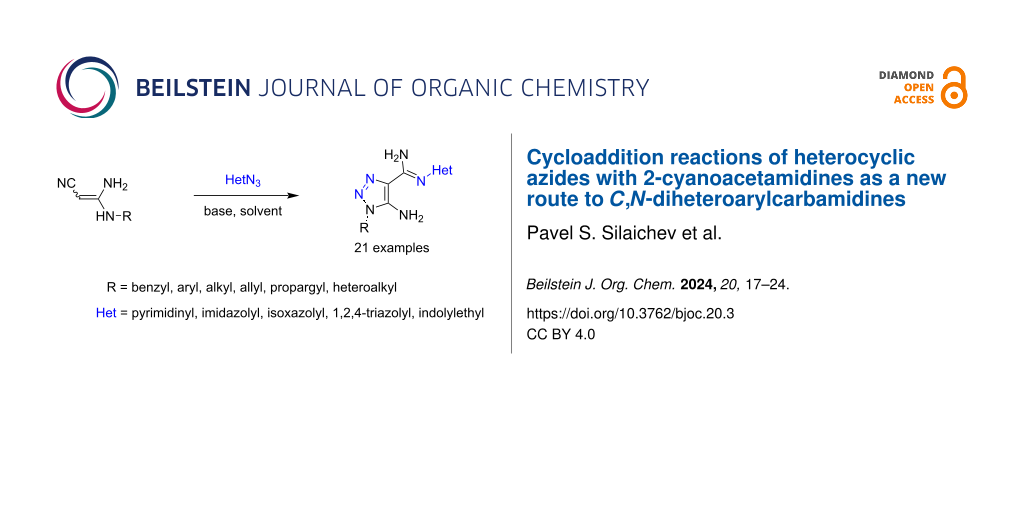
Abstract
A novel and efficient base-catalyzed, transition-metal-free method for the synthesis of diheterocyclic compounds connected by an amidine linker, including apart from the common 1,2,3-triazole ring, either an additional pyrimidinedione, 4-nitroimidazole, isoxazole, 1,3,4-triazole, 2-oxochromone or thiazole ring, has been developed. The process was facilitated by a strong base and includes the cycloaddition reaction of 3,3-diaminoacrylonitriles (2-cyanoacetamidines) to heterocyclic azides followed by a Cornforth-type rearrangement to the final products. The reaction is tolerant to various N-monosubstituted 3,3-diaminoacrylonitriles and to different heterocyclic azides. The developed method has a broad scope and can be applied to obtain a variety of N-heteroaryl-1,2,3-triazole-4-carbimidamides with alkyl, allyl, propargyl, benzyl, cycloalkyl, and indolyl substituents at the N1 position .

Graphical Abstract
Introduction
Heteroaryl amidines are widely used in the synthesis of various nitrogen-containing heterocyclic compounds and have a variety of biological activities [1-4]. After the discovery of click chemistry [5,6] involving the CuAAC method of 1,2,3-triazole synthesis [7,8], there has been great interest of studing the chemical and biological properties of the triazoles thus obtained [9-12]. It should be noted that the synthesis of amidines containing other heterocycles in addition to 1,2,3-triazole in the molecule has not been described in the literature. In this regard, it is of interest to develop an effective method for the synthesis of hybrids of 1,2,3-triazole with other heterocycles and to identify biologically active compounds among the synthesized compounds.
It is known that the cycloaddition reaction of azidopyrimidinediones with enamines [13] represents an effective method for the synthesis of pyrimidinyl amidines [14] (Scheme 1A). A few years ago, we discovered that the replacement of enamines with 2-cyano-N,N-dialkylethanethioamides in this reaction [15] led to the formation of amidines containing two heterocycles: pyrimidine-2,4-dione and 1,2,3-thiadiazole (Scheme 1B).
![[1860-5397-20-3-i1]](/bjoc/content/inline/1860-5397-20-3-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of heteroaryl amidines.
Scheme 1: Synthesis of heteroaryl amidines.
The idea came to our mind to construct a system comprising pyrimidine and 1,2,3-triazole rings connected with an amidine linker by the cycloaddition reaction of 3,3-diaminoacrylonitriles with 6-azidopyrimidine (Scheme 1C). Based on this idea, an efficient and novel method with wide scope has been developed which was then applied to obtain a variety of previously unknown 5-amino-1-substituted 1,2,3-triazole-N-heteroaryl-4-carboximidamides 3 bearing alkyl, allyl, propargyl, benzyl, cycloalkyl, and various heteroaryl substituents at the N1 postion of the carbimidamide group. Herein, we disclose our results on the cycloaddition reaction of 3,3-diaminoacrylonitriles 1 with heteroaryl azides (HetN3) 2 [16] leading to 5-amino-1,2,3-triazole-4-N-heteroarylcarbimidamides 3 (Scheme 1C).
