Abstract
The marine sponge Suberea ianthelliformis was investigated for new chemistry after the recent discovery that polyamines ianthelliformisamines A–C (1–3) – originally sourced from this Australian sponge – act as Pseudomonas aeruginosa biofilm inhibitors and antibiotic enhancers. Large-scale extraction and isolation studies resulted in the discovery of four new and minor natural products, ianthelliformisamines D–G (4–7) and the known steroid, aplysterol (8). Compounds 4–7 were fully characterised following 1D/2D NMR, MS and UV data analyses. All compounds were assessed for their inhibition on planktonic growth of P. aeruginosa PAO1 in addition to their ability to inhibit the formation of biofilms. None of the tested natural products inhibited planktonic growth or biofilm formation of PAO1 when screened at 50 µM. Ianthelliformisamine D (4) contains a rare N-(3-aminopropyl)-2-pyrrolidone moiety only found in <30 natural products. Owing to the novelty of compound 4, we undertook the first total synthesis of this natural product, which was achieved in three steps.

Graphical Abstract
Introduction
The marine environment covers over two thirds of the earth’s surface and it encompasses a wide range of complex ecosystems that are highly variable in their physical attributes including pressure, salinity, temperature, and light availability. Both flora and fauna have evolved over billions of years to survive within this unique environment [1,2], which has led to the production and diversification of unique and novel metabolites with specialised biological functions [3,4]. The richest diversity of marine metabolites are derived from invertebrates; most of which are sessile and lack the ability to physically defend themselves from both predators and competitors and thus rely on chemical mechanisms for their defense [1]. Metabolites derived from marine sponges contribute more than half of all compounds identified from marine invertebrates [1,5], therefore it is no surprise that sponges are highly sought after for novel bioactive metabolites and have been a major focus of marine natural product drug discovery for over 70 years.
The identification of ianthelliformisamines A–C from the Australian marine sponge Suberea ianthelliformis, which displayed activity against both Pseudomonas aeruginosa and Staphylococcus aureus, has contributed to a surge in the interest of polyamines as new antibacterial leads [6]. To date the total synthesis of ianthelliformisamines A–C (1–3) has been described [7] and numerous synthetically related analogues have been published with their antibiotic assessment against numerous bacterial species reported [8-11]. Our recent publication showed that of the naturally occurring metabolites, compound 3 inhibits the formation of P. aeruginosa PAO1 biofilms (MIC 53.1 µg/mL), whilst 1 and 2 enhanced the antibiotic effect of ciprofloxacin when used in combination [12]. In efforts to further examine the chemistry of S. ianthelliformis and potentially discover new antibiotic leads, we undertook a scaled-up chemical investigation on the original specimen from which ianthelliformisamines A–C were isolated.
Herein, we describe the large-scale extraction, isolation, and structure elucidation of four new metabolites, ianthelliformisamines D–G (4–7). Additionally, we report the isolation of the known natural products, aplysterol (8) and ianthelliformisamines A–C (1–3). Owing to the novelty of ianthelliformisamine D (4) we undertook the first total synthesis of this natural product, which was successfully achieved in only three steps and respectable yield. All newly identified metabolites were subsequently assessed for their ability to inhibit the growth of planktonic P. aeruginosa PAO1 and formation of biofilms.
Results and Discussion
For a more comprehensive chemical investigation into the chemistry of Suberea ianthelliformis, a new aliquot of the freeze-dried and ground sample was extracted exhaustively with n-hexane, CH2Cl2, and MeOH. The CH2Cl2 and MeOH extracts were combined then subjected to phenyl-bonded reversed-phase HPLC (RP-HPLC), which led to the purification of the known metabolites, ianthelliformisamines A–C (1–3) [7] and aplysterol (8) [13], and four new natural products, ianthelliformisamines D–G (4–7) (Figure 1). This extraction and isolation process was repeated twice to obtain sufficient quantities of the minor and previously undescribed natural products for full characterisation studies and biological assessment. The spectroscopic and spectrometric data of all known compounds isolated during our studies matched well with the literature values [7,14].
![[1860-5397-20-266-1]](/bjoc/content/figures/1860-5397-20-266-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of ianthelliformisamines A–G (1–7) and aplysterol (8).
Figure 1: Chemical structures of ianthelliformisamines A–G (1–7) and aplysterol (8).
Ianthelliformisamine D (4) was isolated as a stable brown gum. The LRESIMS of 4 indicated the presence of two bromine atoms due to a 1:2:1 ion cluster at m/z 459/461/463 [M + H]+, whilst the HRESIMS data allowed a molecular formula of C17H20Br2N2O3 to be assigned. The 1H NMR (Table 1) and edited HSQC spectra of 4 in DMSO-d6 indicated the presence of one methoxy group (δH 3.81), six methylene signals (δH 3.33, 3.20, 3.14, 2.21, 1.91, 1.63) and one exchangeable proton triplet (δH 8.04) that was indicative of a secondary amide [7]. Additionally, signals for one isolated trans olefin (δH 7.33, 6.66) and one aromatic signal (δH 7.89) that integrated for two protons indicating the presence of a symmetrical aromatic moiety, were observed. The 13C NMR (Table 1) spectrum of 4 showed two carbonyls (δC 164.4, 173.9), with the carbonyl at δC 164.4 readily assigned to an acrylamide group, which is present in all previously published ianthelliformisamine molecules [7]. Methylenes resonating at δH 3.14, 1.64 and 3.20 were assigned to a three-carbon alkyl spin system that was flanked by nitrogen atoms [NH–(CH2)3–N] based on COSY correlations from the amide proton at δH 8.04. A terminal pyrrolidone was assigned based on COSY data for the three remaining methylene protons (δH 3.33, 1.91, 2.21), and a three-bond HMBC correlation from δH 3.33, and a two-bond correlation from δH 2.20 to the carbonyl resonating at δC 173.9 [15]. The observation of a 3JCH correlation from δH 3.20 to the nitrogen-bearing carbon at δC 46.4 and to δC 173.9 supported the assignment of the pyrrolidone moiety. A 1,3,4,5-tetrasubstituted aromatic ring was assigned based on 3JCH correlations from the isolated aromatic signal (δH 7.89) to its symmetrically placed carbon (δC 131.6) and to an oxygen-bearing carbon (δC 153.9) along with 2JCH correlations to a brominated (δC 118.0) and sp2 phenyl carbons (δC 134.5) (see Figure 2). Observation of three-bond correlations in the HMBC spectrum between the olefin protons and aromatic ring carbons (δH 7.33 to δC 131.6 and δH 6.66 to δC 134.5) linked the olefin moiety to the symmetrical 1,3,4,5-tetrasubstituted phenyl system. ROESY and HMBC correlations from the amide proton linked the propyl-2-pyrrolidine moiety to the brominated acrylamide fragment and thus the full chemical structure of 4 was elucidated and assigned to ianthelliformisamine D. A refined literature search using Scifinder Scholar [16] revealed that compound 4 contains a novel scaffold due to the N-(3-aminopropyl)-2-pyrrolidone moiety, which is rarely found in natural products with <30 metabolites reported to date.
