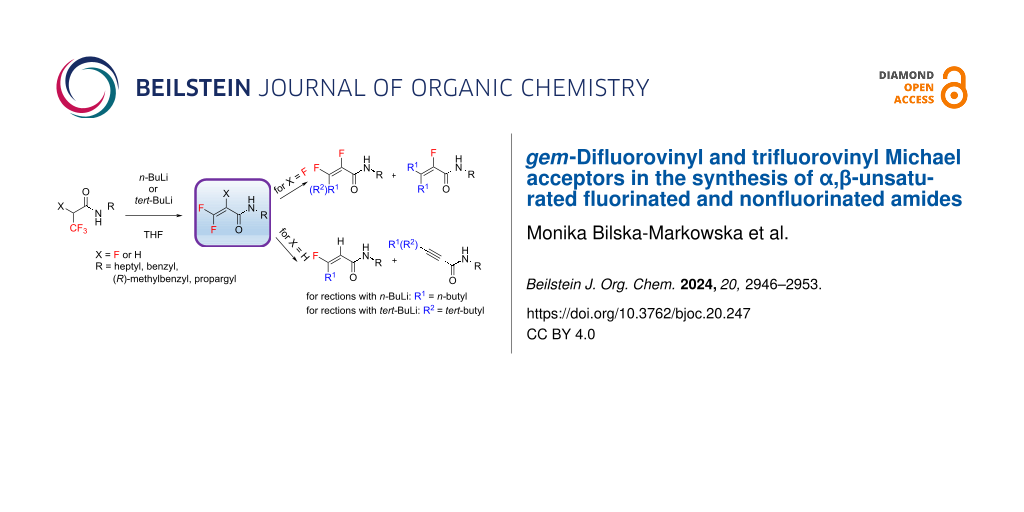
Abstract
The incorporation of fluorine atoms within the structure of organic compounds is known to exert a significant impact on their electronic properties, thereby modulating their reactivity in diverse chemical transformations. In the context of our investigation, we observed a striking illustration of this phenomenon. A Michael addition involving gem-difluorovinyl and trifluorovinyl acceptors was successfully achieved, demonstrating high stereoselectivity. This selectivity was further elucidated through theoretical calculations. Using this methodology, a series of new α,β-unsaturated amides, both fluorinated and nonfluorinated, were synthesized.

Graphical Abstract
Introduction
The Michael reaction, characterized by the addition of stable carbon nucleophiles to unsaturated compounds with electron-withdrawing groups, is a cornerstone in constructing carbon–carbon and carbon–heteroatom bonds [1]. It is instrumental in synthesizing natural products [2-5] and pharmaceuticals [6], underlining its significance in organic chemistry. Recent advancements have broadened the scope of Michael donors and acceptors to encompass fluorine-containing compounds, enhancing the reaction's utility in synthesizing fluorinated derivatives [7,8]. Shibata and colleagues pioneered the use of fluorinated Michael donors, notably achieving enantioselective addition of 1-fluorobis(phenylsulfonyl)methane (FBSM) to α,β-unsaturated ketones with cinchona alkaloids [9]. Fluorinated Michael acceptors usually contain one fluorine atom or a trifluoromethyl group in the structure [10-12]. There are also known examples of gem-difluoroalkenes being used as Michael acceptors [13-17]. The Michael addition with fluorinated acceptors finds application in the synthesis of, among others, fluorinated amino acids, which can be a structural motif in many biologically active compounds [18]. There are also known studies on the incorporation of highly reactive fluorinated Michael acceptors into peptide structures, which can act as the link between an active molecule and its cellular target [19,20]. Such endeavors hint at the potential of fluorinated acceptors in designing fluorinated peptidomimetics, an area attracting global research interest [21-24].
In our laboratory, we have explored the synthesis of 3,3,3-trifluoro- and 2,3,3,3-tetrafluoro-N-substituted propanamides, contributing to the field of fluorinated amides [25]. We have also investigated deprotonation at the α position of other fluorinated carbonyl derivatives as a route to new building blocks [26]. Despite the known instability of trifluoromethylated carbanions [27], their catalytic application has yielded valuable products [28-30]. gem-Difluoroalkenes and trifluoroalkenes are excellent acceptors in the Michael addition reactions. There are also known examples of the use of gem-difluoroalkenes and trifluoroalkenes in reactions with Grignard reagents [13,31]. Although, similar compounds are reported to be unstable molecules that are prone to decomposition under reaction conditions [32,33].
The goal of this work was the formation of gem-difluoro- and trifluorovinyl Michael acceptors by using organolithium reagents (Scheme 1), revealing new avenues in fluorinated unsaturated amide synthesis, which are present in numerous natural products, pharmaceuticals, and polymers [34-38]. The obtained α,β-unsaturated amides may represent promising structural motifs for further synthesis, e.g., via pericyclic reactions or nucleophilic additions.
