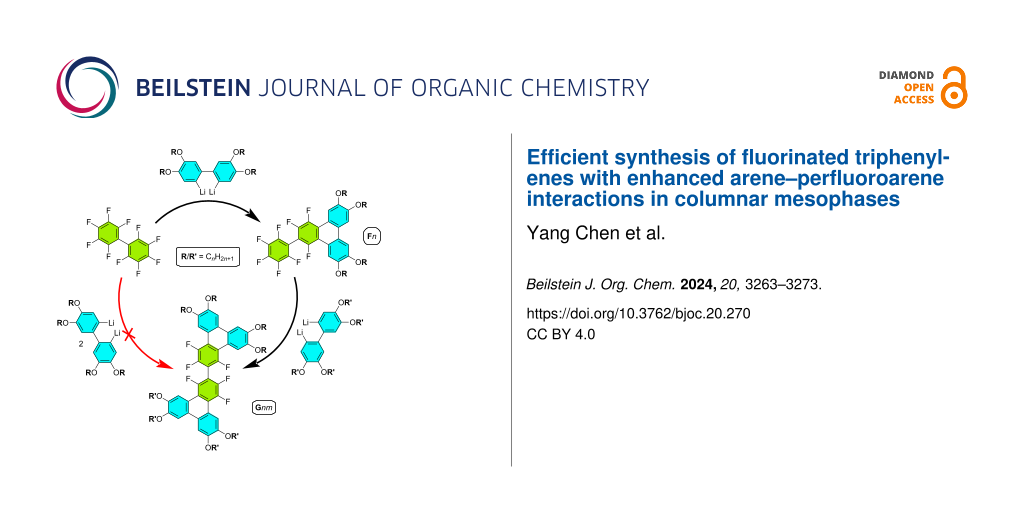
Abstract
The high potential of non-covalent arene–fluoroarene intermolecular interactions in the design of liquid crystals lies in their ability to strongly promote self-assembly, improve the order and stability of the supramolecular mesophases, and enable tuneability of the optical and electronic properties, which can potentially be exploited for advanced applications in display technologies, photonic devices, sensors, and organic electronics. We recently successfully reported the straightforward synthesis of several mesogens containing four lateral aliphatic chains and derived from the classical triphenylene core self-assembling in columnar mesophases based on this paradigm. These mesogenic compounds were simply obtained in good yields by the nucleophilic substitution (SNFAr) of various types of commercially available fluoroarenes with the electrophilic organolithium derivatives 2,2'-dilithio-4,4',5,5'-tetraalkoxy-1,1'-biphenyl (2Li-BPn). In a continuation of this study, aiming at testing the limits of the reaction and providing a large diversity of structures, a structurally related series of compounds is reported here, namely 1,2,4-trifluoro-6,7,10,11-tetraalkoxy-3-(perfluorophenyl)triphenylenes (Fn). They were obtained by reacting the above mentioned 2,2’-dilithiobiphenyl derivatives with decafluorobiphenyl, C6F5–C6F5. These compounds differ from the previously reported series, 1,2,4-trifluoro-6,7,10,11-tetraalkoxy-3-aryltriphenylenes (PHn), solely by the substitution of the terminal phenyl ring with a pentafluorophenyl ring. Thus, as expected, they display a Colhex mesophase over large temperature ranges, with only small differences in the mesophase stability and transition temperatures. Furthermore, the presence of the terminal fluorophenyl group enables a subsequent second annulation, yielding a new series of extended polyaromatic mesomorphic compounds, i.e., 1,1',3,3',4,4'-hexafluoro-6,6',7,7',10,10',11,11'-octaalkoxy-2,2'-bitriphenylene (Gnm) which were found to display a Colrec mesophase. The specific nucleophilic substitution patterns of the Fn derivatives and the antiparallel stacking mode into columnar structures stabilized by arene–perfluoroarene intermolecular interactions were confirmed by the single-crystal structure of the alkoxy-free side chain analog, i.e., 1,2,4-trifluoro-3-(perfluorophenyl)triphenylene (F). UV–vis absorption and fluorescence emission spectroscopies reveal green photoluminescence with fluorescence quantum yields of up to 33% for the Fn derivatives. The J-aggregation for the inner fluorine-substituted dimers Gnm is energetically and stereoelectronically more favorable and G66 exhibits thin-film fluorescence with a large red-shift of the emission peak.

Graphical Abstract
Introduction
Non-covalent arene–fluoroarene intermolecular interactions [1,2] are drawing increasing attention due to their critical role in the engineering of functional and complex supramolecular assemblies [3-11], ranging from rigid crystalline architectures [3-8] to soft liquid crystalline materials [9-11]. Their unique properties originate from the high electronegativity of the fluorine atoms, inserted in the aromatic rings, which considerably modifies the dipole moment of the corresponding fluorinated aromatic rings with respect to their hydrogenated homologs, thus influencing their behavior, binding affinities, and optoelectronic properties. These interactions already represent an effective tool for the design of liquid crystalline materials [3-8]. Rod-like liquid crystalline molecules with fluorine-substituted arenes are ubiquitous in the displays industry [12]. They are also gaining importance in the design of π-conjugated polycyclic aromatic discotic liquid crystals (F-DLCs) [13-17] of interest for organic electronics and optical advanced materials, as they tend to promote more efficient molecular stacking into columns than their purely hydrogenated counterparts [18,19], thereby improving one-dimensional charge transport properties [20-22] in combination with tunable absorption and emission of visible light. Polar nematic phase [23] and chiral columnar phase materials [24] based on polar fluorobenzene rings have also recently emerged as interesting new classes of fluorous materials, revealing their enormous potential in the high-tech fields.
Although, F-DLCs seem to show unique and advantageous physical properties, their numbers and structural variation types are very limited due partially to several synthetic challenges [25-34]. Their syntheses usually are based on the direct transformation of commercial perfluoroarene chemical blocks and reagents, involving catalyzed C–F-bond activation and cross-coupling reactions, usually requiring precious transition-metal catalysts and tedious synthetic routes [28-34]. Therefore, low-cost and facile synthetic strategies are desired to increase their structural and functional diversity. In the modern organic synthetic tool-box, the fluoroarene nucleophilc substitution (SNFAr) reaction possesses many outstanding advantages in the synthesis of π-conjugated functional molecules: the electrophiles are plentiful and include cheaply available perfluorobenzene, perfluoropyridine, perfluoronaphthalene, decafluorobiphenyl, and many other synthesized perfluoroarenes, and the nucleophiles are also abundant and contain aryllithium, conjugated organic dilithium reagents, phenols and benzenethiols, etc. [35-43].
We recently reported the high versatility of these intermolecular interactions in the design of several Janus-like discotic mesogens (Figure 1) [44-47]. A first study dealt with the synthesis of two sets of compounds, namely 1,2,3,4-tetrafluoro-6,7,10,11-tetraalkxoytriphenylenes (4F-TPn) and 9,10,11,12,13,14-hexafluoro-2,3,6,7-tetraalkoxybenzo[f]tetraphenes (6F-BTPn) [44], obtained by the straightforward nucleophilic substitution of fluoroarenes (SNFAr) between 2,2'-dilithio-4,4',5,5'-tetraalkoxy-1,1'-biphenyl (2Li-BPn) derivatives and hexafluorobenzene, C6F6 (4F-TPn), on the one hand, and octafluoronaphthalene, C10F8 (6F-BTPn), on the other. With only four alkoxy chains, these polar “Janus” mesogens [33,44] display a columnar hexagonal mesophase over broader temperature ranges and higher mesophase stability than the archetypical 2,3,6,7,10,11-hexa(alkoxy)triphenylene counterparts [48], whereas the corresponding hydrogenated 2,3,6,7-tetraalkoxytriphenylene counterparts (TPn) were not mesomorphic. Testing further this approach to evaluate the persistence of mesomorphism in this family of compounds, another set of related compounds but with inhomogeneous chain substitution patterns, namely 7,10-dialkoxy-1,2,3,4-tetrafluoro-6,11-dimethoxytriphenylene (p-TPFn) and 6,11-dialkoxy-1,2,3,4-tetrafluoro-7,10-dimethoxytriphenylene (m-TPFn), were synthesized by this method [46]. Both isomers also displayed liquid crystalline properties, despite an even larger deficit of alkyl chains, although the inhomogeneous chain distribution had a net impact on both stability and nature of the mesophases. The versatility of this synthetic approach allows us to synthesize another set of mesomorphic compounds, based on a triphenylene core, 1,2,4-trifluoro-6,7,10,11-tetra(alkyloxy)-3-phenyltriphenylenes (PHn, and extended to other aryl derivatives) by reacting lipophilic 2,2’-dilithiobiphenyl derivatives with the bulkier pentafluorobiphenyl, C6F5–C6H5. All these compounds display large mesomorphic ranges again, with the final phenyl ring being immersed with both the aliphatic continuum and the columns of stacked aromatic cores [45]. All these structural investigations revealed the great resilience of such a molecular system to important structural changes, and the essential role of the fluorinated phenyl moieties in the induction and stability of liquid crystalline mesophases.