Table 1: NMR data of ianthelliformisamines D (4) and E (5) in DMSO-d6.a
| Ianthelliformisamine D (4) | Ianthelliformisamine E (5)b | ||||
| position | δC, type | δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | |
| 1 | 134.5, C | 134.3, C | |||
| 2 | 131.6, CH | 7.89, s | 131.6, CH | 7.89, s | |
| 3 | 118.0, C | 118.0, C | |||
| 4 | 153.9, C | 153.9, C | |||
| 4-OCH3 | 60.6, CH3 | 3.81, s | 60.6, CH3 | 3.82, s | |
| 5 | 118.0, C | 118.0, C | |||
| 6 | 131.6, CH | 7.89, s | 131.6, CH | 7.89, s | |
| 7 | 135.2, CH | 7.33, d (15.8) | 135.5, CH | 7.36, d (15.8) | |
| 8 | 124.5, CH | 6.66, d (15.8) | 124.1, CH | 6.65, d (15.8) | |
| 9 | 164.4, C | 164.9, C | |||
| 9-NH | 8.04, t (5.7) | 8.23, t (5.7) | |||
| 10 | 36.5, CH2 | 3.14 dt (5.7, 6.5) | 35.9, CH2 | 3.25 dt (5.7, 6.5) | |
| 11 | 27.0, CH2 | 1.63, m | 26.1, CH2 | 1.77, m | |
| 12 | 39.7, CH2 | 3.20, t (7.2) | 44.8, CH2 | 2.92, m | |
| 12-NH | 8.35, brs | ||||
| 13 | 46.4, CH2 | 3.33, m | 46.3, CH2 | 2.92, m | |
| 14 | 17.5, CH2 | 1.91, m | 21.1, CH2 | 1.79, m | |
| 15 | 30.5, CH2 | 2.21, t (8.0) | 30.4, CH2 | 2.36, t (7.3) | |
| 16 | 173.9, C | 173.6, C | |||
| 16-OH | c | ||||
aSpectra recorded at 25 °C (800 MHz for 1H NMR and 200 MHz for 13C NMR); bisolated as a TFA salt; cnot observed.
![[1860-5397-20-266-2]](/bjoc/content/figures/1860-5397-20-266-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2:
Key COSY (![[Graphic 1]](/bjoc/content/inline/1860-5397-20-266-i2.svg?max-width=637&scale=1.18182) ), HMBC (
), HMBC (![[Graphic 2]](/bjoc/content/inline/1860-5397-20-266-i3.svg?max-width=637&scale=1.18182) ) and ROESY (
) and ROESY (![[Graphic 3]](/bjoc/content/inline/1860-5397-20-266-i4.svg?max-width=637&scale=1.18182) ) correlations for ianthelliformisamines D (4) and E (5).
) correlations for ianthelliformisamines D (4) and E (5).
Figure 2:
Key COSY (), HMBC () and ROESY () correlations for ianthelliformisamines D (4) and E (5).
The TFA salt of ianthelliformisamine E (5) was obtained as a stable brown gum. The LRESIMS of 5 indicated the presence of two bromine atoms, displaying a 1:2:1 ion cluster at m/z 477/479/481 [M + H]+. Similarly to compound 4, the 1H NMR (Table 1) and edited HSQC spectra of 5 displayed six methylene signals (δH 3.25, 2.92, 2.36, 1.79, 1.77). Additionally, a broad exchangeable proton (δH 8.35) was observed that was indicative of a protonated dialkylated amino group [7]. The 13C NMR (Table 1) data of 5 displayed six aliphatic carbons (δC 35.9, 26.1, 44.8, 46.3, 21.1, 30.4) and two carbonyl signals (δC 164.9, 173.6). Similarly to the other ianthelliformisamines, the aromatic (δC 131.6) and olefin (δC 124.5, 135.2) carbons were observed [7]. COSY correlations associated with the methylene signals of 5 enabled the assignment of an NH–(CH2)3–NH–(CH2)3 spin system, which was confirmed by HMBC and ROESY correlations. The carboxyl group at δC 173.6 was positioned at the end of the alkyl chain based on HMBC correlations from the methylene protons (δH 2.36, 1.79) to this carbon signal. Although a downfield exchangeable CO2H proton was not observed in the 1H NMR spectrum of 5, a carboxylic acid moiety was assigned based on the 13C NMR shift value (δC 173.6) [17], and analysis of the HRESIMS ion at m/z 477.0022 [M + H]+, which confirmed the molecular formula to be C17H22Br2N2O4.
Ianthelliformisamine F (6) was isolated as a stable brown gum. The LRESIMS of 6 indicated the presence of two bromine atoms due to a 1:2:1 ion cluster at m/z 334/336/338 [M + H]+, whilst the HRESIMS data enabled a molecular formula of C10H9Br2NO2 to be assigned. Comparison of the 1D NMR data of 6 to metabolites 4 and 5 and the previously characterised ianthelliformisamines A–C readily allowed the assignment of dibromo-4-methoxyphenyl and propenamide moieties. However, the 1H NMR spectrum of 6 displayed two broad exchangeable signals (δH 7.19, 7.46) indicating the presence of a primary amide [18], thus enabling the full chemical structure of ianthelliformisamine F to be determined.
The TFA salt of ianthelliformisamine G (7) was also purified as a stable brown gum. In a similar manner to compound 6, the LRESIMS indicated the presence of two bromine atoms; these data also indicated that 7 had an additional 57 amu (atomic mass units) compared to 6. Comparison of the 1H NMR (Table 2) and edited HSQC spectra with 7 showed that 6 contained three additional methylene signals (δH 3.25, 2.81, 1.73). Two exchangeable protons were observed in 7, including a broad singlet (δH 7.73) that integrated for three protons and a triplet (δH 8.23) that were indicative of a protonated terminal amino group and a secondary amide functionality, respectively [7]. 13C NMR shifts and COSY correlations associated with the methylene signals of 7 enabled the assignment of an NH–(CH2)3–NH2 moiety, which was confirmed by HMBC correlations (Figure 3). This fragment was connected to the previously assigned moiety through a HMBC correlation from δH 3.25 to the amide carbonyl (δC 164.9), thus enabling the full chemical structure of ianthelliformisamine G (7) to be assigned.