![[1860-5397-20-247-i1]](/bjoc/content/inline/1860-5397-20-247-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Generation of gem-difluorovinyl and trifluorovinyl Michael acceptors and their use in the synthesis of α,β-unsaturated fluoroamides.
Scheme 1: Generation of gem-difluorovinyl and trifluorovinyl Michael acceptors and their use in the synthesis...
Results and Discussion
We commenced our research by screening the nature of the base to generate carbanion at alpha position. We chose 2,3,3,3-tetrafluoro-N-heptylpropanamide, obtained according to the procedure developed earlier in our laboratory [25], for our model reaction. The reactions were carried out under inert gas conditions in anhydrous solvents (THF or DCM) at −78 °C for 3 h, using several bases and electrophiles (Table 1). The use of electrophiles in the first test reactions was to confirm the generation of a carbanion, which was to be evidenced by a substitution reaction at the alpha position. We started testing the different bases with lithium bis(trimethylsilyl)amide [39]. The reactions did not take place in the presence of LiHMDS (Table 1, entries 1 and 2), using either benzyl bromide or methyl iodide as electrophiles. Next, TiCl4 as metal enolate mediator was applied. In the presence of both, Et3N as well as N,N,N′,N′-tetramethyl-1,3-propanediamine no reaction was observed (Table 1, entries 3–6) [40]. With titanium chloride and n-BuLi, low conversion of the starting material and obtained product Z-9a was characteristic (Table 1, entry 7). A slightly higher reactivity was achieved when the BF3·(OEt2) was used instead of TiCl4 (Table 1, entry 8) [28]. The reactions were monitored by 19F NMR of the crude mixtures. The full conversion was reached by applying exclusively n-BuLi, but the formed product was not the anticipated α-substituted compound (Table 1, entry 9). The NMR analysis revealed that the obtained compounds were Michael addition products. The formation of the presented compounds (Table 1) was due to the earlier generation of gem-difluoroalkenes by the elimination of one of the fluorine atoms from the CF3 group, proving that both gem-difluoroalkenes and the double bond of product Z-9a were excellent Michael acceptors. This confirmed that electrophiles were not involved in the reaction. We therefore focused only on using n-BuLi, which, as it turned out, acted as both the base and Michael's donor (Table 1, entry 10).
Table 1: Optimization of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-247-i7.svg?max-width=637&scale=1.0)
|
||||
| Entry | Base/Lewis acid, base |
Electrophile
RX, RCHO |
Solvent | Result |
| 1 | LiHMDS, 2 equiv | BnBr, 2 equiv | THF | n. r.a |
| 2 | LiHMDS, 2 equiv | MeI, 2 equiv | THF | n. r. |
| 3 |
Et3N, 3 equiv
TiCl4, 1.2 equiv |
MeI, 2 equiv | DCM | n. r. |
| 4 |
TMPDA, 1.2 equiv
TiCl4, 1.2 equiv |
MeI, 2 equiv | DCM | n. r. |
| 5 |
Et3N, 3 equiv
TiCl4, 1.2 equiv |
PhCHO, 2 equiv | DCM | n. r. |
| 6 |
TMPDA, 1.2 equiv
TiCl4, 1.2 equiv |
PhCHO, 2 equiv | DCM | n. r. |
| 7 |
n-BuLi, 4 equiv
TiCl4, 1.2 equiv |
PhCHO, 2 equiv | THF | traces of Z-9ab |
| 8 |
n-BuLi, 4 equiv
BF3·OEt2, 1.2 equiv |
PhCHO, 2 equiv | THF | 1:2.8:0.3:traces (1a/Z-9a/10a/E-9a)b |
| 9 | n-BuLi, 4 equiv | MeI, 2 equiv | THF | 0:1:0.15:traces (1a/Z-9a/10a/E-9a)b |
| 10 | n-BuLi, 4 equiv | – | THF | 0:1:0.1:0 (1a/Z-9a/10a/E-9a)b |
aNo reaction. bDetermined by 19F NMR spectroscopy of crude reaction mixtures.