![[1860-5397-20-270-1]](/bjoc/content/figures/1860-5397-20-270-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Fluorotriphenylene derivatives and their nonfluorinated homologs obtained by SNFAr from 2,2'-dilithio-4,4',5,5'-tetraalkoxy-1,1'-biphenyl (2Br-BPn → 2Li-BPn) e.g., 4F-TPn [44], p-TPFn [46], m-TPFn [46], PHn [45], and Fn (this work); BTPn was synthesized by a Suzuki–Scholl reaction sequence (Scheme S3, Supporting Information File 1).
Figure 1: Fluorotriphenylene derivatives and their nonfluorinated homologs obtained by SNFAr from 2,2'-dilith...
The present study focuses on synthesizing new, structurally-related series of π-conjugated aromatic compounds (Figure 1) based on a simple triphenylene core and evaluating the mesomorphic and optical properties. Specifically 1,2,4-trifluoro-6,7,10,11-tetraalkoxy-3-(perfluorophenyl)triphenylenes (Fn, Figure 1), bearing a terminal fluoroarene ring was obtained using the same reaction as previously reported for PHn (Figure 1) [45], between 2,2’-dilithiobiphenyl derivatives but this time with the electrophile decafluorobiphenyl C6F5–C6F5 instead of C6H5–C6F5 (Fn, n = 3–12, Scheme 1). The presence of the terminal fluoroarene group in Fn enables to exploit further the SNFAr procedure, as demonstrated for 4F-TPn and 6F-BTPn [44], through a subsequent second annulation reaction. This results in a second series of extended π-conjugated aromatic mesomorphic compounds, 1,1',3,3',4,4'-hexafluoro-6,6',7,7',10,10',11,11'-octaalkoxy-2,2'-bitriphenylene (Gnm, n,m = 4, 5, 6, Scheme 1), with the possibility to synthesize molecules with dissymmetrical chain substitution patterns. The investigation has two main objectives. First, it seeks to explore and understand the impact of the fluorination of the pendant ring on both the self-organization and optical properties of these compounds by comparing the properties of Fn with those of partially fluorinated PHn and non-fluorinated BTPn. Such comparison is critical for optimizing materials for specific applications. Second, the presence of this terminal fluoroarene group provides a basic platform to expand this chemistry, enabling access to new π-extended molecular systems that might be challenging to synthesize through conventional synthetic routes. This dual focus highlights the study’s potential to advance both the design of functional materials and the development of innovative synthetic methodologies.
![[1860-5397-20-270-i1]](/bjoc/content/inline/1860-5397-20-270-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis, yields, and nomenclature of 1,2,4-trifluoro-6,7,10,11-tetraalkoxy-3-(perfluorophenyl)triphenylene (Fn, n = 3–12) and corresponding 1,1',3,3',4,4'-hexafluoro-6,6',7,7',10,10',11,11'-octakisalkoxy-2,2'-bitriphenylene dimers (G55, G66 and G48).
Scheme 1: Synthesis, yields, and nomenclature of 1,2,4-trifluoro-6,7,10,11-tetraalkoxy-3-(perfluorophenyl)tri...
The mesomorphous properties of these two sets of compounds were investigated by polarized optical microscopy (POM), differential scanning calorimetry (DSC), and small-and wide-angle X-ray scattering (S/WAXS) and compared to related fluorinated and non-fluorinated homologs. The results showed that the Janus Fn derivatives exhibit a hexagonal columnar liquid crystal phase (Colhex), with clearing temperatures decreasing gradually with the elongation of the alkoxy side-chains, from nearly 200 °C for the shortest homologs down to ca. 100 °C for the dodecyloxy derivative. The larger lath-like compounds, Gnm, exhibit a rectangular columnar phase (Colrec) also over large temperature ranges from ambient up to 183 and 164 °C, respectively. The unsymmetrically alkoxy-substituted derivative, G48, also displays a Colrec over a similar temperature range. The compounds’ photophysical properties have also been studied in various solutions and thin film: G66 emitted green light in solution with an absolute photoluminescent quantum yield of ca. 33%.
Results and Discussion
Synthesis and characterization
We have recently generalized a very efficient “palladium-free” synthesis for the preparation of a variety of triphenylene derivatives (Figure 1) [44-47] based on the nucleophilic substitution of various electrophilic perfluoroarenes, including C6F2H4, C6F6, C10F8, and C6F5-Ph, by nucleophilic organolithium reagents. e.g., 2,2’-dilithio-4,4’,5,5’-tetrakis(alkoxy)-1,1’-biphenyls, 2Li-BPn, prepared in situ from the reaction between 3,3’,4,4’-tetra(alkoxy)-2,2’-dibromo-1,1’-biphenyl, 2Br-BPn, and n-BuLi at low temperature. All new Fn compounds were prepared as previously described by reaction of 2Li-BPn with decafluorobiphenyl, C6F5-C6F5 (Scheme 1). The starting materials 2Br-BPn were prepared in high yield via FeCl3-oxidative coupling of 1,2-dialkoxy-4-bromobenzene (Scholl reaction). The new triphenylene derivatives, F3–F12, were prepared in moderate to high yields (51–73%). In addition, three bitriphenylene derivatives were synthesized in a subsequent annulation step from Fn derivatives: the in situ prepared 2Li-BP5/6 was reacted with F5/6 to yield the symmetrical discotic dimers, G55/G66 respectively, in an average yield of 42–45% (Scheme 1). The reaction of 2Li-BP8 with F4 was also successfully tested, and allowed the preparation of the unsymmetrical discotic dimer G48, obtained in 50% yield, opening great possibilities in structural variations. The facile synthesis of G55, G66, and G48 demonstrates again the versatility of this synthetic method. The synthesis and detailed characterization of compounds F6 and G66 have been the subject of a preliminary patent description. This documentation describes the methodologies employed for their preparation, the analytical techniques used to confirm their structures, and the data obtained confirming their identities [49].
Two additional compounds were synthesized to complete this study: the derivative with no alkoxy chains (F) was prepared to grow single crystals suitable for X-ray analysis (Scheme S2 in Supporting Information File 1) in order to confirm the annulation reaction pattern, and 2,3,6,7-tetrakishexyloxy-10-phenyltriphenylene (BTP6, Scheme S3), the non-fluorinated isomer of F6 and PH6 (Figure 1), for probing the effect of fluorinated rings and the decisive role of arene–fluoroarene interactions in both mesomorphism induction and stabilization. Thus, 2Li-BP was reacted with C6F5–C6F5 to produce 2-perfluorophenyl-1,3,4-trifluorotriphenylene (F), in 37% yield and 2,3,6,7-tetrakishexyloxy-10-phenyltriphenylene (BTP6) was synthesized via consecutive Suzuki coupling and Scholl reaction in a total yield of 77%.
The synthesis, molecular structures, nomenclature, and synthetic yields of all compounds are shown in general Scheme 1. All prepared molecules were fully characterized by NMR (1H, 19F and 13C), HRMS, and CHN analysis (Figures S1–S32, Supporting Information File 1), and all the results agree with the proposed molecular structures.
Single-crystal structure of F
Suitable single crystals of compound F for X-ray analysis were obtained by slow evaporation of an ethyl acetate/ethanol solution (Figure 2, and Supporting Information File 1, Figures S33, S34 and Tables S1–S3). The crystal structure unequivocally confirms that the reaction pattern between 2Li-BP and perfluorobiphenyl, effectively yielded the desired 1,2,4-trifluoro-3-(perfluorophenyl)triphenylene and that the annulation occurred at the expected positions. The similarity of the 19F NMR spectra of F and alkoxy-substituted derivatives, Fn, showing 6 single peaks, corresponding to the 8 different fluorine atoms at almost identical positions (Figures S8–S14 and S21 in Supporting Information File 1), confirms that the pattern of the annulation reaction is the same for all triphenylene derivatives. Thus, compound F crystallizes in an orthorhombic crystallographic system (Pccn space group, no. 56) [50] with unit cell dimensions a = 13.2645(2) Å, b = 5.5284(1) Å, and c = 22.7571(3) Å; the unit cell contains 8 molecules, which gives a calculated molecular density of 1.688 g cm−3.
![[1860-5397-20-270-2]](/bjoc/content/figures/1860-5397-20-270-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Single crystal structure of 1,2,4-trifluoro-3-(perfluorophenyl)triphenylene (F) viewed along the main axes: ORTEP diagram showing 50% thermal ellipsoid probability: carbon (gray), fluorine (green), and hydrogen (white).
Figure 2: Single crystal structure of 1,2,4-trifluoro-3-(perfluorophenyl)triphenylene (F) viewed along the ma...