Table 2: NMR data of ianthelliformisamines F (6) and G (7) in DMSO-d6.a
| Ianthelliformisamine F (6) | Ianthelliformisamine G (7)b | ||||
| position | δC, type | δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | |
| 1 | 134.4, C | 134.3, C | |||
| 2 | 131.6, CH | 7.88, s | 131.6, CH | 7.89, s | |
| 3 | 118.0, C | 118.0, C | |||
| 4 | 153.9, C | 153.9, C | |||
| 4-OCH3 | 60.6, CH3 | 3.81, s | 60.6, CH3 | 3.82, s | |
| 5 | 118.0, C | 118.0, C | |||
| 6 | 131.6, CH | 7.88, s | 131.6, CH | 7.89, s | |
| 7 | 135.7, CH | 7.32, d (15.8) | 135.4, CH | 7.35, d (15.8) | |
| 8 | 124.6, CH | 6.65, d (15.8) | 124.2, CH | 6.65, d (15.8) | |
| 9 | 166.1, C | 164.9, C | |||
| 9-NH2 | 7.19, brs | ||||
| 7.46, brs | |||||
| 9-NH | 8.23, t (5.9) | ||||
| 10 | 35.9, CH2 | 3.25, dt (5.9, 6.8) | |||
| 11 | 27.5, CH2 | 1.73, tt (6.8, 7.5) | |||
| 12 | 36.9, CH2 | 2.81, m | |||
| 12-NH2 | 7.73, brs | ||||
aSpectra recorded at 25 °C (800 MHz for 1H NMR and 200 MHz for 13C NMR); bisolated as a TFA salt.
![[1860-5397-20-266-3]](/bjoc/content/figures/1860-5397-20-266-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3:
Key COSY (![[Graphic 4]](/bjoc/content/inline/1860-5397-20-266-i5.svg?max-width=637&scale=1.18182) ) and HMBC (
) and HMBC (![[Graphic 5]](/bjoc/content/inline/1860-5397-20-266-i6.svg?max-width=637&scale=1.18182) ) correlations for ianthelliformisamines F (6) and G (7).
) correlations for ianthelliformisamines F (6) and G (7).
Figure 3:
Key COSY () and HMBC () correlations for ianthelliformisamines F (6) and G (7).
Compounds 6 and 7 are both commercially available [16] but neither of these compounds has been fully characterised with only the total synthesis of 7 being reported in the literature [8]. Our study reports the first identification of 6 and 7 from a natural origin and the first full characterisation of these molecules using NMR, UV, and MS data.
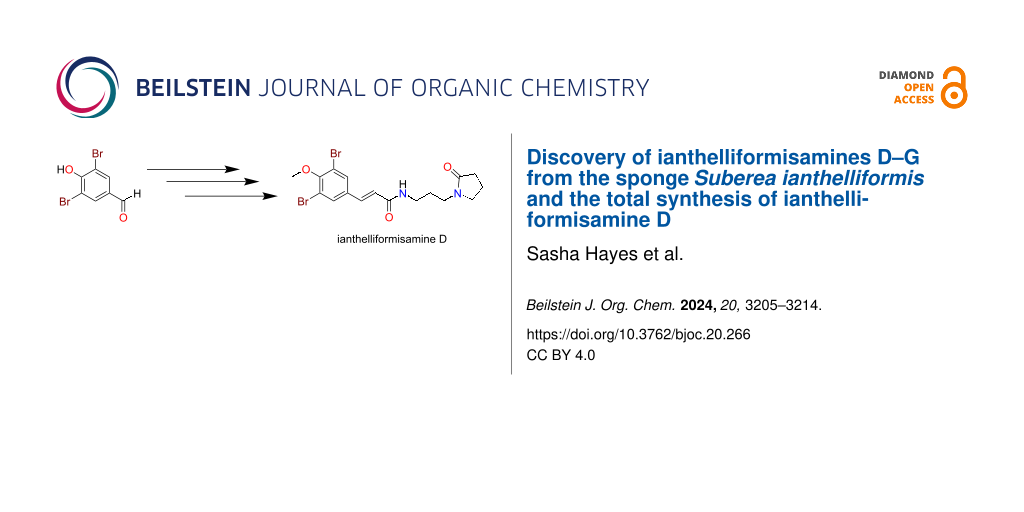
Owing to the novelty of ianthelliformisamine D (4), we attempted the first total synthesis of this natural product (Scheme 1). In a similar, but modified manner to that reported by Gan et al. [19] we firstly methylated the commercially sourced 3,5-dibromo-4-hydroxybenzaldehyde using TMSCHN2 in MeOH/CH2Cl2 at room temperature (17% yield). Subjecting the methoxylated benzaldehyde intermediate 9 to a Doebner–Knoevenagel condensation with malonic acid and pyridine afforded the brominated cinnamic acid analogue 10 in 54% yield [19]. Amidation chemistry using carbonyldiimidazole (CDI) [18] and the commercially available primary amine, 1-(3-aminopropyl)pyrrolidin-2-one completed the total synthesis of the natural product in an overall yield of 1.5%. The NMR data comparison of the natural product and our synthetic compound was essentially identical.
![[1860-5397-20-266-i1]](/bjoc/content/inline/1860-5397-20-266-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Total synthesis of ianthelliformisamine D (4).
Scheme 1: Total synthesis of ianthelliformisamine D (4).
Due to our interest in the identification of potential new leads against Pseudomonas aeruginosa and supported by numerous reports of favourable activity for ianthelliformisamines A–C [7,12] and their synthetic analogues [8-10], we investigated the planktonic and biofilm activity of the new natural products 4–7 in addition to the known metabolite, aplysterol (8) [13]. Our biological assessment of compounds 4–8 showed no inhibition of planktonic growth or biofilm formation for P. aeruginosa when screened at 50 µM. Previously reported antibacterial assessment of ianthelliformisamines A–C (1–3) and their synthetic analogues has generated structure–activity relationship data, leading to speculation over the moieties responsible for their antibiotic effects. A reduction in the number of amines in the polymeric chain and the absence of a primary amine was noted to decrease bioactivity by Xu et al. [7]. Research reported by Khan et al. in 2014 [9] also supported the important role that the polyamine chain length plays in antibacterial activity. Of note, increasing research providing structure–activity relationship analyses shows that polyamines (including spermine) are bacterial membrane disruptors and are beneficial in enhancing activity of known antibacterial agents [20,21]. The absence of polyamine chains in ianthelliformisamines D–G (4–7) could explain the loss of activity seen in our assessment against P. aeruginosa. The synthetic molecule of 7 has been tested by other researchers against Gram-negative bacteria including P. aeruginosa with reported MIC values of >200 µg/mL [8], which is consistent with the data we report in this paper.
Finally, and of note, our studies described here report the first isolation of aplysterol (8) from the genus Suberea. To date, there has been no evaluation of this compound’s antibiotic potential towards P. aeruginosa, including biofilm inhibition. Furthermore, this is the first report of any biological assessment of aplysterol (8) as a pure compound, since prior studies have only tested a mixture of 8 with 24,28-didehydroaplysterol, where it was found to inhibit DNA topoisomerase II-α (MIC = 50 µM) [22].