Having the optimized conditions in hand, we subjected other 2,3,3,3-tetrafluoropropanamides to the same process. Both amides substituted by electron-withdrawing and electron-donating groups proved to be suitable substrates for this reaction, providing the corresponding Michael addition products. These highly stable compounds were isolated after purification on silica gel in good yields (Scheme 2) and characterized by spectroscopic methods. The reaction proceeded with very high Z-stereoselectivity (Scheme 2, compounds 9a–d). In the 19F NMR spectra of crude mixtures, only trace amounts of E-isomer of products 9 were identified. The fluorine atom signals of 9a–d were located at approximately −112 ppm (triplets, J ≈ 27 Hz, Fβ) and at −155 to −159 ppm (multiplets, Fα). The stereochemistry was determined by 19F{1H} NMR spectroscopy. The observed coupling constants J ≈ 2 Hz between vinylic fluorine atoms were typical and confirmed that the Z isomers were obtained predominantly [41]. The vicinal coupling constant between the Fβ and Hγ atoms amounted approximately 27 Hz in cases of 9a–d. Based on these findings, we concluded that the dihedral angle between the Fβ and Hγ atoms is approximately 150° [42-45]. These data were consistent with DFT calculations (see Supporting Information File 1). The reaction of the amide 1d with n-BuLi resulted in a surprising outcome. In this case, product 9d and only traces of the expected product 10d were received, as indicated by the 19F NMR spectrum of the crude reaction mixture. Interestingly, for this reaction we also observed that the Z-9d/E-9d products were obtained in a ratio of 1:0.2. However, the main Z-isomer was only isolated and fully characterized.
![[1860-5397-20-247-i2]](/bjoc/content/inline/1860-5397-20-247-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Formation of α,β-difluorinated and α-fluorinated α,β-unsaturated amides.
Scheme 2: Formation of α,β-difluorinated and α-fluorinated α,β-unsaturated amides.
In our subsequent investigation of the 3,3,3-trifluoropropanamides substrate 2a–d scope, we observed that gem-difluoroalkenes produced β-fluoro-unsaturated amides 11a–d (Scheme 3). In these reactions, we used conditions previously optimised for derivatives 1a–d (n-BuLi 4 equiv, THF, −78 °C, 3 h). The amides 11a–d preferred HF elimination over engaging in another Michael reaction, leading to the formation of products 12a–d as illustrated in Scheme 3. This outcome suggests a significant role of the fluorine atom at the alpha position, where its electron-withdrawing effect likely influenced the feasibility of the following Michael addition for compounds 9a–d (Scheme 2). Interestingly, such a reaction pathway was absent for derivatives 11a–d, where the alpha-positioned proton exhibited a low pKa, favouring an easy elimination reaction. This is supported by the higher yields of products 12a–d compared to their 11a–d counterparts. The exclusive formation of E isomers in compounds 11a–d was confirmed by the observed coupling constants (J ≈ 21 Hz) between the vinylic proton and the fluorine atom [41]. Moreover, the vicinal coupling constants between the Fβ and Hγ atoms ranging from 25–26 Hz for 11a–d suggest a dihedral angle of approximately 170° between these atoms [42-45]. These findings are in alignment with DFT calculations (see Supporting Information File 1) and corroborate data for compounds 9a–d.
![[1860-5397-20-247-i3]](/bjoc/content/inline/1860-5397-20-247-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Formation of β-fluorinated and nonfluorinated α,β-unsaturated amides.
Scheme 3: Formation of β-fluorinated and nonfluorinated α,β-unsaturated amides.
We further decided to use tert-BuLi in our research, considering its role as a stronger base and simultaneously as a weak nucleophile. We first performed the reaction with 2 equiv of tert-BuLi, which did not yield the expected results. The substrate was still observed in the reaction mixture. Only the use of 4 equiv of base gave the desired findings. The treatment of compounds 1a–d and 2a–d, respectively, with tert-BuLi induced the carbanion formation followed by an addition–elimination reaction, affording the corresponding fluorinated 13a–d (Scheme 4) and nonfluorinated 14a–d (Scheme 5) unsaturated products. Also this time, for compounds 13a–d, the formation of only Z isomers was observed (Scheme 4). The stereochemistry was determined by 19F{1H} NMR spectroscopy methods by the observed coupling constants J ≈ 6 Hz between vinylic fluorine atoms [41]. Due to the steric hindrance, these compounds did not serve as good Michael acceptors for the next step.
![[1860-5397-20-247-i4]](/bjoc/content/inline/1860-5397-20-247-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Michael addition of 1a–d with tert-BuLi.
Scheme 4: Michael addition of 1a–d with tert-BuLi.
Only elimination products 14a–d were obtained from trifluorinated amides 2a–d, showing good yields (Scheme 5).
![[1860-5397-20-247-i5]](/bjoc/content/inline/1860-5397-20-247-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Michael addition of 2a–d with tert-BuLi.
Scheme 5: Michael addition of 2a–d with tert-BuLi.
We also tried to perform a substitution reaction by treating compounds 1a and 2a with tert-BuLi, employing methyl iodide as the electrophile. However, similar to previous reactions, this did not yield substitution products at the alpha position, but to the addition–elimination reaction products. More importantly, the application of 8 equiv of tert-BuLi induced the formation of N-methylation products (Scheme 6).