In more details, the crystal structure reveals two short intramolecular F···H hydrogen contacts in the triphenylene core part with lengths of 2.047 Å and 2.114 Å, respectively (see Figure S33 in Supporting Information File 1). The triphenylene part of the molecule is rather flat, with, however, a substantial planar deviation of the pending perfluorophenyl group, forming a large tilt of ca. 45°. From the view along the b-axis, it can be seen that the flat triphenylene cores stack perfectly on top of each other, but with the pending fluoroarene group being alternately distributed from one side to the other, likely for steric hindrance, thus maximizing the fluoro–arene interactions by superimposing hydrogenated phenyl segments with fluoroarene ones (Figure S34 in Supporting Information File 1). Consequently, the triphenylene core–core distance is 3.83 Å and almost identical to the stacking distance in the columnar mesophase as measured by wide-angle X-ray scattering. Due to the efficient space filling and fluoroarene polar π-interaction, neighboring F molecules stack in an antiparallel way with a slippering distance of 1.697 Å from each other. Of course, with the presence of the lateral aliphatic chains, the cores rotate in order to maximize aliphatic–aromatic segregation whilst preserving fluoro–arene interactions.
Liquid crystal properties
Prior to investigating the mesomorphism of Fn and Gnm compounds, their thermal stability was first examined by thermal gravimetric analysis (TGA) under a N2 atmosphere in the dynamic mode. The TGA curves (Figure S35 and Table S4 in Supporting Information File 1) show that all compounds undergo two thermal decomposition processes; an initial thermal event with a decomposition temperature (Tdec, at weight loss 5%) between 283–332 °C for Fn and above 340 °C for Gnm, probing their excellent thermal stability.
All Fn and Gnm compounds display mesophases at room temperature. Their optical textures and phase-transition behaviors were observed via hot stage polarizing optical microscopy (POM). POM images were systematically captured during the cooling process at a cooling rate of 1 °C/min after the compounds were heated into the isotropic liquid (Figure 3 and Figure S36 in Supporting Information File 1). The liquid crystalline phases of the Janus triphenylenes, F3 to F12, all showed optical textures of a hexagonal columnar mesophase. Slowly cooled from its isotropic liquid state, F6 grew up into broken, fan-shaped color plates among a large dark area, with straight line defects, characteristic of the hexagonal columnar mesophase. The discotic dimers Gnm displayed a dense optical texture with dendritic- and flower-like features, with small fraction of dark area, which could be possibly attributed to a reduction of the mesophase symmetry.
![[1860-5397-20-270-3]](/bjoc/content/figures/1860-5397-20-270-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: POM textures, observed between crossed polarizers of Janus and dimer, F6, F12, G66, and G48, respectively, as representative examples. More images can be seen in Supporting Information File 1 (Figure S36).
Figure 3: POM textures, observed between crossed polarizers of Janus and dimer, F6, F12, G66, and G48, respec...
The results of the differential scanning calorimetry (DSC) are summarized in Figure 4 (see also Supporting Information File 1, Figures S37, S38 and Table S5 for details), confirming the POM observations and their room temperature behaviors (no crystallization is observed even at low temperature, except for F12). Compounds with shorter alkyl chains, F3, F4, and F5, possess almost the same clearing temperatures near 190 °C, whereas from F6 to F12, the clearing temperature gradually lowers from 176 down to 112 °C. When comparing Fn and PHn (Figure 4) [45], both types of compounds show a high-range columnar mesophase at high temperature with almost identical clearing temperatures, which decrease gradually with the elongation of the alkoxy side-chains. The only difference is the emergence of a more ordered, 3D phase for some PHn derivatives observed on cooling at lower temperature. As expected, neither F or BTP6 are mesomorphic.
![[1860-5397-20-270-4]](/bjoc/content/figures/1860-5397-20-270-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Comparative bar graph summarizing the thermal behavior of Fn, BTP6, and PHn derivatives (2nd heating DSC data).
Figure 4: Comparative bar graph summarizing the thermal behavior of Fn, BTP6, and PHn derivatives (2nd heatin...
As for the larger fluorine derivatives Gnm (G55, G66, and G48), they all possess enantiotropic columnar mesophases right from room temperature up to 183 (G55), 164 (G66), and ca. 120 °C (G48). Compared to the previously synthesized non-fluorous pentyloxy homolog, showing a monotropic Colrec phase [51,52], core fluorination surely plays a positive role in the induction of mesomorphism.
The mesophases of Fn and Gnm compounds were fully characterized by small- and wide-angle X-ray scattering (S/WAXS) at several temperatures (Figure 5, Figures S39–S41 and Table S6 in Supporting Information File 1). The X-ray patterns of the Fn compounds exhibit one main, sharp, and intense reflection in the small-angle range, and an additional small peak indexed as (20) for F3 and F4, or two peaks indexed as (11) and (20), respectively, for F10 and F12, confirming unambiguously the hexagonal symmetry (F5, F6, and F8 only show the intense small-angle reflection). In addition, they all show a broad diffuse scattering and a sharp and intense diffraction peak in the wide-angle region, assigned respectively to as hch, for the disordered chains, and hπ, for the long-range core–core stacking resulting from strong polar–π interactions. The correlation length of the stacking was calculated by the Debye–Scherrer formula, which correspond to ca. 15–25 stacked molecules (Table 1). This coincides with the high clearing temperatures and high stability of the columnar mesophase of Fn. Overall, the behaviors of Fn and PHn compounds are very similar, with only minor deviations of the isotropic temperatures.
![[1860-5397-20-270-5]](/bjoc/content/figures/1860-5397-20-270-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Representative S/WAXS patterns of Fn and Gnm compounds.
Figure 5: Representative S/WAXS patterns of Fn and Gnm compounds.
Table 1: Mesophases’ parameters.
| Cpds | Phase | Temp.a | ab/bb | Ab | hπ(ξ)/hchc | Vmold/ρd | hmole |
| F3 | Colhex | 120 | 16.73 | 242.44 | 3.53 (67)/4.41 | 895/1.26 | 3.69 |
| F3 | Colhex | 180 | 16.71 | 241.77 | 3.64 (56)/4.40 | 938/1.20 | 3.88 |
| F4 | Colhex | 50 | 17.57 | 267.38 | 3.45 (64)/4.42 | 968/1.26 | 3.62 |
| F4 | Colhex | 120 | 17.74 | 272.60 | 3.55 (65)/4.31 | 1010/1.21 | 3.71 |
| F5 | Colhex | 40 | 18.46 | 295.14 | 3.44 (79)/4.44 | 1074/1.22 | 3.64 |
| F5 | Colhex | 100 | 18.52 | 297.08 | 3.52 (83)/4.38 | 1111/1.18 | 3.74 |
| F5 | Colhex | 160 | 18.57 | 298.57 | 3.62 (59)/4.37 | 1161/1.13 | 3.89 |
| F6 | Colhex | 70 | 19.31 | 322.78 | 3.48 (72)/4.47 | 1203/1.17 | 3.73 |
| F6 | Colhex | 150 | 19.56 | 331.25 | 3.60 (47)/4.58 | 1270/1.11 | 3.83 |
| F8 | Colhex | 50 | 21.43 | 397.59 | 3.46 (79)/4.30 | 1410/1.13 | 3.55 |
| F8 | Colhex | 130 | 21.46 | 398.76 | 3.58 (42)/4.40 | 1485/1.07 | 3.72 |
| F10 | Colhex | 30 | 22.79 | 450.03 Å2 | 3.45 (78)/4.37 | 1612/1.10 | 3.58 |
| F10 | Colhex | 100 | 23.09 | 461.88 Å2 | 3.54 (42)/4.36 | 1629/1.09 | 3.64 |
| F12 | Colhex | 30 | 24.30 | 511.40 Å2 | 3.45 (75)/4.38 | 1829/1.08 | 3.58 |
| F12 | Colhex | 80 | 24.47 | 518.53 Å2 | 3.51 (47)/4.40 | 1886/1.04 | 3.64 |
| G55 | Colrec | 40 | 35.20/25.46 | 896.19 Å2 | 3.57 (–)/4.28 | 1778/1.17 | 4.04 |
| G55 | Colrec | 100 | 35.26/25.55 | 900.89 Å2 | 3.68 (–)/4.45 | 1853/1.12 | 4.11 |
| G55 | Colrec | 160 | 35.32/25.74 | 909.00 Å2 | 3.72 (–)/4.51 | 1952/1.06 | 4.31 |
| G66 | Colrec | 50 | 37.18/26.80 | 996.42 Å2 | 3.56 (–)/4.36 | 2009/1.13 | 4.03 |
| G66 | Colrec | 90 | 37.32/26.65 | 994.58 Å2 | 3.66 (–)/4.48 | 2066/1.10 | 4.15 |
| G66 | Colrec | 140 | 37.46/26.53 | 993.81 Å2 | 3.75 (–)/4.50 | 2152/1.05 | 4.33 |
| G48 | Colrec | 30 | 36.78/26.61 | 978.71 Å2 | 3.54 (67)/4.39 | 1985/1.14 | 4.06 |
| G48 | Colrec | 70 | 36.98/26.54 | 981.45 Å2 | 3.59 (–)/4.45 | 2036/1.11 | 4.15 |
| G48 | Colrec | 110 | 37.14/26.31 | 977.15 Å2 | 3.66 (–)/4.39 | 2098/1.08 | 4.29 |
aTemperature of experiment (°C); blattice parameter, a/b (Å) and area, A (Å2), A = a2√3/2 (for Colhex) = a × b ( for Colrec), Ncol: number of columns per lattice: Ncol = 1 for Colhex, Ncol = 2 for Colrec; caverage face-to-face stacking distance (Å) between consecutive mesogens, hπ, determined from scattering maximum from SWAXS pattern, and ξ, correlation length (Å) calculated by Debye–Scherrer formula; hch, average distance (Å) between molten chains; dmolecular volume (Å3) and density (g·cm−3) calculated from partial volumes of reference substances: Vmol = Var + Vch, the sum of the volume of the aromatic part, Var (from reference compounds) and the volume of the chains, Vch [53], ρ = MW/(NA·Vmol); ecolumnar slice thickness (Å), hmol = NcolVmol/A (Å).