Conclusion
In summary, we report here the discovery and full characterisation of four new natural products, ianthelliformisamines D–G from the marine sponge Suberea ianthelliformis. Furthermore, we describe the first total synthesis of ianthelliformisamine D which contains a rare N-(3-aminopropyl)-2-pyrrolidone moiety. Whilst testing of these natural products against Pseudomonas aeruginosa showed that none of them inhibited planktonic growth or biofilm formation at 50 µM, synthetic efforts has generated sufficient quantities of the novel compound ianthelliformisamine D that will enable additional biological profiling.
Experimental
General experimental procedures
Specific rotations were recorded using a JASCO P-1020 polarimeter. UV data was recorded on a JASCO V-650 UV–vis spectrophotometer. NMR spectra were recorded at 25 °C on a Bruker Avance III HD 800 MHz NMR spectrometer equipped with a cryoprobe. The 1H and 13C chemical shifts were referenced to solvent peaks for DMSO-d6 at δH 2.50 and δC 39.52, respectively. LRESIMS data was recorded on an Ultimate 3000 RS UHPLC coupled to a Thermo Fisher Scientific ISQEC single quadruple ESI mass spectrometer. HRESIMS data was acquired on a Bruker maXis II ETD ESI-qTOF. Alltech Davisil (30–40 µm, 60 Å) C8-bonded silica and Alltech Davisil (30–40 µm, 60 Å) diol-bonded silica were used for pre-adsorption work before RP- or NP-HPLC, respectively. The pre-adsorbed material was subsequently packed into an Alltech stainless steel guard cartridge (10 × 30 mm) then attached to a HPLC column prior to fractionation. A Waters 600 pump fitted with a Waters 996 photodiode array detector fitted with a Gilson 717-plus autosampler were used for RP-HPLC separations. A Thermo Fisher Scientific Dionex Ultimate 3000 UHPLC was used for NP-HPLC separations. Thermo Betasil phenyl-bonded silica (5 μm, 100 Å, 150 × 21.2 mm) and Phenomenex Luna C18 column (5 µm, 90–110 Å, 10 mm × 250 mm) were used for RP-HPLC separations. For NP-HPLC, a YMC diol-bonded silica (5 μm, 120 Å, 150 × 20 mm) column was used. The frozen marine sponge was dried using a Dynamic FD12 freeze dryer and ground using a Fritsch Universal Cutting Mill Pulverisette 19. The ground marine sponge was extracted at room temperature using an Edwards Instrument Company Bioline orbital shaker set to 200 rpm. Solvents were removed from extracts with a Büchi R-144 rotary evaporator and from HPLC fractions using a GeneVac XL4 centrifugal evaporator. All solvents used for chromatography, UV, MS, and [α]D were Honeywell Burdick & Jackson or Lab-Scan HPLC grade. H2O was filtered using a Sartorius Stedium Arium Pro VF ultrapure water system. Reagents used for the total synthesis of Ianthelliformisamine D were purchased from either Sigma or Aaron Chemicals.
Sponge material
The sponge sample was obtained from the NatureBank [23] biota library housed at the Institute for Biomedicine and Glycomics, Griffith University, Australia. A voucher specimen of Suberea ianthelliformis (NB6014998; phylum Porifera, class Demospongiae, order Verongida, family Aplysinellidae) has been previously lodged (G322255) at the Queensland Museum, South Brisbane, Queensland, Australia [7].
Extraction and isolation
In a manner similar to that reported by Xu et al. [7], the freeze-dried and ground specimen of S. ianthelliformis (10 g) was extracted sequentially with n-hexane (250 mL, 2 h), CH2Cl2 (250 mL, 2 h), and MeOH (250 mL, 2 h; 250 mL, 16 h). The n-hexane extract was discarded (as it contained only highly lipophilic material) and the CH2Cl2 and MeOH extracts were combined to produce a brown gum (173.3 mg) that was pre-adsorbed to C8-bonded silica (≈1 g), packed into a stainless-steel guard cartridge, and subjected to phenyl semipreparative RP-HPLC separation. Isocratic conditions of 90% H2O (0.1% TFA)/10% MeOH (0.1% TFA) were initially employed for the first 10 min, then a linear gradient to 100% MeOH (0.1% TFA) was run over 40 min, followed by isocratic conditions of 100% MeOH (0.1% TFA) for a further 10 min, all at a flowrate of 9 mL/min; 60 fractions (60 × 1 min) were collected. This first HPLC fractionation afforded ianthelliformisamine B (2, 10.4 mg, tR 36–37 min, 0.104% dry wt) and several other UV-active fractions that contained mixtures of polyamine-type alkaloids, which were combined for further isolation work. The following describes the purification of these fractions. Fractions 34–35 (5.0 mg) were purified by semipreparative phenyl RP-HPLC using isocratic conditions of 70% H2O (0.1% TFA)/30% MeOH (0.1% TFA) for the first 10 min, then a linear gradient to 10% H2O (0.1% TFA)/90% MeOH (0.1% TFA) was run over 50 min at a flowrate of 9 mL/min; 60 fractions (60 × 1 min) were collected and resulted in the purification of ianthelliformisamine A (1, 1.0 mg, tR 16 min, 0.010% dry wt). Fractions 38–40 were combined (45.0 mg) and purified by semipreparative phenyl RP-HPLC using the same solvent gradient as above and afforded the new natural products, ianthelliformisamines G (7, 1.2 mg, tR 19 min, 0.012% dry wt) and E (5, 1.5 mg, tR 23 min, 0.015% dry wt). Fractions 41–42 were combined (22.4 mg) and purified by semipreparative RP-HPLC using the same solvent gradient and afforded the new natural products, ianthelliformisamines F (6, 1.2 mg, tR 19 min, 0.012% dry wt) and D (4, 1.3 mg, tR 41 min, 0.013% dry wt). Fractions 43–45 were combined (28.7 mg) and purified by semipreparative RP-HPLC using the same solvent gradient and afforded the known metabolite, ianthelliformisamine C (3, 1.8 mg, tR 38 min, 0.018% dry wt). Fractions 46–60 (20.1 mg) was purified by semipreparative NP-HPLC. Isocratic conditions of 100% n-hexane were initially employed for the first 10 min, then a linear gradient to 80% n-hexane/20% iPrOH was run over 50 min at a flow rate of 9 mL/min; 60 fractions (60 × 1 min) were collected and resulted in the purification of the known sterol, aplysterol (8, 0.9 mg, tR 12–13 min, 0.009% dry wt). The extraction and isolation process described above was repeated twice more (identical scale) to obtain sufficient quantities of the minor natural products for characterisation and biological assessment with similar recoveries obtained.