![[1860-5397-20-247-i6]](/bjoc/content/inline/1860-5397-20-247-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Formation of N-methylation products.
Scheme 6: Formation of N-methylation products.
Compounds 15a (15a’) and 16a (16a’) existed as two rotamers, in ratios of 1:1.15 and 1:1.76, respectively, with the predominant cisoid isomer. Transoid (trans 15a(16a)) isomers contained a larger substituent at nitrogen located in the opposite direction to the carbonyl group, while cisoid (cis 15a’ (16a’)) isomers featured a smaller substituent at nitrogen located in the opposite direction to the carbonyl group. We determined the quantitative ratio as well as the cis/trans configuration of isomers by analyzing the differences in the chemical shift values in the 1H NMR spectra for NCH3 and NCH2- proton groups, based on our previous studies concerning fluorinated amides [46,47].
Conclusion
In this study, we have established that tri- and tetrafluorinated amides, featuring a CF3 group at the α position, serve as effective motifs for designing stable gem-difluorovinyl and trifluorovinyl Michael acceptors. To our knowledge, this represents the inaugural instance of employing potent bases such as n-BuLi and tert-BuLi to fulfill dual roles as both base catalysts and Michael donors. The reactions exhibited remarkable stereoselectivity, a finding elucidated by DFT analysis. These results mark significant progress toward the synthesis of novel fluorinated building blocks. Our team is currently exploring the application of this methodology to amino acid substrates, aiming to contribute further to the burgeoning field of fluorinated peptidomimetics.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, DFT calculations, characterization data, and copies of 1H, 13C, 19F NMR and 1H−13C HSQC spectra. | ||
| Format: PDF | Size: 11.1 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Reznikov, A. N.; Klimochkin, Y. N. Synthesis 2020, 52, 781–795. doi:10.1055/s-0039-1690044
Return to citation in text: [1] -
Kaplan, W.; Khatri, H. R.; Nagorny, P. J. Am. Chem. Soc. 2016, 138, 7194–7198. doi:10.1021/jacs.6b04029
Return to citation in text: [1] -
Hui, C.; Pu, F.; Xu, J. Chem. – Eur. J. 2017, 23, 4023–4036. doi:10.1002/chem.201604110
Return to citation in text: [1] -
Li, L.; Yang, Q.; Wang, Y.; Jia, Y. Angew. Chem., Int. Ed. 2015, 54, 6255–6259. doi:10.1002/anie.201411338
Return to citation in text: [1] -
Zhang, Y.; Wang, W. Catal. Sci. Technol. 2012, 2, 42–53. doi:10.1039/c1cy00334h
Return to citation in text: [1] -
Santos, P. P.; Veiros, L. F. Tetrahedron 2020, 76, 131373. doi:10.1016/j.tet.2020.131373
Return to citation in text: [1] -
Valero, G.; Companyó, X.; Rios, R. Chem. – Eur. J. 2011, 17, 2018–2037. doi:10.1002/chem.201001546
Return to citation in text: [1] -
Barron, B.; Edge, C.; Fenner, S.; Shrives, H.; Sollis, S.; Whiting, M.; Valette, D. Org. Lett. 2024, 26, 1533–1538. doi:10.1021/acs.orglett.3c03694
Return to citation in text: [1] -
Furukawa, T.; Shibata, N.; Mizuta, S.; Nakamura, S.; Toru, T.; Shiro, M. Angew. Chem., Int. Ed. 2008, 47, 8051–8054. doi:10.1002/anie.200802904
Return to citation in text: [1] -
Huang, X.; Besset, T.; Jubault, P.; Couve-Bonnaire, S. J. Org. Chem. 2022, 87, 9210–9221. doi:10.1021/acs.joc.2c00937