The single structure shows that the flat triphenylene cores almost stack perfectly on top of each other, with an alternation of the pending fluorinated phenyl groups by superimposition of one of the hydrogenated rings of the triphenylene segment above the trifluoroarene one, in order to maximize fluoro–arene intermolecular interactions between the molecules (Figure S34 in Supporting Information File 1). It also shows that the pending fluorophenyl makes an angle of ca. 45° with the triphenylene plane (see also DFT below). Despite this conformational distortion, the molecular thickness, hmol, obtained by dividing the molecular volume with the columnar cross section, is not drastically increased (Table 1); hmol ranges between 3.62 and 3.73 Å, very close to the stacking distance between consecutive cores measured by S/WAXS (3.42 Å ≤ hπ ≤ 3.62 Å), confirming that the triphenylene mesogens pile up in the columns with no or little tilt, reminiscent of the stacking observed in the crystal structure. It would thus consist of the piling of the triphenylene cores with the protruding fluorophenyl segments partly mixed with the aliphatic medium, with specific orientations of the triphenylenes (multiple of 120° orientations which contribute to the average circular cross-section of the columns) in order to maximize both the intermolecular interactions through the superimposition of fluoroarene segments and (alkylated) arene ones, partly similar to the crystal structure, and the homogeneous distribution of the chains around the columns, in agreement with the hexagonal symmetry. Both sets Fn and PHn show rather similar variation of the cross-sectional area, increasing homogeneously with n, as well as similar molecular thickness throughout (A and hmol, Figure S41 in Supporting Information File 1). Consequently, both the packing of Fn and of PHn in the Colhex phase must be very similar, with no effect of the pending group nature (C6H5 vs C6F5) on the mesophase stability. A proposed model for the supramolecular organization of Fn in the Colhex phase is shown in Figure S42 of Supporting Information File 1.
The bitriphenylene Gnm materials exhibit different X-ray patterns, with a multitude of sharp peaks, that could be indexed according to a rectangular lattice (p2gg symmetry) [50], confirming the reduction of the phase symmetry and well-defined interfaces between aliphatic continuum, hydrogenated aromatics, and fluorinated arenes. The S/WAXS patterns of the three compounds are also identical, independently of the chain length or the chain distribution, and the lattice expansion with temperature is not significative. However, the stacking appears to be less effective than for the Janus derivatives as the signal corresponding to core–core stacking is not as sharp and intense, corresponding to a decrease of fluoroarene–arene interactions. This agrees with an increase of the molecular thickness (see hmol values, Table 1), likely due to some electrostatic and steric repulsion between fluorine atoms within the inner core. DFT shows that both triphenylene segments do not lie in the same plane and that the overall molecule is slightly twisted. Nevertheless, the molecules still stack on top of each other in columns, maintaining the segregation between the various regions as in polyphilic molecules [9,10,54] and, since the molecules have a more anisotropic shape, the columnar cross-section cannot adopt a circular shape but rather an elliptical one, hence their arrangement into a rectangular lattice (Figure S43 in Supporting Information File 1). For Gnm, the mesomorphism is thus essentially driven by microsegregation between the various molecular constituents.
Photophysical properties: UV–vis absorption and photoluminescence
UV–vis absorption and fluorescence spectra of the synthesized fluorine polycyclic aromatic hydrocarbons Fn and Gnm were measured in solution (in various solvents) and thin film and the results are summarized in Figure 6 and Table S7 in Supporting Information File 1; F6 and G66 were chosen as representative examples.
![[1860-5397-20-270-6]](/bjoc/content/figures/1860-5397-20-270-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Absorption (a) and emission (b) spectra of F6 and G66, measured in different solvents at a concentration of 1 × 10−5 mol/L and in solid-state thin film.
Figure 6: Absorption (a) and emission (b) spectra of F6 and G66, measured in different solvents at a concentr...
F6 and G66 show almost identical UV–vis absorption spectra, both of them possessing a very broad absorption band below 400 nm with two maxima at ca. 380 nm for the strongest peak, and at ca. 360 nm for the smaller one (λabs = 284 nm for PH8), with no or little influence of the solvent polarity (Figure 6a). This may suggest that the sigma-bonded F6 and G66 have no conjugation in their ground states, the electron density being located on one triphenylene moiety (see DFT). G66 shows a stronger absorption band than F6, with expected ε values in the range of 104 and 105 L·mol−1·cm−1, respectively, as G66 possesses two triphenylene units whereas F6 only one.
Both compounds, however, display substantially different photoluminescent behavior. Both F6 and G66 show a single, broad emission band with a peak maximum around 410–430 nm for F6 (λem = 402 nm for PH8) and 460–500 nm for G66, with absolute quantum yields of 30% and 32%, respectively. Further, their fluorescence spectra in solutions show some solvent polarity influence, with a substantial red shift as the solvent polarity is increased. In thin film, the single luminescence maximum is shifted to 450 nm (518 nm) and 610 nm for F6 (PH8) and G66, respectively. The thin film emission of G66 is red-shifted by about 120 nm compared with its solution, while the one of F6 is red-shifted only by 30 nm. This huge difference in thin film emission can be explained via their intermolecular slipped J-type aggregation, supported by S/WAXS and proposed the model of Colrec mesophase [55-57].
DFT computation
For a deeper understanding of the electronic properties of these fluorine triphenylenes, we performed some DFT computation of F1 and G11 with the shortest methyl chain, and the results are summarized in Figure 7 and Figure S44 in Supporting Information File 1. First, the theoretically optimized molecular structure of F1 agrees well with the single crystal structure of F: the triphenylene core is planar and the side arm perfluorophenyl group is rotated by a few degrees due to the F···F repulsion on different rings. G11 exhibits a similar twist between the two triphenylene cores. Further, the calculated HOMO electron cloud of F1 (−5.8 eV; −5.89 eV for PH1) and G11 (−5.60 eV) both are located on a triphenylene core, which explains the similarity of their UV–vis absorption spectra. Their LUMO electron density maps distribute across to the side arm for F1 (−1.88 eV; −1.78 eV for PH1) and to the other triphenylene core for the dimer G11 (−1.78 eV). The π-conjugation of excited states results in a difference of their HOMO and LUMO energy levels: the fluorine dimer G11 possesses higher HOMO and LUMO energy levels than that of the monomer F1, with a smaller HOMO–LUMO energy gap (3.92 eV for F1 versus 4.11 eV for PH1). The DFT results thus agree pretty well with the fluorescence spectra in solution: G66 shows a fluorescence peak at 470–500 nm, while F6 has peaks located between 410–430 nm. It is noted that G66 shows deeper red-shifted fluorescence than F6, with peaks of 610 nm and 450 nm, respectively. The J-type aggregation for G66 in thin film and liquid crystalline state is energetically favorable for the arene–perfluoroarene overlap stacking and related stereoelectronic effects, which results in more than 100 nm red-shift of the fluorescence peak.
![[1860-5397-20-270-7]](/bjoc/content/figures/1860-5397-20-270-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: DFT calculated frontier molecular orbitals and optimized molecular structures for F1 and G11.
Figure 7: DFT calculated frontier molecular orbitals and optimized molecular structures for F1 and G11.
Conclusion
We have successfully prepared seven fluorine triphenylenes (F3 to F12) with various alkyl chain lengths and three bitriphenylene dimers (G55, G66, and G48) with different molecular symmetry by the SNAr reaction. This “palladium-free” reaction between 2Li-BPn and C6F5–C6F5 possesses several advantages: easily available starting chemicals, low cost, efficient, and versatile, displaying the potential to synthesize more complicated fluoroarene molecules and polymers. These fluorine-containing triphenylenes Fn and dimers Gnm display Colhex and Colrec mesophases, respectively, with high stability of the columnar mesophases due to strong arene–perfluoroarene polar π-interactions and related stereoelectronic effects. These Janus-type Fn compounds exhibit high clearing temperatures, and no crystalline phase, thus a very broad columnar mesophase range. Further, the sigma-bonded triphenylene dimers Gnm display an enantiotropic Colrec mesophase including room temperature. These π-conjugated materials are advantageous for further investigation in device applications. The aromatic core fluorination changes the electronic structures of the triphenylenes, and their supramolecular arene–perfluoroarene slipped stacks (J-aggregate) result in G66 with orange-yellow color fluorescence in the solid state.