Results and Discussion
Optimization of the reaction of amidine 1a with azide 2a
We initiated the study by investigating a model reaction involving the cycloaddition of 3-amino-3-(benzylamino)acrylonitrile (1a) to 6-azidopyrimidine-2,4-dione 2a (Table 1). To our surprise we obtained (Z)-5-amino-1-benzyl-N'-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)-1H-1,2,3-triazole-4-carboximidamide (3a) in 93% yield as the major product with 5-amino-1-benzyl-1H-1,2,3-triazole-4-carbonitrile (4) in 5% yield when the reaction was carried out at room temperature in 1,4-dioxane in the presence of an equivalent amount of TEA (Table 1, entry 1). This result is in contrast to our previous findings where the reaction of compound 1a with sulfonyl azides led to 5-amino-1,2,3-triazole-4-N-sulfonylamidines selectively [17].
Table 1: Optimization of the reaction of amidine 1a with 6-azidopyrimidine-2,4-dione 2a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-3-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent | Base (mol %) | Т | Ratio 3:4 (%) | Yield triazole 3a (%)b |
| 1 | 1,4-dioxane | Et3N (100) | rt | 95:5 | 93 |
| 2 | EtOH | DBU (100) | rt | 91:9 | 87 |
| 3 | DCM | DBU (100) | rt | 90:10 | 83 |
| 4 | 1,4-dioxane | DBU (100) | rt | 99:1 | 95 |
| 5 | 1,4-dioxane | DBU (100) | rt | 100:0 | 98c |
| 6 | 1,4-dioxane | DBU (80) | rt | 90:10 | 85 |
| 7 | 1,4-dioxane | DBU (120) | rt | 100:0 | 98 |
| 8 | 1,4-dioxane | no base | rt | 40:60 | 38 |
| 9 | 1,4-dioxane | pyridine (100) | rt | 37:63 | 35 |
| 10 | EtOH | NaOH (100) | rt | 94:6 | 90 |
| 11 | EtOH | NaOH (100) | 0 °С | 97:3 | 95c |
aConditions: 1a (0.5 mmol), 2a (0.5 mmol), solvent (2 mL), 5–10 min; bdetermined by 1H NMR spectroscopy; cisolated yield.
The following screening of organic (Table 1, entries 2‒7 and 8) and inorganic (Table 1, entry 10) bases at room temperature revealed that using DBU resulted in the highest yield of triazole 3a. Analysis of experiments with 100 mol %, 120 mol %, and 80 mol % of DBU (Table 1, entries 2‒7) showed that the use of 100 mol % of DBU is optimal for the selective synthesis of triazole 3a in high yield (Table 1, entries 4 and 5). A study of the reaction medium revealed that common organic solvents are highly efficient for this cascade reaction (Table 1, entries 1‒11). Among the solvents screened, 1,4-dioxane was found the best solvent in terms of yield of the target product, solubility of the reagents, and ease of separation of the product.
Thus, the optimal conditions found were reacting amidine 1 with azide 2 in the presence of DBU in a 1:1:1 ratio in 1,4-dioxane at room temperature. A similar yield of the target product 3a was obtained by the reaction in ethanol in the presence of NaOH at 0 °С (entry 11 in Table 1). The latter conditions can be an alternative for the synthesis of compounds 3.
Synthesis of N-heteroaryl amidines 3
We hypothesized that both 3,3-diaminoacrylonitriles 1 and azides 2 can serve as three atom building blocks (Figure 1), affording 5-amino-1,2,3-triazole-4-carbimidamides A (Scheme 1C), which could be suitable precursors for the desired compounds 3.
![[1860-5397-20-3-1]](/bjoc/content/figures/1860-5397-20-3-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of starting compounds.
Figure 1: Structures of starting compounds.
With the starting 3,3-diaminoacrylonitriles 1 and azides 2 at hands they were subjected to the optimized reaction conditions to obtain the desired 5-amino-1,2,3-triazole-4-N-heteroarylcarbimidamides 3. In this way, 21 pyrimidine (3a–k), azolyl (3l–s), and chromenone (3t and 3u) derivatives were successfully prepared (Scheme 2 and Supporting Information File 1).