Return to citation in text: [1] -
Huang, X.; Besset, T.; Jubault, P.; Couve‐Bonnaire, S. Adv. Synth. Catal. 2023, 365, 2467–2486. doi:10.1002/adsc.202300565
Return to citation in text: [1] -
Ramb, D. C.; Lerchen, A.; Kischkewitz, M.; Beutel, B.; Fustero, S.; Haufe, G. Eur. J. Org. Chem. 2016, 1751–1759. doi:10.1002/ejoc.201600088
Return to citation in text: [1] -
Adam, A. T.; Fronczek, F. R.; Colby, D. A. Org. Lett. 2020, 22, 2630–2633. doi:10.1021/acs.orglett.0c00599
Return to citation in text: [1] [2] -
Ma, Y.; Mao, K.; Chen, Y.; Lv, L.; Li, Z. Tetrahedron Lett. 2022, 100, 153902. doi:10.1016/j.tetlet.2022.153902
Return to citation in text: [1] -
Gao, G.; Li, Z. New J. Chem. 2023, 47, 6171–6175. doi:10.1039/d2nj06338g
Return to citation in text: [1] -
Zong, Y.; Tsui, G. C. Org. Lett. 2024, 26, 1261–1264. doi:10.1021/acs.orglett.4c00095
Return to citation in text: [1] -
Li, M.; Tsui, G. C. Org. Lett. 2024, 26, 376–379. doi:10.1021/acs.orglett.3c04037
Return to citation in text: [1] -
Aparici, I.; Guerola, M.; Dialer, C.; Simón-Fuentes, A.; Sánchez-Roselló, M.; del Pozo, C.; Fustero, S. Org. Lett. 2015, 17, 5412–5415. doi:10.1021/acs.orglett.5b02759
Return to citation in text: [1] -
Zhou, H.; Schmidt, D. M. Z.; Gerlt, J. A.; van der Donk, W. A. ChemBioChem 2003, 4, 1206–1215. doi:10.1002/cbic.200300654
Return to citation in text: [1] -
Volonterio, A.; Chiva, G.; Fustero, S.; Piera, J.; Sanchez Rosello, M.; Sani, M.; Zanda, M. Tetrahedron Lett. 2003, 44, 7019–7022. doi:10.1016/s0040-4039(03)01787-8
Return to citation in text: [1] -
Molteni, M.; Pesenti, C.; Sani, M.; Volonterio, A.; Zanda, M. J. Fluorine Chem. 2004, 125, 1735–1743. doi:10.1016/j.jfluchem.2004.09.014
Return to citation in text: [1] -
Altman, R. A.; Sharma, K. K.; Rajewski, L. G.; Toren, P. C.; Baltezor, M. J.; Pal, M.; Karad, S. N. ACS Chem. Neurosci. 2018, 9, 1735–1742. doi:10.1021/acschemneuro.8b00085
Return to citation in text: [1] -
Yang, M.-H.; Matikonda, S. S.; Altman, R. A. Org. Lett. 2013, 15, 3894–3897. doi:10.1021/ol401637n
Return to citation in text: [1] -
Van der Veken, P.; Senten, K.; Kertèsz, I.; De Meester, I.; Lambeir, A.-M.; Maes, M.-B.; Scharpé, S.; Haemers, A.; Augustyns, K. J. Med. Chem. 2005, 48, 1768–1780. doi:10.1021/jm0495982
Return to citation in text: [1] -
Bilska-Markowska, M.; Patyk-Kaźmierczak, E.; Lusina, A. Eur. J. Org. Chem. 2022, e202101378. doi:10.1002/ejoc.202101378
Return to citation in text: [1] [2] -
Bilska-Markowska, M.; Jankowski, W.; Hoffmann, M.; Kaźmierczak, M. Molecules 2022, 27, 5404. doi:10.3390/molecules27175404
Return to citation in text: [1] -
Uneyama, K.; Katagiri, T.; Amii, H. Acc. Chem. Res. 2008, 41, 817–829. doi:10.1021/ar7002573
Return to citation in text: [1] -
Shimada, T.; Yoshioka, M.; Konno, T.; Ishihara, T. Org. Lett. 2006, 8, 1129–1131. doi:10.1021/ol0531435
Return to citation in text: [1] [2] -
Brewitz, L.; Arteaga, F. A.; Yin, L.; Alagiri, K.; Kumagai, N.; Shibasaki, M. J. Am. Chem. Soc. 2015, 137, 15929–15939. doi:10.1021/jacs.5b11064
Return to citation in text: [1] -
Sun, Z.; Sun, B.; Kumagai, N.; Shibasaki, M. Org. Lett. 2018, 20, 3070–3073. doi:10.1021/acs.orglett.8b01109
Return to citation in text: [1] -
Watanabe, S.; Fujita, T.; Sakamoto, M.; Hosokawa, O.; Ikeda, N. Int. J. Mater. Prod. Technol. 1990, 5, 213–219.