Supporting Information
Synthesis (Schemes S1–S3) and characterization, 1H, 13C, and 19F NMR (Figures S1–S21), HRMS (Figures S22–S32), EA, single crystal X-ray structures (Figures S33, S34, Tables S1–S3), TGA (Figure S35, Table S4), POM (Figure S36), DSC (Figures S37, S38, Table S5), S/WAXS (Figures S39–S43, Table S6), optical properties (Table S7), and DFT (Figure S44, Table S8).
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 6.5 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Wang, W.; Wu, W. X.; Zhang, Y.; Jin, W. J. Chem. Phys. Rev. 2024, 5, 031303. doi:10.1063/5.0205540
Return to citation in text: [1] -
Pace, C. J.; Gao, J. Acc. Chem. Res. 2013, 46, 907–915. doi:10.1021/ar300086n
Return to citation in text: [1] -
Roesner, E. K.; Asheghali, D.; Kirillova, A.; Strauss, M. J.; Evans, A. M.; Becker, M. L.; Dichtel, W. R. Chem. Sci. 2022, 13, 2475–2480. doi:10.1039/d1sc05932g
Return to citation in text: [1] [2] [3] -
Braunecker, W. A.; Hurst, K. E.; Ray, K. G.; Owczarczyk, Z. R.; Martinez, M. B.; Leick, N.; Keuhlen, A.; Sellinger, A.; Johnson, J. C. Cryst. Growth Des. 2018, 18, 4160–4166. doi:10.1021/acs.cgd.8b00630
Return to citation in text: [1] [2] [3] -
Neitz, H.; Bessi, I.; Kachler, V.; Michel, M.; Höbartner, C. Angew. Chem., Int. Ed. 2023, 62, e202214456. doi:10.1002/anie.202214456
Return to citation in text: [1] [2] [3] -
Liu, B.; Gao, J.; Hao, A.; Xing, P. Angew. Chem., Int. Ed. 2023, 62, e202305135. doi:10.1002/anie.202305135
Return to citation in text: [1] [2] [3] -
Zhang, H.; Han, J.; Jin, X.; Duan, P. Angew. Chem., Int. Ed. 2021, 60, 4575–4580. doi:10.1002/anie.202014891
Return to citation in text: [1] [2] [3] -
Cheng, Q.; Hao, A.; Xing, P. Chem. Sci. 2024, 15, 618–628. doi:10.1039/d3sc05212e
Return to citation in text: [1] [2] [3] -
Tschierske, C. Top. Curr. Chem. 2011, 318, 1–108. doi:10.1007/128_2011_267
Return to citation in text: [1] [2] [3] -
Hird, M. Chem. Soc. Rev. 2007, 36, 2070–2095. doi:10.1039/b610738a
Return to citation in text: [1] [2] [3] -
Kishikawa, K. Isr. J. Chem. 2012, 52, 800–808. doi:10.1002/ijch.201200028
Return to citation in text: [1] [2] -
Kirsch, P.; Bremer, M. Angew. Chem., Int. Ed. 2000, 39, 4216–4235. doi:10.1002/1521-3773(20001201)39:23<4216::aid-anie4216>3.0.co;2-k
Return to citation in text: [1] -
Wöhrle, T.; Wurzbach, I.; Kirres, J.; Kostidou, A.; Kapernaum, N.; Litterscheidt, J.; Haenle, J. C.; Staffeld, P.; Baro, A.; Giesselmann, F.; Laschat, S. Chem. Rev. 2016, 116, 1139–1241. doi:10.1021/acs.chemrev.5b00190
Return to citation in text: [1] -
O’Neill, M.; Kelly, S. M. Adv. Mater. (Weinheim, Ger.) 2011, 23, 566–584. doi:10.1002/adma.201002884
Return to citation in text: [1] -
Sergeyev, S.; Pisula, W.; Geerts, Y. H. Chem. Soc. Rev. 2007, 36, 1902–1929. doi:10.1039/b417320c
Return to citation in text: [1] -
Kaafarani, B. R. Chem. Mater. 2011, 23, 378–396. doi:10.1021/cm102117c
Return to citation in text: [1] -
Kumar, S. Chemistry of discotic liquid crystals: from monomer to polymers; CRC Press: Boca Raton, FL, 2011. doi:10.1201/b10457
Return to citation in text: [1] -
Boden, N.; Bushby, R. J.; Cammidge, A. N.; Duckworth, S.; Headdock, G. J. Mater. Chem. 1997, 7, 601–605. doi:10.1039/a606447g
Return to citation in text: [1] -
Yardley, R. E.; Paquette, J. A.; Taing, H.; Gaebler, H. M.; Eichhorn, S. H.; Hamilton, I. P.; Maly, K. E. Org. Lett. 2019, 21, 10102–10105. doi:10.1021/acs.orglett.9b04091
Return to citation in text: [1] -
Bushby, R. J.; Donovan, K. J.; Kreouzis, T.; Lozman, O. R. Opto-Electron. Rev. 2005, 13, 269–279.
Return to citation in text: [1] -
Sasada, Y.; Monobe, H.; Ueda, Y.; Shimizu, Y. Mol. Cryst. Liq. Cryst. 2010, 525, 153–157. doi:10.1080/15421401003799060
Return to citation in text: [1] -
Zhao, K.-Q.; Du, J.-Q.; Long, X.-H.; Jing, M.; Wang, B.-Q.; Hu, P.; Monobe, H.; Henrich, B.; Donnio, B. Dyes Pigm. 2017, 143, 252–260. doi:10.1016/j.dyepig.2017.04.048
Return to citation in text: [1] -
Cruickshank, E. ChemPlusChem 2024, 89, e202300726. doi:10.1002/cplu.202300726
Return to citation in text: [1] -
Concellón, A.; Lu, R.-Q.; Yoshinaga, K.; Hsu, H.-F.; Swager, T. M. J. Am. Chem. Soc. 2021, 143, 9260–9266. doi:10.1021/jacs.1c05268
Return to citation in text: [1] -
Weck, M.; Dunn, A. R.; Matsumoto, K.; Coates, G. W.; Lobkovsky, E. B.; Grubbs, R. H. Angew. Chem., Int. Ed. 1999, 38, 2741–2745. doi:10.1002/(sici)1521-3773(19990917)38:18<2741::aid-anie2741>3.0.co;2-1
Return to citation in text: [1] -
Kishikawa, K.; Oda, K.; Aikyo, S.; Kohmoto, S. Angew. Chem., Int. Ed. 2007, 46, 764–768. doi:10.1002/anie.200603594
Return to citation in text: [1] -
Sasada, Y.; Monobe, H.; Ueda, Y.; Shimizu, Y. Chem. Commun. 2008, 1452–1454. doi:10.1039/b716920e
Return to citation in text: [1] -
Zhao, K.-Q.; Gao, Y.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Heinrich, B.; Donnio, B. Eur. J. Org. Chem. 2016, 2802–2814. doi:10.1002/ejoc.201600270
Return to citation in text: [1] [2] -
Zhao, K.-Q.; Jing, M.; An, L.-L.; Du, J.-Q.; Wang, Y.-H.; Hu, P.; Wang, B.-Q.; Monobe, H.; Heinrich, B.; Donnio, B. J. Mater. Chem. C 2017, 5, 669–682. doi:10.1039/c6tc04530h
Return to citation in text: [1] [2] -
Zhang, Q.; Prins, P.; Jones, S. C.; Barlow, S.; Kondo, T.; An, Z.; Siebbeles, L. D. A.; Marder, S. R. Org. Lett. 2005, 7, 5019–5022. doi:10.1021/ol051972k
Return to citation in text: [1] [2] -
Ahmida, M.; Larocque, R.; Ahmed, M. S.; Vacaru, A.; Donnio, B.; Guillon, D.; Eichhorn, S. H. J. Mater. Chem. 2010, 20, 1292–1303. doi:10.1039/b917169j
Return to citation in text: [1] [2] -
Vieira, A. A.; Farias, G.; Costa, W. C.; Eccher, J.; Bechtold, I. H.; Durola, F.; Bock, H. Chem. – Eur. J. 2021, 27, 9003–9010. doi:10.1002/chem.202005456
Return to citation in text: [1] [2] -
Li, Z.; Powers, M.; Ivey, K.; Adas, S.; Ellman, B.; Bunge, S. D.; Twieg, R. J. Mater. Adv. 2022, 3, 534–546. doi:10.1039/d1ma00606a
Return to citation in text: [1] [2] [3] -
Guragain, P.; Powers, M.; Portman, J.; Ellman, B.; Twieg, R. J. Mater. Adv. 2023, 4, 4129–4137. doi:10.1039/d3ma00281k
Return to citation in text: [1] [2] -