Ianthelliformisamine D (4): Stable brown gum; UV (MeOH) λmax, nm (log ε): 226 (4.01), 278 (3.86); 1H and 13C NMR data (DMSO-d6), see Table 1; LRESIMS (m/z): 459/461/463 [M + H]+; HRESIMS (m/z): [M + H]+ calcd for C17H2179Br2N2O3, 458.9913; found, 458.9917.
Ianthelliformisamine E TFA salt (5): Stable brown gum; UV (MeOH) λmax, nm (log ε): 229 (4.25), 279 (4.17); 1H and 13C NMR data (DMSO-d6), see Table 1; LRESIMS (m/z): 477/479/481 [M + H]+; HRESIMS (m/z): [M + H]+ calcd for C17H2379Br2N2O4, 477.0019; found, 477.0022.
Ianthelliformisamine F (6): Stable brown gum; UV (MeOH) λmax, nm (log ε): 229 (3.83), 280 (3.71); 1H and 13C NMR data (DMSO-d6), see Table 2; LRESIMS (m/z): 334/336/338 [M + H]+; HRESIMS (m/z): [M + Na]+ calcd for C10H979Br2NO2Na, 355.8892; found, 355.8896.
Ianthelliformisamine G TFA salt (7): Stable brown gum; UV (MeOH) λmax, nm (log ε): 229 (3.63), 280 (3.57); 1H and 13C NMR data (DMSO-d6), see Table 2; LRESIMS (m/z): 391/393/395 [M + H]+; HRESIMS (m/z): [M + H]+ calcd for C13H1779Br2N2O2, 390.9651; found, 390.9647.
Aplysterol (8): Stable white powder; ![[Graphic 6]](/bjoc/content/inline/1860-5397-20-266-i7.svg?max-width=637&scale=1.18182) −46.7 (c 0.025, CHCl3), lit.
−46.7 (c 0.025, CHCl3), lit. ![[Graphic 7]](/bjoc/content/inline/1860-5397-20-266-i8.svg?max-width=637&scale=1.18182) −25 (c not specified, CHCl3) [24]; HRESIMS (m/z): [M + Na]+ calcd for C29H50ONa, 437.3754; found, 437.3741.
−25 (c not specified, CHCl3) [24]; HRESIMS (m/z): [M + Na]+ calcd for C29H50ONa, 437.3754; found, 437.3741.
Methylation of 3,5-dibromo-4-hydroxybenzaldehyde
3,5-Dibromo-4-hydroxybenzaldehyde (279.0 mg, 1.0 mmol) was dissolved in CH2Cl2/MeOH 1:1 (1 mL) then a solution of TMSCHN2 in hexanes (2.0 M, 1.5 mL, 11 mmol) was slowly added and the mixture stirred at room temperature for 6 h. The reaction crude was pre-adsorbed to C8-bonded silica, packed into a stainless steel guard cartridge then purified by Luna C18 semipreparative RP-HPLC using isocratic conditions of 50% H2O (0.1% TFA)/50% MeOH (0.1% TFA) for the first 5 min, then a linear gradient to 100% MeOH (0.1% TFA) was run over 20 min and held at 100% MeOH (0.1% TFA) for a further 5 min at a flowrate of 4 mL/min; 30 fractions (30 × 1 min) were collected and resulted in the purification of 3,5-dibromo-4-methoxybenzaldehyde (9, 50.1 mg, tR 18–22 min, 17% yield) [19].
3,5-Dibromo-4-methoxybenzaldehyde (9): White powder; 1H NMR (DMSO-d6, 800 MHz) δH 9.89 (s, 1H, H-7), 8.17 (s, 2H, H-2, H-6), 3.88 (s, 3H, 4-OCH3); 13C NMR (DMSO-d6, 200 MHz) δC 190.1 (C-7), 158.1 (C-4), 134.5 (C-1), 133.8 (2C, C-2, C-6), 118.6 (2C, C-3, C-5), 60.8 (4-OCH3); LRESIMS (m/z): 293/295/297 [M + H]+.
Doebner–Knoevenagel condensation of 3,5-dibromo-4-methoxybenzaldehyde with malonic acid
3,5-Dibromo-4-methoxybenzaldehyde (9, 80.0 mg, 0.27 mmol), malonic acid (56.0 mg, 0.54 mmol) and dry pyridine (1 mL) were refluxed at 100 °C for 5 h. The reaction crude was pre-adsorbed to C8-bonded silica, packed into a stainless steel guard cartridge then purified by Betasil semipreparative RP-HPLC using isocratic conditions of 90% H2O (0.1% TFA)/10% MeOH (0.1% TFA) that were initially employed for the first 10 min, then a linear gradient to 100% MeOH (0.1% TFA) was run over 40 min, followed by isocratic conditions of 100% MeOH (0.1% TFA) for a further 10 min, all at a flowrate of 9 mL/min; 60 fractions (60 × 1 min) were collected resulting in the purification of (E)-3-(3,5-dibromo-4-methoxyphenyl)acrylic acid (10, 49.0 mg, tR 46–48 min, 54% yield) [19].
(E)-3-(3,5-Dibromo-4-methoxyphenyl)acrylic acid (10): White powder; 1H NMR (DMSO-d6, 800 MHz) δH 8.06 (s, 2H, H-2, H-6), 7.50 (d, J = 16.0 Hz, 1H, H-7), 6.62 (d, J = 16.0 Hz, 1H, H-8), 3.82 (s, 3H, 4-OCH3); 13C NMR (DMSO-d6, 200 MHz) δC 167.3 (C-9), 154.4 (C-4), 140.5 (C-7), 133.7 (C-1), 132.4 (2C, C-2, C-6), 121.5 (C-8), 118.0 (2C, C-3, C-5) 60.6 (4-OCH3) ; LRESIMS (m/z): 333/335/337 [M − H]−.
Synthesis of Ianthelliformisamine D
(E)-3-(3,5-Dibromo-4-methoxyphenyl)acrylic acid (10, 49.0 mg, 0.15 mmol) and 1,1′-carbonyldiimadazole (24.0 mg, 0.15 mmol) was added to a reaction vial with dry CH3CN (400 µL) and stirred at room temperature for 10 min. 1-(3-Aminopropyl)pyrrolidin-2-one (100 µL, 0.60 mmol) was then added dropwise and the resulting solution was stirred at room temperature for 3 h. The reaction crude was pre-adsorbed to C8-bonded silica, packed into a stainless steel guard cartridge then purified by Luna semipreparative RP-HPLC using isocratic conditions of 50% H2O (0.1% TFA)/50% MeOH (0.1% TFA) for the first 5 min, then a linear gradient to 100% MeOH (0.1% TFA) was run over 20 min and held at 100% MeOH (0.1% TFA) for a further 5 minutes at a flowrate of 4 mL/min; 30 fractions (30 × 1 min) were collected and resulted in the purification of synthetic Ianthelliformisamine D (4, 11.1 mg, tR 21 min, 16% yield). The 1H and 13C NMR spectra of the synthetic compound matched that of the natural product.