Return to citation in text: [1] -
Yu, J.-S.; Noda, H.; Kumagai, N.; Shibasaki, M. Synlett 2019, 30, 488–492. doi:10.1055/s-0037-1611642
Return to citation in text: [1] -
Matsuzawa, A.; Noda, H.; Kumagai, N.; Shibasaki, M. J. Org. Chem. 2017, 82, 8304–8308. doi:10.1021/acs.joc.7b01381
Return to citation in text: [1] -
Shi, G.-q.; Cai, W.-l. J. Org. Chem. 1995, 60, 6289–6295. doi:10.1021/jo00125a013
Return to citation in text: [1] -
Gaikwad, N.; Nanduri, S.; Madhavi, Y. V. Eur. J. Med. Chem. 2019, 181, 111561. doi:10.1016/j.ejmech.2019.07.064
Return to citation in text: [1] -
Li, S.-N.; Li, B.; Yu, Z.-R.; Dai, S.-W.; Shen, S.-C.; Mao, M.; Gong, L.-X.; Feng, Y.; Jia, D.; Zhou, Y.; Tang, L.-C. ACS Appl. Polym. Mater. 2020, 2, 1874–1885. doi:10.1021/acsapm.0c00106
Return to citation in text: [1] -
Rozsar, D.; Formica, M.; Yamazaki, K.; Hamlin, T. A.; Dixon, D. J. J. Am. Chem. Soc. 2022, 144, 1006–1015. doi:10.1021/jacs.1c11898
Return to citation in text: [1] -
Verma, S.; Singh, V.; Jat, J. L.; Tiwari, B. J. Org. Chem. 2024, 89, 8201–8207. doi:10.1021/acs.joc.3c02478
Return to citation in text: [1] -
Skibinska, M.; Warowicka, A.; Koroniak, H.; Cytlak, T.; Crousse, B. Org. Lett. 2024, 26, 692–696. doi:10.1021/acs.orglett.3c04094
Return to citation in text: [1] -
Chen, J.-L.; You, Z.-W.; Qing, F.-L. J. Fluorine Chem. 2013, 155, 143–150. doi:10.1016/j.jfluchem.2013.07.017
Return to citation in text: [1] -
Dolbier, W. R., Jr. Guide to Fluorine NMR for Organic Chemists; John Wiley & Sons: Hoboken, NJ, USA, 2009. doi:10.1002/9780470483404
Return to citation in text: [1] [2] [3] -
Thibaudeau, C.; Plavec, J.; Chattopadhyaya, J. J. Org. Chem. 1998, 63, 4967–4984. doi:10.1021/jo980144k
Return to citation in text: [1] [2] -
San Fabián, J.; Guilleme, J.; Dı́ez, E. J. Magn. Reson. 1998, 133, 255–265. doi:10.1006/jmre.1998.1465
Return to citation in text: [1] [2] -
Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Eur. J. Org. Chem. 2018, 3844–3852. doi:10.1002/ejoc.201800631
Return to citation in text: [1] [2] -
Govil, G. Mol. Phys. 1971, 21, 953–957. doi:10.1080/00268977100102111
Return to citation in text: [1] [2] -
Bilska-Markowska, M.; Rapp, M.; Siodła, T.; Katrusiak, A.; Hoffmann, M.; Koroniak, H. New J. Chem. 2014, 38, 3819–3830. doi:10.1039/c4nj00317a
Return to citation in text: [1] -
Bilska-Markowska, M.; Siodla, T.; Patyk-Kaźmierczak, E.; Katrusiak, A.; Koroniak, H. New J. Chem. 2017, 41, 12631–12644. doi:10.1039/c7nj02986a
Return to citation in text: [1]
| 1. | Reznikov, A. N.; Klimochkin, Y. N. Synthesis 2020, 52, 781–795. doi:10.1055/s-0039-1690044 |
| 9. | Furukawa, T.; Shibata, N.; Mizuta, S.; Nakamura, S.; Toru, T.; Shiro, M. Angew. Chem., Int. Ed. 2008, 47, 8051–8054. doi:10.1002/anie.200802904 |
| 13. | Adam, A. T.; Fronczek, F. R.; Colby, D. A. Org. Lett. 2020, 22, 2630–2633. doi:10.1021/acs.orglett.0c00599 |
| 31. | Watanabe, S.; Fujita, T.; Sakamoto, M.; Hosokawa, O.; Ikeda, N. Int. J. Mater. Prod. Technol. 1990, 5, 213–219. |
| 7. | Valero, G.; Companyó, X.; Rios, R. Chem. – Eur. J. 2011, 17, 2018–2037. doi:10.1002/chem.201001546 |
| 8. | Barron, B.; Edge, C.; Fenner, S.; Shrives, H.; Sollis, S.; Whiting, M.; Valette, D. Org. Lett. 2024, 26, 1533–1538. doi:10.1021/acs.orglett.3c03694 |