Górski, K.; Ostojić, T.; Banasiewicz, M.; Ouellette, E. T.; Grisanti, L.; Gryko, D. T. Chem. – Eur. J. 2023, 29, e202203464. doi:10.1002/chem.202203464
Return to citation in text: [1] -
See, Y. Y.; Morales-Colón, M. T.; Bland, D. C.; Sanford, M. S. Acc. Chem. Res. 2020, 53, 2372–2383. doi:10.1021/acs.accounts.0c00471
Return to citation in text: [1] -
Wang, Z.; Wang, C.; Xi, Z. Tetrahedron Lett. 2006, 47, 4157–4160. doi:10.1016/j.tetlet.2006.04.035
Return to citation in text: [1] -
Cho, D. M.; Parkin, S. R.; Watson, M. D. Org. Lett. 2005, 7, 1067–1068. doi:10.1021/ol050019c
Return to citation in text: [1] -
Wang, Y.; Parkin, S. R.; Gierschner, J.; Watson, M. D. Org. Lett. 2008, 10, 3307–3310. doi:10.1021/ol8003468
Return to citation in text: [1] -
Morrison, D. J.; Trefz, T. K.; Piers, W. E.; McDonald, R.; Parvez, M. J. Org. Chem. 2005, 70, 5309–5312. doi:10.1021/jo0506231
Return to citation in text: [1] -
Leroux, F.; Mangano, G.; Schlosser, M. Eur. J. Org. Chem. 2005, 5049–5054. doi:10.1002/ejoc.200500514
Return to citation in text: [1] -
Kaga, A.; Iida, H.; Tsuchiya, S.; Saito, H.; Nakano, K.; Yorimitsu, H. Chem. – Eur. J. 2021, 27, 4567–4572. doi:10.1002/chem.202005223
Return to citation in text: [1] -
Li, H.; Wang, X.-Y.; Wei, B.; Xu, L.; Zhang, W.-X.; Pei, J.; Xi, Z. Nat. Commun. 2014, 5, 4508. doi:10.1038/ncomms5508
Return to citation in text: [1] -
Zhou, M.-M.; He, J.; Pan, H.-M.; Zeng, Q.; Lin, H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Eur. J. 2023, 29, e202301829. doi:10.1002/chem.202301829
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Pan, H.-M.; He, J.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Zhao, K.-Q.; Donnio, B. J. Mater. Chem. C 2023, 11, 14695–14704. doi:10.1039/d3tc02569a
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Zhang, K.-L.; Yu, W.-H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Asian J. 2024, 19, e202301080. doi:10.1002/asia.202301080
Return to citation in text: [1] [2] [3] [4] [5] -
He, J.; Chen, Y.; Hu, P.; Wang, B.-Q.; Zhao, K.-Q.; Donnio, B. J. Mol. Liq. 2024, 414, 126218. doi:10.1016/j.molliq.2024.126218
Return to citation in text: [1] [2] -
Monobe, H.; Shimizu, Y.; Okamoto, S.; Enomoto, H. Mol. Cryst. Liq. Cryst. 2007, 476, 31/[277]–41/[287]. doi:10.1080/15421400701732324
Return to citation in text: [1] -
Zhao, K.-Q.; He, J.; Zhao, K.-X.; Hu, P.; Wang, B.-Q. Synthesis of discotic perfluorophenyl-trifluorotriphenylene and hexafluorobitriphenylene. Chin. Pat. Appl. CN115477573 A, Nov 16, 2022.
Return to citation in text: [1] -
Aroyo, M. I., Ed. International Tables for Crystallography Volume A: Space-group symmetry, 2nd ed.; John Wiley & Sons, 2016. doi:10.1107/97809553602060000114
Return to citation in text: [1] [2] -
Zhao, K.-C.; Du, J.-Q.; Wang, H.-F.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Monobe, H.; Heinrich, B.; Donnio, B. Chem. – Asian J. 2019, 14, 462–470. doi:10.1002/asia.201801483
Return to citation in text: [1] -
Lin, H.; Zhao, K.-X.; Jing, M.; Long, X.-H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Lei, P.; Zeng, Q.-D.; Donnio, B. J. Mater. Chem. C 2022, 10, 14453–14470. doi:10.1039/d2tc02441a
Return to citation in text: [1] -
Donnio, B.; Heinrich, B.; Allouchi, H.; Kain, J.; Diele, S.; Guillon, D.; Bruce, D. W. J. Am. Chem. Soc. 2004, 126, 15258–15268. doi:10.1021/ja0471673
Return to citation in text: [1] -
Kishikawa, K.; Aikyo, S.; Akiyama, S.; Inoue, T.; Takahashi, M.; Yagai, S.; Aonuma, H.; Kohmoto, S. Soft Matter 2011, 7, 5176–5187. doi:10.1039/c0sm01459a
Return to citation in text: [1] -
Milián-Medina, B.; Gierschner, J. J. Phys. Chem. Lett. 2017, 8, 91–101. doi:10.1021/acs.jpclett.6b02495
Return to citation in text: [1] -
Hayashi, S.; Asano, A.; Kamiya, N.; Yokomori, Y.; Maeda, T.; Koizumi, T. Sci. Rep. 2017, 7, 9453. doi:10.1038/s41598-017-09848-0
Return to citation in text: [1] -
Bischof, D.; Tripp, M. W.; Hofmann, P. E.; Ip, C.-H.; Ivlev, S. I.; Gerhard, M.; Koert, U.; Witte, G. Chem. – Eur. J. 2022, 28, e202103653. doi:10.1002/chem.202103653
Return to citation in text: [1]
| 49. | Zhao, K.-Q.; He, J.; Zhao, K.-X.; Hu, P.; Wang, B.-Q. Synthesis of discotic perfluorophenyl-trifluorotriphenylene and hexafluorobitriphenylene. Chin. Pat. Appl. CN115477573 A, Nov 16, 2022. |
| 50. | Aroyo, M. I., Ed. International Tables for Crystallography Volume A: Space-group symmetry, 2nd ed.; John Wiley & Sons, 2016. doi:10.1107/97809553602060000114 |
| 45. | Pan, H.-M.; He, J.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Zhao, K.-Q.; Donnio, B. J. Mater. Chem. C 2023, 11, 14695–14704. doi:10.1039/d3tc02569a |
| 1. | Wang, W.; Wu, W. X.; Zhang, Y.; Jin, W. J. Chem. Phys. Rev. 2024, 5, 031303. doi:10.1063/5.0205540 |
| 2. | Pace, C. J.; Gao, J. Acc. Chem. Res. 2013, 46, 907–915. doi:10.1021/ar300086n |
| 3. | Roesner, E. K.; Asheghali, D.; Kirillova, A.; Strauss, M. J.; Evans, A. M.; Becker, M. L.; Dichtel, W. R. Chem. Sci. 2022, 13, 2475–2480. doi:10.1039/d1sc05932g |
| 4. | Braunecker, W. A.; Hurst, K. E.; Ray, K. G.; Owczarczyk, Z. R.; Martinez, M. B.; Leick, N.; Keuhlen, A.; Sellinger, A.; Johnson, J. C. Cryst. Growth Des. 2018, 18, 4160–4166. doi:10.1021/acs.cgd.8b00630 |
| 5. | Neitz, H.; Bessi, I.; Kachler, V.; Michel, M.; Höbartner, C. Angew. Chem., Int. Ed. 2023, 62, e202214456. doi:10.1002/anie.202214456 |
| 6. | Liu, B.; Gao, J.; Hao, A.; Xing, P. Angew. Chem., Int. Ed. 2023, 62, e202305135. doi:10.1002/anie.202305135 |
| 7. | Zhang, H.; Han, J.; Jin, X.; Duan, P. Angew. Chem., Int. Ed. 2021, 60, 4575–4580. doi:10.1002/anie.202014891 |
| 8. | Cheng, Q.; Hao, A.; Xing, P. Chem. Sci. 2024, 15, 618–628. doi:10.1039/d3sc05212e |
| 44. | Zhou, M.-M.; He, J.; Pan, H.-M.; Zeng, Q.; Lin, H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Eur. J. 2023, 29, e202301829. doi:10.1002/chem.202301829 |
| 45. | Pan, H.-M.; He, J.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Zhao, K.-Q.; Donnio, B. J. Mater. Chem. C 2023, 11, 14695–14704. doi:10.1039/d3tc02569a |