![[1860-5397-20-3-i2]](/bjoc/content/inline/1860-5397-20-3-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of 3,3-diaminoacrylonitriles 1 and heterocyclic azides 2. Reaction conditions: 1 (0.5 mmol), 2 (0.5 mmol), DBU (0.5 mmol), 1,4-dioxane (2 mL), rt, 30 min (method A)a; 1 (0.5 mmol), 2 (0.5 mmol), NaOH (0.5 mmol), EtOH (2 mL), 0 °C → rt, 30 min (method B)b.
Scheme 2: Scope of 3,3-diaminoacrylonitriles 1 and heterocyclic azides 2. Reaction conditions: 1 (0.5 mmol), 2...
Generally, the reaction proceeded in moderate to high yields (46–98%), ranging between 79–98% for reactions of 3,3-diaminoacrylonitriles 1 with azidoheterocycles 2. Compared with azidopyrimidines 2a,b and azidochromene 2g, the use of azidoazoles 2c–f delivered the expected amidines 3l–r in somewhat lower yields. No definite electronic effect of the substituents in azides 2 was observed. We assume the higher yield of pyrimidine containing triazoles 3a–k by their lower solubility in 1,4-dioxane compared with the solubility of azole containing compounds 3l–s in both 1,4-dioxane and ethanol.
With regard to the scope of diaminoacrylonitriles 1a–j, various substituents at the nitrogen were tolerated including aryl, (substituted) benzyl and alkyl substituents (including tryptamine, cyclohexyl, propargyl, and allyl) and furnished the N-heteroaryl amidines 3a–u in high or moderate yields (46–98%) (Scheme 2). There was no apparent substituent effect on the yield of the final compounds 3 observed.
The structures of compounds 3a–u were confirmed by IR, 1H and 13C NMR spectroscopy (Figures S1‒S44 in Supporting Information File 1) as well as by high-resolution mass spectrometry (HRMS). X-ray data obtained for compound 3g gave us final proof of the structure of the prepared compounds.
To explain the outcome of the tandem reaction of 3,3-diaminoacrylonitriles to heterocyclic azides, a tentative mechanism for the formation of 1,2,3-triazoles 3 from acrylonitriles 1 and azides 2 is shown in Scheme 3.
![[1860-5397-20-3-i3]](/bjoc/content/inline/1860-5397-20-3-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism for the formation of triazoles 3.
Scheme 3: Proposed mechanism for the formation of triazoles 3.
Firstly, treatment with a base, leads to deprotonation of acrylonitriles 1 to form anion 1′. The subsequent formal cycloaddition of anion 1′ with azides 2 then affords triazolines 5, which aromatize through a 1,3-H-shift to afford the triazoles 6. Two pathways towards the isomeric triazoles 3 can be proposed: the first involving the electrocyclic ring opening of triazole 6 to form diazo compound anti-7, followed by rotation around the C‒C bond of the amidine group to furnish rotamer syn-7 which then undergoes 1,5-dipolar cyclization to products 3. The second path involves a Dimroth-type cyclization to form products 3′, which however, were not experimentally observed.
The base-catalyzed cycloaddition of 3,3-diaminoacrylonitriles 1 to azides 2, thus proceeds in a Cornforth-type fashion, which involves a triazole–triazole isomerization through ring opening, rotation of the amidine substituent around the single bond, and cyclization.
Cytotoxic activity
The cytotoxic activity of seven synthesized compounds 3b,d,e,g,h,l,m was studied. According to the results of the MTT test, IC50 values were calculated for these compounds (Table 2). The results obtained indicated that compounds 3b,e,g,h did not have a pronounced effect on the viability of cultured cells within the concentration range. This indicates the low cytotoxicity of these compounds. Meanwhile, compounds 3m,l are characterized by a borderline inhibition of cell culture viability. Attention is drawn to the increased toxic effect of 3m on cells of tumor origin, in comparison with normal human embryonic cells. Identification of the cause of the registered selective toxicity of the tested compounds requires further study.
Table 2: Cytotoxicity index (IC50 ± SE) in µM of the studied compounds on human embryo kidney cells (HEK-293), glioblastoma (A-172), and osteosarcoma (HOS) cells.