| 32. | Yu, J.-S.; Noda, H.; Kumagai, N.; Shibasaki, M. Synlett 2019, 30, 488–492. doi:10.1055/s-0037-1611642 |
| 33. | Matsuzawa, A.; Noda, H.; Kumagai, N.; Shibasaki, M. J. Org. Chem. 2017, 82, 8304–8308. doi:10.1021/acs.joc.7b01381 |
| 6. | Santos, P. P.; Veiros, L. F. Tetrahedron 2020, 76, 131373. doi:10.1016/j.tet.2020.131373 |
| 27. | Uneyama, K.; Katagiri, T.; Amii, H. Acc. Chem. Res. 2008, 41, 817–829. doi:10.1021/ar7002573 |
| 2. | Kaplan, W.; Khatri, H. R.; Nagorny, P. J. Am. Chem. Soc. 2016, 138, 7194–7198. doi:10.1021/jacs.6b04029 |
| 3. | Hui, C.; Pu, F.; Xu, J. Chem. – Eur. J. 2017, 23, 4023–4036. doi:10.1002/chem.201604110 |
| 4. | Li, L.; Yang, Q.; Wang, Y.; Jia, Y. Angew. Chem., Int. Ed. 2015, 54, 6255–6259. doi:10.1002/anie.201411338 |
| 5. | Zhang, Y.; Wang, W. Catal. Sci. Technol. 2012, 2, 42–53. doi:10.1039/c1cy00334h |
| 28. | Shimada, T.; Yoshioka, M.; Konno, T.; Ishihara, T. Org. Lett. 2006, 8, 1129–1131. doi:10.1021/ol0531435 |
| 29. | Brewitz, L.; Arteaga, F. A.; Yin, L.; Alagiri, K.; Kumagai, N.; Shibasaki, M. J. Am. Chem. Soc. 2015, 137, 15929–15939. doi:10.1021/jacs.5b11064 |
| 30. | Sun, Z.; Sun, B.; Kumagai, N.; Shibasaki, M. Org. Lett. 2018, 20, 3070–3073. doi:10.1021/acs.orglett.8b01109 |
| 19. | Zhou, H.; Schmidt, D. M. Z.; Gerlt, J. A.; van der Donk, W. A. ChemBioChem 2003, 4, 1206–1215. doi:10.1002/cbic.200300654 |
| 20. | Volonterio, A.; Chiva, G.; Fustero, S.; Piera, J.; Sanchez Rosello, M.; Sani, M.; Zanda, M. Tetrahedron Lett. 2003, 44, 7019–7022. doi:10.1016/s0040-4039(03)01787-8 |
| 25. | Bilska-Markowska, M.; Patyk-Kaźmierczak, E.; Lusina, A. Eur. J. Org. Chem. 2022, e202101378. doi:10.1002/ejoc.202101378 |
| 18. | Aparici, I.; Guerola, M.; Dialer, C.; Simón-Fuentes, A.; Sánchez-Roselló, M.; del Pozo, C.; Fustero, S. Org. Lett. 2015, 17, 5412–5415. doi:10.1021/acs.orglett.5b02759 |
| 26. | Bilska-Markowska, M.; Jankowski, W.; Hoffmann, M.; Kaźmierczak, M. Molecules 2022, 27, 5404. doi:10.3390/molecules27175404 |
| 13. | Adam, A. T.; Fronczek, F. R.; Colby, D. A. Org. Lett. 2020, 22, 2630–2633. doi:10.1021/acs.orglett.0c00599 |
| 14. | Ma, Y.; Mao, K.; Chen, Y.; Lv, L.; Li, Z. Tetrahedron Lett. 2022, 100, 153902. doi:10.1016/j.tetlet.2022.153902 |
| 15. | Gao, G.; Li, Z. New J. Chem. 2023, 47, 6171–6175. doi:10.1039/d2nj06338g |
| 16. | Zong, Y.; Tsui, G. C. Org. Lett. 2024, 26, 1261–1264. doi:10.1021/acs.orglett.4c00095 |
| 17. | Li, M.; Tsui, G. C. Org. Lett. 2024, 26, 376–379. doi:10.1021/acs.orglett.3c04037 |
| 10. | Huang, X.; Besset, T.; Jubault, P.; Couve-Bonnaire, S. J. Org. Chem. 2022, 87, 9210–9221. doi:10.1021/acs.joc.2c00937 |
| 11. | Huang, X.; Besset, T.; Jubault, P.; Couve‐Bonnaire, S. Adv. Synth. Catal. 2023, 365, 2467–2486. doi:10.1002/adsc.202300565 |
| 12. | Ramb, D. C.; Lerchen, A.; Kischkewitz, M.; Beutel, B.; Fustero, S.; Haufe, G. Eur. J. Org. Chem. 2016, 1751–1759. doi:10.1002/ejoc.201600088 |
| 21. | Molteni, M.; Pesenti, C.; Sani, M.; Volonterio, A.; Zanda, M. J. Fluorine Chem. 2004, 125, 1735–1743. doi:10.1016/j.jfluchem.2004.09.014 |
| 22. | Altman, R. A.; Sharma, K. K.; Rajewski, L. G.; Toren, P. C.; Baltezor, M. J.; Pal, M.; Karad, S. N. ACS Chem. Neurosci. 2018, 9, 1735–1742. doi:10.1021/acschemneuro.8b00085 |