| 46. | Zhang, K.-L.; Yu, W.-H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Asian J. 2024, 19, e202301080. doi:10.1002/asia.202301080 |
| 47. | He, J.; Chen, Y.; Hu, P.; Wang, B.-Q.; Zhao, K.-Q.; Donnio, B. J. Mol. Liq. 2024, 414, 126218. doi:10.1016/j.molliq.2024.126218 |
| 9. | Tschierske, C. Top. Curr. Chem. 2011, 318, 1–108. doi:10.1007/128_2011_267 |
| 10. | Hird, M. Chem. Soc. Rev. 2007, 36, 2070–2095. doi:10.1039/b610738a |
| 11. | Kishikawa, K. Isr. J. Chem. 2012, 52, 800–808. doi:10.1002/ijch.201200028 |
| 44. | Zhou, M.-M.; He, J.; Pan, H.-M.; Zeng, Q.; Lin, H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Eur. J. 2023, 29, e202301829. doi:10.1002/chem.202301829 |
| 3. | Roesner, E. K.; Asheghali, D.; Kirillova, A.; Strauss, M. J.; Evans, A. M.; Becker, M. L.; Dichtel, W. R. Chem. Sci. 2022, 13, 2475–2480. doi:10.1039/d1sc05932g |
| 4. | Braunecker, W. A.; Hurst, K. E.; Ray, K. G.; Owczarczyk, Z. R.; Martinez, M. B.; Leick, N.; Keuhlen, A.; Sellinger, A.; Johnson, J. C. Cryst. Growth Des. 2018, 18, 4160–4166. doi:10.1021/acs.cgd.8b00630 |
| 5. | Neitz, H.; Bessi, I.; Kachler, V.; Michel, M.; Höbartner, C. Angew. Chem., Int. Ed. 2023, 62, e202214456. doi:10.1002/anie.202214456 |
| 6. | Liu, B.; Gao, J.; Hao, A.; Xing, P. Angew. Chem., Int. Ed. 2023, 62, e202305135. doi:10.1002/anie.202305135 |
| 7. | Zhang, H.; Han, J.; Jin, X.; Duan, P. Angew. Chem., Int. Ed. 2021, 60, 4575–4580. doi:10.1002/anie.202014891 |
| 8. | Cheng, Q.; Hao, A.; Xing, P. Chem. Sci. 2024, 15, 618–628. doi:10.1039/d3sc05212e |
| 28. | Zhao, K.-Q.; Gao, Y.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Heinrich, B.; Donnio, B. Eur. J. Org. Chem. 2016, 2802–2814. doi:10.1002/ejoc.201600270 |
| 29. | Zhao, K.-Q.; Jing, M.; An, L.-L.; Du, J.-Q.; Wang, Y.-H.; Hu, P.; Wang, B.-Q.; Monobe, H.; Heinrich, B.; Donnio, B. J. Mater. Chem. C 2017, 5, 669–682. doi:10.1039/c6tc04530h |
| 30. | Zhang, Q.; Prins, P.; Jones, S. C.; Barlow, S.; Kondo, T.; An, Z.; Siebbeles, L. D. A.; Marder, S. R. Org. Lett. 2005, 7, 5019–5022. doi:10.1021/ol051972k |
| 31. | Ahmida, M.; Larocque, R.; Ahmed, M. S.; Vacaru, A.; Donnio, B.; Guillon, D.; Eichhorn, S. H. J. Mater. Chem. 2010, 20, 1292–1303. doi:10.1039/b917169j |
| 32. | Vieira, A. A.; Farias, G.; Costa, W. C.; Eccher, J.; Bechtold, I. H.; Durola, F.; Bock, H. Chem. – Eur. J. 2021, 27, 9003–9010. doi:10.1002/chem.202005456 |
| 33. | Li, Z.; Powers, M.; Ivey, K.; Adas, S.; Ellman, B.; Bunge, S. D.; Twieg, R. J. Mater. Adv. 2022, 3, 534–546. doi:10.1039/d1ma00606a |
| 34. | Guragain, P.; Powers, M.; Portman, J.; Ellman, B.; Twieg, R. J. Mater. Adv. 2023, 4, 4129–4137. doi:10.1039/d3ma00281k |
| 55. | Milián-Medina, B.; Gierschner, J. J. Phys. Chem. Lett. 2017, 8, 91–101. doi:10.1021/acs.jpclett.6b02495 |
| 56. | Hayashi, S.; Asano, A.; Kamiya, N.; Yokomori, Y.; Maeda, T.; Koizumi, T. Sci. Rep. 2017, 7, 9453. doi:10.1038/s41598-017-09848-0 |
| 57. | Bischof, D.; Tripp, M. W.; Hofmann, P. E.; Ip, C.-H.; Ivlev, S. I.; Gerhard, M.; Koert, U.; Witte, G. Chem. – Eur. J. 2022, 28, e202103653. doi:10.1002/chem.202103653 |
| 3. | Roesner, E. K.; Asheghali, D.; Kirillova, A.; Strauss, M. J.; Evans, A. M.; Becker, M. L.; Dichtel, W. R. Chem. Sci. 2022, 13, 2475–2480. doi:10.1039/d1sc05932g |
| 4. | Braunecker, W. A.; Hurst, K. E.; Ray, K. G.; Owczarczyk, Z. R.; Martinez, M. B.; Leick, N.; Keuhlen, A.; Sellinger, A.; Johnson, J. C. Cryst. Growth Des. 2018, 18, 4160–4166. doi:10.1021/acs.cgd.8b00630 |
| 5. | Neitz, H.; Bessi, I.; Kachler, V.; Michel, M.; Höbartner, C. Angew. Chem., Int. Ed. 2023, 62, e202214456. doi:10.1002/anie.202214456 |
| 6. | Liu, B.; Gao, J.; Hao, A.; Xing, P. Angew. Chem., Int. Ed. 2023, 62, e202305135. doi:10.1002/anie.202305135 |
| 7. | Zhang, H.; Han, J.; Jin, X.; Duan, P. Angew. Chem., Int. Ed. 2021, 60, 4575–4580. doi:10.1002/anie.202014891 |
| 8. | Cheng, Q.; Hao, A.; Xing, P. Chem. Sci. 2024, 15, 618–628. doi:10.1039/d3sc05212e |
| 9. | Tschierske, C. Top. Curr. Chem. 2011, 318, 1–108. doi:10.1007/128_2011_267 |
| 10. | Hird, M. Chem. Soc. Rev. 2007, 36, 2070–2095. doi:10.1039/b610738a |
| 11. | Kishikawa, K. Isr. J. Chem. 2012, 52, 800–808. doi:10.1002/ijch.201200028 |
| 35. | Górski, K.; Ostojić, T.; Banasiewicz, M.; Ouellette, E. T.; Grisanti, L.; Gryko, D. T. Chem. – Eur. J. 2023, 29, e202203464. doi:10.1002/chem.202203464 |
| 36. | See, Y. Y.; Morales-Colón, M. T.; Bland, D. C.; Sanford, M. S. Acc. Chem. Res. 2020, 53, 2372–2383. doi:10.1021/acs.accounts.0c00471 |
| 37. | Wang, Z.; Wang, C.; Xi, Z. Tetrahedron Lett. 2006, 47, 4157–4160. doi:10.1016/j.tetlet.2006.04.035 |
| 38. | Cho, D. M.; Parkin, S. R.; Watson, M. D. Org. Lett. 2005, 7, 1067–1068. doi:10.1021/ol050019c |
| 39. | Wang, Y.; Parkin, S. R.; Gierschner, J.; Watson, M. D. Org. Lett. 2008, 10, 3307–3310. doi:10.1021/ol8003468 |
| 40. | Morrison, D. J.; Trefz, T. K.; Piers, W. E.; McDonald, R.; Parvez, M. J. Org. Chem. 2005, 70, 5309–5312. doi:10.1021/jo0506231 |
| 41. | Leroux, F.; Mangano, G.; Schlosser, M. Eur. J. Org. Chem. 2005, 5049–5054. doi:10.1002/ejoc.200500514 |
| 42. | Kaga, A.; Iida, H.; Tsuchiya, S.; Saito, H.; Nakano, K.; Yorimitsu, H. Chem. – Eur. J. 2021, 27, 4567–4572. doi:10.1002/chem.202005223 |
| 43. | Li, H.; Wang, X.-Y.; Wei, B.; Xu, L.; Zhang, W.-X.; Pei, J.; Xi, Z. Nat. Commun. 2014, 5, 4508. doi:10.1038/ncomms5508 |
| 20. | Bushby, R. J.; Donovan, K. J.; Kreouzis, T.; Lozman, O. R. Opto-Electron. Rev. 2005, 13, 269–279. |
| 21. | Sasada, Y.; Monobe, H.; Ueda, Y.; Shimizu, Y. Mol. Cryst. Liq. Cryst. 2010, 525, 153–157. doi:10.1080/15421401003799060 |
| 22. | Zhao, K.-Q.; Du, J.-Q.; Long, X.-H.; Jing, M.; Wang, B.-Q.; Hu, P.; Monobe, H.; Henrich, B.; Donnio, B. Dyes Pigm. 2017, 143, 252–260. doi:10.1016/j.dyepig.2017.04.048 |
| 24. | Concellón, A.; Lu, R.-Q.; Yoshinaga, K.; Hsu, H.-F.; Swager, T. M. J. Am. Chem. Soc. 2021, 143, 9260–9266. doi:10.1021/jacs.1c05268 |
| 50. | Aroyo, M. I., Ed. International Tables for Crystallography Volume A: Space-group symmetry, 2nd ed.; John Wiley & Sons, 2016. doi:10.1107/97809553602060000114 |