Synthetic Ianthelliformisamine D (4): Clear film: 1H NMR (DMSO-d6, 800 MHz) δH 8.04 (t, J = 5.7 Hz, 1H, 9-NH), 7.88 (s, 2H, H-2, H-6), 7.33 (d, J = 15.7 Hz, 1H, H-7), 6.66 (d, J = 15.7 Hz, 1H, H-8), 3.81 (s, 3H, 4-OCH3), 3.33 (t, J = 7.1 Hz, 2H, H-13), 3.20 (t, J = 7.2 Hz, 2H, H-12), 3.14 (dt, J = 5.7, 6.8 Hz, 2H, H-10), 2.21 (t, J = 8.1 Hz, 2H, H-15), 1.91 (tt, J = 7.1, 8.1 Hz, 2H, H-14), 1.63 (tt, J = 6.8, 7.2 Hz, 2H, H-11); 13C NMR (DMSO-d6, 200 MHz) δC 173.9 (C-16), 164.4 (C-9), 153.9 (C-4), 135.2 (C-7), 134.5 (C-1), 131.6 (2C, C-2, C-6), 124.5 (C-8), 118.0 (2C, C-3, C-5) 60.5 (4-OCH3), 46.4 (C-13), 39.6 (C-12), 36.5 (C-10), 30.5 (C-15), 26.9 (C-11), 17.5 (C-14); LRESIMS (m/z): 459/461/463 [M + H]+.
Bacterial strains and media
Wild-type Pseudomonas aeruginosa strain PAO1 (prototrophic wild-type) [25] was used in this study. Bacteria were grown in Luria–Bertani (LB) medium (10 g/L tryptone, 10 g/L sodium chloride and 5 g/L yeast extract).
Biofilm inhibition assay
The overnight cultures at 37 °C in LB medium were washed once with sterile saline 0.9% (w/v) and adjusted to an OD600 of 0.05. Then, 1% inoculum was transferred into fresh LB medium. Following the incubation at 37 °C, 200 rpm for 5–6 h to reach the mid-log phase, the cells were washed once with sterile saline 0.9% (w/v) and diluted to an OD600 of 0.01. Test compounds (15 µL) were loaded prior to the addition of bacteria (135 µL) into 96-well plates. The plates were incubated for 24 h at 37 °C in static conditions. The effects of compounds on bacterial growth and the viability of biofilm bacteria was determined by the OD600 and resazurin (RSZ) metabolic assay, respectively. The final concentration of DMSO in the assays was 1% (v/v). The negative controls consisted of inoculum and 1% DMSO. The antibiotic tobramycin (Sigma-Aldrich; 16 µg/mL) was used as a positive control. The initial OD600 and final OD600 were read before incubation at 37 °C and after 24 h of incubation, respectively, followed by the assessment of biofilm viability by resazurin metabolic assay. The experiments were carried out with three technical replicates and three biological replicates. The assay was performed as previously described [26]. The cultures were withdrawn, and the plates were washed twice with sterile water. Then, 50 µL of diluted RSZ solution (0.2% w/v) in LB medium was added into each well followed by incubation at 37 °C for 5 h. A microplate reader (iD5 Multi-Mode) was used to measure the fluorescence intensity (excitation 530 nm, emission 590 nm).
Supporting Information
| Supporting Information File 1: NMR data tables for compounds 4–7, 1D and 2D NMR spectra of compounds 4–7, 1H NMR spectra of natural products 1–3 and 8 and 1H and 13C NMR spectra of synthetic compounds 4, 9, and 10. | ||
| Format: PDF | Size: 3.1 MB | Download |
Acknowledgements
We gratefully acknowledge NatureBank [23] for supply of the sponge sample that was investigated during these studies.
Data Availability Statement
Data generated and analyzed during this study is available from the corresponding author upon reasonable request.
References
-
Bernardini, S.; Tiezzi, A.; Laghezza Masci, V.; Ovidi, E. Nat. Prod. Res. 2018, 32, 1926–1950. doi:10.1080/14786419.2017.1356838
Return to citation in text: [1] [2] [3] -
Buss, A. D.; Cox, B.; Waigh, R. D. Natural Products as Leads for New Pharmaceuticals. In Burger's Medicinal Chemistry and Drug Discovery; Abraham, D. J., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2003; pp 847–900. doi:10.1002/0471266949.bmc018
Return to citation in text: [1] -
Carroll, A. R.; Copp, B. R.; Davis, R. A.; Keyzers, R. A.; Prinsep, M. R. Nat. Prod. Rep. 2023, 40, 275–325. doi:10.1039/d2np00083k
Return to citation in text: [1] -
Carroll, A. R.; Copp, B. R.; Grkovic, T.; Keyzers, R. A.; Prinsep, M. R. Nat. Prod. Rep. 2024, 41, 162–207. doi:10.1039/d3np00061c
Return to citation in text: [1] -
Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2020, 83, 770–803. doi:10.1021/acs.jnatprod.9b01285
Return to citation in text: [1] -
Talebi Bezmin Abadi, A.; Rizvanov, A. A.; Haertlé, T.; Blatt, N. L. Bionanosci. 2019, 9, 778–788. doi:10.1007/s12668-019-00658-4
Return to citation in text: [1] -
Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] -
Pieri, C.; Borselli, D.; Di Giorgio, C.; De Méo, M.; Bolla, J.-M.; Vidal, N.; Combes, S.; Brunel, J. M. J. Med. Chem. 2014, 57, 4263–4272. doi:10.1021/jm500194e
Return to citation in text: [1] [2] [3] [4] -
Khan, F. A.; Ahmad, S.; Kodipelli, N.; Shivange, G.; Anindya, R. Org. Biomol. Chem. 2014, 12, 3847–3865. doi:10.1039/c3ob42537a
Return to citation in text: [1] [2] [3] -
Khadake, S. N.; Karamathulla, S.; Jena, T. K.; Monisha, M.; Tuti, N. K.; Khan, F. A.; Anindya, R. Bioorg. Med. Chem. Lett. 2021, 39, 127883. doi:10.1016/j.bmcl.2021.127883
Return to citation in text: [1] [2] -
Allen, R. A.; McCormack, C. E. M.; Wuest, W. M. ChemMedChem 2023, 18, e202300253. doi:10.1002/cmdc.202300253
Return to citation in text: [1] -
Tran, T. M. T.; Addison, R. S.; Davis, R. A.; Rehm, B. H. A. Int. J. Mol. Sci. 2023, 24, 10204. doi:10.3390/ijms241210204
Return to citation in text: [1] [2] -
Ferriol, P.; Marante, F. J. T.; Martin, I. B.; Rodríguez, J. J. S.; Alonso, R. G.; Benkovics, A. G.; Mioso, R. Rec. Nat. Prod. 2018, 12, 470–479. doi:10.25135/rnp.52.17.11.558.204
Return to citation in text: [1] [2] -
Rahelivao, M. P.; Lübken, T.; Gruner, M.; Kataeva, O.; Ralambondrahety, R.; Andriamanantoanina, H.; Checinski, M. P.; Bauer, I.; Knölker, H.-J. Org. Biomol. Chem. 2017, 15, 2593–2608. doi:10.1039/c7ob00191f
Return to citation in text: [1] -
Mizushina, Y.; Suzuki-Fukudome, H.; Takeuchi, T.; Takemoto, K.; Kuriyama, I.; Yoshida, H.; Kamisuki, S.; Sugawara, F. Bioorg. Med. Chem. 2014, 22, 1070–1076. doi:10.1016/j.bmc.2013.12.038
Return to citation in text: [1] -
CAS Scifinder. https://scifinder-n.cas.org/ (accessed Jan 31, 2024).