| 23. | Yang, M.-H.; Matikonda, S. S.; Altman, R. A. Org. Lett. 2013, 15, 3894–3897. doi:10.1021/ol401637n |
| 24. | Van der Veken, P.; Senten, K.; Kertèsz, I.; De Meester, I.; Lambeir, A.-M.; Maes, M.-B.; Scharpé, S.; Haemers, A.; Augustyns, K. J. Med. Chem. 2005, 48, 1768–1780. doi:10.1021/jm0495982 |
| 39. | Skibinska, M.; Warowicka, A.; Koroniak, H.; Cytlak, T.; Crousse, B. Org. Lett. 2024, 26, 692–696. doi:10.1021/acs.orglett.3c04094 |
| 34. | Shi, G.-q.; Cai, W.-l. J. Org. Chem. 1995, 60, 6289–6295. doi:10.1021/jo00125a013 |
| 35. | Gaikwad, N.; Nanduri, S.; Madhavi, Y. V. Eur. J. Med. Chem. 2019, 181, 111561. doi:10.1016/j.ejmech.2019.07.064 |
| 36. | Li, S.-N.; Li, B.; Yu, Z.-R.; Dai, S.-W.; Shen, S.-C.; Mao, M.; Gong, L.-X.; Feng, Y.; Jia, D.; Zhou, Y.; Tang, L.-C. ACS Appl. Polym. Mater. 2020, 2, 1874–1885. doi:10.1021/acsapm.0c00106 |
| 37. | Rozsar, D.; Formica, M.; Yamazaki, K.; Hamlin, T. A.; Dixon, D. J. J. Am. Chem. Soc. 2022, 144, 1006–1015. doi:10.1021/jacs.1c11898 |
| 38. | Verma, S.; Singh, V.; Jat, J. L.; Tiwari, B. J. Org. Chem. 2024, 89, 8201–8207. doi:10.1021/acs.joc.3c02478 |
| 25. | Bilska-Markowska, M.; Patyk-Kaźmierczak, E.; Lusina, A. Eur. J. Org. Chem. 2022, e202101378. doi:10.1002/ejoc.202101378 |
| 41. | Dolbier, W. R., Jr. Guide to Fluorine NMR for Organic Chemists; John Wiley & Sons: Hoboken, NJ, USA, 2009. doi:10.1002/9780470483404 |
| 46. | Bilska-Markowska, M.; Rapp, M.; Siodła, T.; Katrusiak, A.; Hoffmann, M.; Koroniak, H. New J. Chem. 2014, 38, 3819–3830. doi:10.1039/c4nj00317a |
| 47. | Bilska-Markowska, M.; Siodla, T.; Patyk-Kaźmierczak, E.; Katrusiak, A.; Koroniak, H. New J. Chem. 2017, 41, 12631–12644. doi:10.1039/c7nj02986a |
| 41. | Dolbier, W. R., Jr. Guide to Fluorine NMR for Organic Chemists; John Wiley & Sons: Hoboken, NJ, USA, 2009. doi:10.1002/9780470483404 |
| 42. | Thibaudeau, C.; Plavec, J.; Chattopadhyaya, J. J. Org. Chem. 1998, 63, 4967–4984. doi:10.1021/jo980144k |
| 43. | San Fabián, J.; Guilleme, J.; Dı́ez, E. J. Magn. Reson. 1998, 133, 255–265. doi:10.1006/jmre.1998.1465 |
| 44. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Eur. J. Org. Chem. 2018, 3844–3852. doi:10.1002/ejoc.201800631 |
| 45. | Govil, G. Mol. Phys. 1971, 21, 953–957. doi:10.1080/00268977100102111 |
| 41. | Dolbier, W. R., Jr. Guide to Fluorine NMR for Organic Chemists; John Wiley & Sons: Hoboken, NJ, USA, 2009. doi:10.1002/9780470483404 |
| 42. | Thibaudeau, C.; Plavec, J.; Chattopadhyaya, J. J. Org. Chem. 1998, 63, 4967–4984. doi:10.1021/jo980144k |
| 43. | San Fabián, J.; Guilleme, J.; Dı́ez, E. J. Magn. Reson. 1998, 133, 255–265. doi:10.1006/jmre.1998.1465 |
| 44. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Eur. J. Org. Chem. 2018, 3844–3852. doi:10.1002/ejoc.201800631 |
| 45. | Govil, G. Mol. Phys. 1971, 21, 953–957. doi:10.1080/00268977100102111 |
| 40. | Chen, J.-L.; You, Z.-W.; Qing, F.-L. J. Fluorine Chem. 2013, 155, 143–150. doi:10.1016/j.jfluchem.2013.07.017 |
| 28. | Shimada, T.; Yoshioka, M.; Konno, T.; Ishihara, T. Org. Lett. 2006, 8, 1129–1131. doi:10.1021/ol0531435 |
© 2024 Bilska-Markowska et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.