| 18. | Boden, N.; Bushby, R. J.; Cammidge, A. N.; Duckworth, S.; Headdock, G. J. Mater. Chem. 1997, 7, 601–605. doi:10.1039/a606447g |
| 19. | Yardley, R. E.; Paquette, J. A.; Taing, H.; Gaebler, H. M.; Eichhorn, S. H.; Hamilton, I. P.; Maly, K. E. Org. Lett. 2019, 21, 10102–10105. doi:10.1021/acs.orglett.9b04091 |
| 25. | Weck, M.; Dunn, A. R.; Matsumoto, K.; Coates, G. W.; Lobkovsky, E. B.; Grubbs, R. H. Angew. Chem., Int. Ed. 1999, 38, 2741–2745. doi:10.1002/(sici)1521-3773(19990917)38:18<2741::aid-anie2741>3.0.co;2-1 |
| 26. | Kishikawa, K.; Oda, K.; Aikyo, S.; Kohmoto, S. Angew. Chem., Int. Ed. 2007, 46, 764–768. doi:10.1002/anie.200603594 |
| 27. | Sasada, Y.; Monobe, H.; Ueda, Y.; Shimizu, Y. Chem. Commun. 2008, 1452–1454. doi:10.1039/b716920e |
| 28. | Zhao, K.-Q.; Gao, Y.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Heinrich, B.; Donnio, B. Eur. J. Org. Chem. 2016, 2802–2814. doi:10.1002/ejoc.201600270 |
| 29. | Zhao, K.-Q.; Jing, M.; An, L.-L.; Du, J.-Q.; Wang, Y.-H.; Hu, P.; Wang, B.-Q.; Monobe, H.; Heinrich, B.; Donnio, B. J. Mater. Chem. C 2017, 5, 669–682. doi:10.1039/c6tc04530h |
| 30. | Zhang, Q.; Prins, P.; Jones, S. C.; Barlow, S.; Kondo, T.; An, Z.; Siebbeles, L. D. A.; Marder, S. R. Org. Lett. 2005, 7, 5019–5022. doi:10.1021/ol051972k |
| 31. | Ahmida, M.; Larocque, R.; Ahmed, M. S.; Vacaru, A.; Donnio, B.; Guillon, D.; Eichhorn, S. H. J. Mater. Chem. 2010, 20, 1292–1303. doi:10.1039/b917169j |
| 32. | Vieira, A. A.; Farias, G.; Costa, W. C.; Eccher, J.; Bechtold, I. H.; Durola, F.; Bock, H. Chem. – Eur. J. 2021, 27, 9003–9010. doi:10.1002/chem.202005456 |
| 33. | Li, Z.; Powers, M.; Ivey, K.; Adas, S.; Ellman, B.; Bunge, S. D.; Twieg, R. J. Mater. Adv. 2022, 3, 534–546. doi:10.1039/d1ma00606a |
| 34. | Guragain, P.; Powers, M.; Portman, J.; Ellman, B.; Twieg, R. J. Mater. Adv. 2023, 4, 4129–4137. doi:10.1039/d3ma00281k |
| 9. | Tschierske, C. Top. Curr. Chem. 2011, 318, 1–108. doi:10.1007/128_2011_267 |
| 10. | Hird, M. Chem. Soc. Rev. 2007, 36, 2070–2095. doi:10.1039/b610738a |
| 54. | Kishikawa, K.; Aikyo, S.; Akiyama, S.; Inoue, T.; Takahashi, M.; Yagai, S.; Aonuma, H.; Kohmoto, S. Soft Matter 2011, 7, 5176–5187. doi:10.1039/c0sm01459a |
| 13. | Wöhrle, T.; Wurzbach, I.; Kirres, J.; Kostidou, A.; Kapernaum, N.; Litterscheidt, J.; Haenle, J. C.; Staffeld, P.; Baro, A.; Giesselmann, F.; Laschat, S. Chem. Rev. 2016, 116, 1139–1241. doi:10.1021/acs.chemrev.5b00190 |
| 14. | O’Neill, M.; Kelly, S. M. Adv. Mater. (Weinheim, Ger.) 2011, 23, 566–584. doi:10.1002/adma.201002884 |
| 15. | Sergeyev, S.; Pisula, W.; Geerts, Y. H. Chem. Soc. Rev. 2007, 36, 1902–1929. doi:10.1039/b417320c |
| 16. | Kaafarani, B. R. Chem. Mater. 2011, 23, 378–396. doi:10.1021/cm102117c |
| 17. | Kumar, S. Chemistry of discotic liquid crystals: from monomer to polymers; CRC Press: Boca Raton, FL, 2011. doi:10.1201/b10457 |
| 51. | Zhao, K.-C.; Du, J.-Q.; Wang, H.-F.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Monobe, H.; Heinrich, B.; Donnio, B. Chem. – Asian J. 2019, 14, 462–470. doi:10.1002/asia.201801483 |
| 52. | Lin, H.; Zhao, K.-X.; Jing, M.; Long, X.-H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Lei, P.; Zeng, Q.-D.; Donnio, B. J. Mater. Chem. C 2022, 10, 14453–14470. doi:10.1039/d2tc02441a |
| 12. | Kirsch, P.; Bremer, M. Angew. Chem., Int. Ed. 2000, 39, 4216–4235. doi:10.1002/1521-3773(20001201)39:23<4216::aid-anie4216>3.0.co;2-k |
| 23. | Cruickshank, E. ChemPlusChem 2024, 89, e202300726. doi:10.1002/cplu.202300726 |
| 53. | Donnio, B.; Heinrich, B.; Allouchi, H.; Kain, J.; Diele, S.; Guillon, D.; Bruce, D. W. J. Am. Chem. Soc. 2004, 126, 15258–15268. doi:10.1021/ja0471673 |
| 46. | Zhang, K.-L.; Yu, W.-H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Asian J. 2024, 19, e202301080. doi:10.1002/asia.202301080 |
| 33. | Li, Z.; Powers, M.; Ivey, K.; Adas, S.; Ellman, B.; Bunge, S. D.; Twieg, R. J. Mater. Adv. 2022, 3, 534–546. doi:10.1039/d1ma00606a |
| 44. | Zhou, M.-M.; He, J.; Pan, H.-M.; Zeng, Q.; Lin, H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Eur. J. 2023, 29, e202301829. doi:10.1002/chem.202301829 |
| 48. | Monobe, H.; Shimizu, Y.; Okamoto, S.; Enomoto, H. Mol. Cryst. Liq. Cryst. 2007, 476, 31/[277]–41/[287]. doi:10.1080/15421400701732324 |
| 44. | Zhou, M.-M.; He, J.; Pan, H.-M.; Zeng, Q.; Lin, H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Eur. J. 2023, 29, e202301829. doi:10.1002/chem.202301829 |
| 44. | Zhou, M.-M.; He, J.; Pan, H.-M.; Zeng, Q.; Lin, H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Eur. J. 2023, 29, e202301829. doi:10.1002/chem.202301829 |
| 45. | Pan, H.-M.; He, J.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Zhao, K.-Q.; Donnio, B. J. Mater. Chem. C 2023, 11, 14695–14704. doi:10.1039/d3tc02569a |
| 46. | Zhang, K.-L.; Yu, W.-H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Asian J. 2024, 19, e202301080. doi:10.1002/asia.202301080 |
| 47. | He, J.; Chen, Y.; Hu, P.; Wang, B.-Q.; Zhao, K.-Q.; Donnio, B. J. Mol. Liq. 2024, 414, 126218. doi:10.1016/j.molliq.2024.126218 |
| 45. | Pan, H.-M.; He, J.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Zhao, K.-Q.; Donnio, B. J. Mater. Chem. C 2023, 11, 14695–14704. doi:10.1039/d3tc02569a |
| 45. | Pan, H.-M.; He, J.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Zhao, K.-Q.; Donnio, B. J. Mater. Chem. C 2023, 11, 14695–14704. doi:10.1039/d3tc02569a |
| 46. | Zhang, K.-L.; Yu, W.-H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Asian J. 2024, 19, e202301080. doi:10.1002/asia.202301080 |
| 46. | Zhang, K.-L.; Yu, W.-H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Asian J. 2024, 19, e202301080. doi:10.1002/asia.202301080 |
| 45. | Pan, H.-M.; He, J.; Yu, W.-H.; Hu, P.; Wang, B.-Q.; Zhao, K.-Q.; Donnio, B. J. Mater. Chem. C 2023, 11, 14695–14704. doi:10.1039/d3tc02569a |
| 44. | Zhou, M.-M.; He, J.; Pan, H.-M.; Zeng, Q.; Lin, H.; Zhao, K.-Q.; Hu, P.; Wang, B.-Q.; Donnio, B. Chem. – Eur. J. 2023, 29, e202301829. doi:10.1002/chem.202301829 |
© 2024 Chen et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.