Return to citation in text: [1] [2] -
Pretsch, E.; Bühlmann, P.; Affolter, C. Structure determination of organic compounds; Springer: Berlin, Heidelberg, 2000. doi:10.1007/978-3-662-04201-4
Return to citation in text: [1] -
Davis, R. A.; Watters, D.; Healy, P. C. Tetrahedron Lett. 2005, 46, 919–921. doi:10.1016/j.tetlet.2004.12.057
Return to citation in text: [1] [2] -
Gan, H.; Huang, Y.; Feng, W.; Zhu, W.; Guo, K. J. Chem. Res. 2015, 39, 336–339. doi:10.3184/174751915x14326563172262
Return to citation in text: [1] [2] [3] [4] -
Chen, D.; Cadelis, M. M.; Rouvier, F.; Troia, T.; Edmeades, L. R.; Fraser, K.; Gill, E. S.; Bourguet-Kondracki, M.-L.; Brunel, J. M.; Copp, B. R. Int. J. Mol. Sci. 2023, 24, 5882. doi:10.3390/ijms24065882
Return to citation in text: [1] -
Sue, K.; Cadelis, M. M.; Rouvier, F.; Bourguet-Kondracki, M.-L.; Brunel, J. M.; Copp, B. R. Biomolecules 2024, 14, 261. doi:10.3390/biom14030261
Return to citation in text: [1] -
Lira, N. S.; Monte-Neto, R. L.; Marchi, J. G. B.; Lins, A. C. d. S.; Tavares, J. F.; Silva, M. S. d.; Dias, C. d. S.; Barbosa-Filho, J. M.; Santos, C. F. d.; Cunha, E. V. L. d.; Pinheiro, U. d. S.; Braz-Filho, R. Quim. Nova 2012, 35, 2189–2193. doi:10.1590/s0100-40422012001100017
Return to citation in text: [1] -
NatureBank; Griffith University. https://www.griffith.edu.au/institute-drug-discovery/unique-resources/naturebank.
Return to citation in text: [1] [2] -
De Luca, P.; De Rosa, M.; Minale, L.; Sodano, G. J. Chem. Soc., Perkin Trans. 1 1972, 2132–2135. doi:10.1039/p19720002132
Return to citation in text: [1] -
Holloway, B. W.; Matsumoto, H.; Phibbs, P. V., Jr. Acta Microbiol. Pol. 1986, 35, 161–164.
Return to citation in text: [1] -
Paytubi, S.; de La Cruz, M.; Tormo, J. R.; Martín, J.; González, I.; González-Menendez, V.; Genilloud, O.; Reyes, F.; Vicente, F.; Madrid, C.; Balsalobre, C. Front. Microbiol. 2017, 8, 326. doi:10.3389/fmicb.2017.00326
Return to citation in text: [1]
| 13. | Ferriol, P.; Marante, F. J. T.; Martin, I. B.; Rodríguez, J. J. S.; Alonso, R. G.; Benkovics, A. G.; Mioso, R. Rec. Nat. Prod. 2018, 12, 470–479. doi:10.25135/rnp.52.17.11.558.204 |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 9. | Khan, F. A.; Ahmad, S.; Kodipelli, N.; Shivange, G.; Anindya, R. Org. Biomol. Chem. 2014, 12, 3847–3865. doi:10.1039/c3ob42537a |
| 1. | Bernardini, S.; Tiezzi, A.; Laghezza Masci, V.; Ovidi, E. Nat. Prod. Res. 2018, 32, 1926–1950. doi:10.1080/14786419.2017.1356838 |
| 2. | Buss, A. D.; Cox, B.; Waigh, R. D. Natural Products as Leads for New Pharmaceuticals. In Burger's Medicinal Chemistry and Drug Discovery; Abraham, D. J., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2003; pp 847–900. doi:10.1002/0471266949.bmc018 |
| 6. | Talebi Bezmin Abadi, A.; Rizvanov, A. A.; Haertlé, T.; Blatt, N. L. Bionanosci. 2019, 9, 778–788. doi:10.1007/s12668-019-00658-4 |
| 24. | De Luca, P.; De Rosa, M.; Minale, L.; Sodano, G. J. Chem. Soc., Perkin Trans. 1 1972, 2132–2135. doi:10.1039/p19720002132 |
| 1. | Bernardini, S.; Tiezzi, A.; Laghezza Masci, V.; Ovidi, E. Nat. Prod. Res. 2018, 32, 1926–1950. doi:10.1080/14786419.2017.1356838 |
| 5. | Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2020, 83, 770–803. doi:10.1021/acs.jnatprod.9b01285 |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 19. | Gan, H.; Huang, Y.; Feng, W.; Zhu, W.; Guo, K. J. Chem. Res. 2015, 39, 336–339. doi:10.3184/174751915x14326563172262 |
| 1. | Bernardini, S.; Tiezzi, A.; Laghezza Masci, V.; Ovidi, E. Nat. Prod. Res. 2018, 32, 1926–1950. doi:10.1080/14786419.2017.1356838 |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 3. | Carroll, A. R.; Copp, B. R.; Davis, R. A.; Keyzers, R. A.; Prinsep, M. R. Nat. Prod. Rep. 2023, 40, 275–325. doi:10.1039/d2np00083k |
| 4. | Carroll, A. R.; Copp, B. R.; Grkovic, T.; Keyzers, R. A.; Prinsep, M. R. Nat. Prod. Rep. 2024, 41, 162–207. doi:10.1039/d3np00061c |
| 15. | Mizushina, Y.; Suzuki-Fukudome, H.; Takeuchi, T.; Takemoto, K.; Kuriyama, I.; Yoshida, H.; Kamisuki, S.; Sugawara, F. Bioorg. Med. Chem. 2014, 22, 1070–1076. doi:10.1016/j.bmc.2013.12.038 |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 14. | Rahelivao, M. P.; Lübken, T.; Gruner, M.; Kataeva, O.; Ralambondrahety, R.; Andriamanantoanina, H.; Checinski, M. P.; Bauer, I.; Knölker, H.-J. Org. Biomol. Chem. 2017, 15, 2593–2608. doi:10.1039/c7ob00191f |
| 22. | Lira, N. S.; Monte-Neto, R. L.; Marchi, J. G. B.; Lins, A. C. d. S.; Tavares, J. F.; Silva, M. S. d.; Dias, C. d. S.; Barbosa-Filho, J. M.; Santos, C. F. d.; Cunha, E. V. L. d.; Pinheiro, U. d. S.; Braz-Filho, R. Quim. Nova 2012, 35, 2189–2193. doi:10.1590/s0100-40422012001100017 |
| 12. | Tran, T. M. T.; Addison, R. S.; Davis, R. A.; Rehm, B. H. A. Int. J. Mol. Sci. 2023, 24, 10204. doi:10.3390/ijms241210204 |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 23. | NatureBank; Griffith University. https://www.griffith.edu.au/institute-drug-discovery/unique-resources/naturebank. |
| 8. | Pieri, C.; Borselli, D.; Di Giorgio, C.; De Méo, M.; Bolla, J.-M.; Vidal, N.; Combes, S.; Brunel, J. M. J. Med. Chem. 2014, 57, 4263–4272. doi:10.1021/jm500194e |
| 9. | Khan, F. A.; Ahmad, S.; Kodipelli, N.; Shivange, G.; Anindya, R. Org. Biomol. Chem. 2014, 12, 3847–3865. doi:10.1039/c3ob42537a |
| 10. | Khadake, S. N.; Karamathulla, S.; Jena, T. K.; Monisha, M.; Tuti, N. K.; Khan, F. A.; Anindya, R. Bioorg. Med. Chem. Lett. 2021, 39, 127883. doi:10.1016/j.bmcl.2021.127883 |
| 11. | Allen, R. A.; McCormack, C. E. M.; Wuest, W. M. ChemMedChem 2023, 18, e202300253. doi:10.1002/cmdc.202300253 |
| 20. | Chen, D.; Cadelis, M. M.; Rouvier, F.; Troia, T.; Edmeades, L. R.; Fraser, K.; Gill, E. S.; Bourguet-Kondracki, M.-L.; Brunel, J. M.; Copp, B. R. Int. J. Mol. Sci. 2023, 24, 5882. doi:10.3390/ijms24065882 |
| 21. | Sue, K.; Cadelis, M. M.; Rouvier, F.; Bourguet-Kondracki, M.-L.; Brunel, J. M.; Copp, B. R. Biomolecules 2024, 14, 261. doi:10.3390/biom14030261 |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 13. | Ferriol, P.; Marante, F. J. T.; Martin, I. B.; Rodríguez, J. J. S.; Alonso, R. G.; Benkovics, A. G.; Mioso, R. Rec. Nat. Prod. 2018, 12, 470–479. doi:10.25135/rnp.52.17.11.558.204 |
| 8. | Pieri, C.; Borselli, D.; Di Giorgio, C.; De Méo, M.; Bolla, J.-M.; Vidal, N.; Combes, S.; Brunel, J. M. J. Med. Chem. 2014, 57, 4263–4272. doi:10.1021/jm500194e |
| 18. | Davis, R. A.; Watters, D.; Healy, P. C. Tetrahedron Lett. 2005, 46, 919–921. doi:10.1016/j.tetlet.2004.12.057 |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 19. | Gan, H.; Huang, Y.; Feng, W.; Zhu, W.; Guo, K. J. Chem. Res. 2015, 39, 336–339. doi:10.3184/174751915x14326563172262 |
| 17. | Pretsch, E.; Bühlmann, P.; Affolter, C. Structure determination of organic compounds; Springer: Berlin, Heidelberg, 2000. doi:10.1007/978-3-662-04201-4 |
| 25. | Holloway, B. W.; Matsumoto, H.; Phibbs, P. V., Jr. Acta Microbiol. Pol. 1986, 35, 161–164. |
| 26. | Paytubi, S.; de La Cruz, M.; Tormo, J. R.; Martín, J.; González, I.; González-Menendez, V.; Genilloud, O.; Reyes, F.; Vicente, F.; Madrid, C.; Balsalobre, C. Front. Microbiol. 2017, 8, 326. doi:10.3389/fmicb.2017.00326 |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 12. | Tran, T. M. T.; Addison, R. S.; Davis, R. A.; Rehm, B. H. A. Int. J. Mol. Sci. 2023, 24, 10204. doi:10.3390/ijms241210204 |
| 8. | Pieri, C.; Borselli, D.; Di Giorgio, C.; De Méo, M.; Bolla, J.-M.; Vidal, N.; Combes, S.; Brunel, J. M. J. Med. Chem. 2014, 57, 4263–4272. doi:10.1021/jm500194e |
| 9. | Khan, F. A.; Ahmad, S.; Kodipelli, N.; Shivange, G.; Anindya, R. Org. Biomol. Chem. 2014, 12, 3847–3865. doi:10.1039/c3ob42537a |
| 10. | Khadake, S. N.; Karamathulla, S.; Jena, T. K.; Monisha, M.; Tuti, N. K.; Khan, F. A.; Anindya, R. Bioorg. Med. Chem. Lett. 2021, 39, 127883. doi:10.1016/j.bmcl.2021.127883 |
| 19. | Gan, H.; Huang, Y.; Feng, W.; Zhu, W.; Guo, K. J. Chem. Res. 2015, 39, 336–339. doi:10.3184/174751915x14326563172262 |
| 18. | Davis, R. A.; Watters, D.; Healy, P. C. Tetrahedron Lett. 2005, 46, 919–921. doi:10.1016/j.tetlet.2004.12.057 |
| 8. | Pieri, C.; Borselli, D.; Di Giorgio, C.; De Méo, M.; Bolla, J.-M.; Vidal, N.; Combes, S.; Brunel, J. M. J. Med. Chem. 2014, 57, 4263–4272. doi:10.1021/jm500194e |
| 19. | Gan, H.; Huang, Y.; Feng, W.; Zhu, W.; Guo, K. J. Chem. Res. 2015, 39, 336–339. doi:10.3184/174751915x14326563172262 |
| 7. | Xu, M.; Davis, R. A.; Feng, Y.; Sykes, M. L.; Shelper, T.; Avery, V. M.; Camp, D.; Quinn, R. J. J. Nat. Prod. 2012, 75, 1001–1005. doi:10.1021/np300147d |
| 23. | NatureBank; Griffith University. https://www.griffith.edu.au/institute-drug-discovery/unique-resources/naturebank. |
© 2024 Hayes et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.