| Entry | Compound | Type of cell studied | ||
| НЕK-293 | A-172 | HOS | ||
| 1 | 3b | >256 | >256 | >256 |
| 2 | 3d | >1024 | >1024 | >1024 |
| 3 | 3e | >1024 | >1024 | >1024 |
| 4 | 3g | >512 | >512 | >512 |
| 5 | 3h | 471.98 ± 106.54 | >512 | >512 |
| 6 | 3l | 174.62 ± 43.72 | >256 | 224.28 ± 67.36 |
| 7 | 3m | 184.60 ± 51.06 | 95.71 ± 13.29 | 83.86 ± 17.13 |
| 8 | cisplatin | 59.90 ± 19.97 | 39.51 ± 6.39 | 38.14 ± 5.93 |
Conclusion
Thus, we have introduced an effective base-catalyzed tandem reaction including a Cornforth-type rearrangement of 1-heteroaryl-1,2,3-triazole-4-carboximidamides and formal cycloaddition reaction of readily available heterocyclic azides with 3,3-diaminoacrylonitriles. The reaction represents a novel method for the preparation of 1,2,3-triazoles bearing an N-hetaryl amidine moiety and thus this reaction offers a novel method for the preparation of new types of 1-substituted-1,2,3-triazoles, widening the synthetic applications of both azides and derivatives of acrylonitrile. Some of the prepared compounds exhibited a mild toxic effect on tumor cells in comparison with normal human embryonic cells.
Experimental
3,3-Diaminoacrylonitriles 1b and 1f were synthesized from ethyl 2-cyanoacetimidate and corresponding amines according to the literature procedure [17] and compounds 1a,c–e,h–j are commercially available. Azides 2a–d,f,g were synthesized according to the literature procedures [18-23], and azide 2e is commercially available.
Preparation of triazoles 3
Method A. In a manner similar to [17], DBU (0.5 mmol) was added to the solution of amidine 1 (0.5 mmol) in 1,4-dioxane (2 mL) at room temperature and azide 2 (0.5 mmol) was added to the resulting solution 5 min later. The reaction mixture was stirred for 30 min at room temperature, then water (6 mL) was added and the resulting solution was stirred for additional 5 min. Then, acetic acid (34 µL) was added to the reaction mixture, the formed precipitate was filtered off, washed with water, and dried in a desiccator over P4O10.
Method B. Amidine 1 (0.5 mmol) was added into a solution of sodium hydroxide, freshly prepared from sodium hydroxide (20 mg, 0.5 mmol) and ethanol (2 mL), and the resulting mixture was stirred at room temperature for 5–10 min. Then, the mixture was cooled to 0 °C, the corresponding azide 2 (0.5 mmol) was added and the resulting mixture were stirred for 30 min, after which cooling was removed. The reaction mixture was allowed to warm to ambient temperature under stirring and water (8 mL) was added to the mixture. The resulting solution was stirred for 5 min, after which acetic acid (34 µL) was added. The formed precipitate was filtered off, washed with water, and dried in a desiccator over P4O10.
(Z)-5-Amino-1-benzyl-N'-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)-1H-1,2,3-triazole-4-carboximidamide (3a). Compound 3a was obtained in 98% yield (174 mg) according to the general procedure A (DBU: 76 mg, 75 µL, 0.5 mmol; amidine 1a: 86 mg, 0.5 mmol; azide 2a: 90 mg, 0.5 mmol; 1,4-dioxane (2 mL)) as a colorless powder; mp 225–226 °C; 1H NMR (400 MHz, DMSO-d6) δ 3.16 (s, 3H), 3.21 (s, 3H), 5.08 (s, 1H), 5.46 (s, 2H), 6.52 (s, 1H), 6.53 (s, 1H), 7.19 (br. s, 2H), 7.25–7.39 (m, 5H); 13C NMR (101 MHz, DMSO-d6) δ 27.1, 29.8, 48.4, 87.4, 120.9, 127.4, 127.6, 128.5, 135.7, 143.8, 152.3, 152.9, 157.3, 162.4; IR (ATR, KBr, cm−1): ν 3402, 3316, 3201, 1700, 1688, 1649, 1629, 1594, 1568, 1520, 1497, 1476, 1454, 1444, 1426, 1386, 1358, 1335, 1311, 1276, 1264, 1248, 1194, 1090, 1057, 1028; HRMS–ESI-TOF (m/z): [M + H]+ calcd for C16H19N8O2+, 355.1625; found: 355.1628.
(Z)-5-Amino-1-benzyl-N'-(thiazol-2-yl)-1H-1,2,3-triazole-4-carboximidamide (3l). Compound 3l was obtained in 88% yield (117 mg) according to the general procedure B (NaOH: 20 mg, 0.5 mmol; amidine 1a: 77 mg, 0.5 mmol; azide 2c: 56 mg, 0.5 mmol; ethanol (2 mL)) as light yellow needles; mp 199–200 °C; 1H NMR (400 MHz, DMSO-d6) δ 5.48 (s, 2H), 6.68 (s, 1H), 6.69 (s, 1H), 7.10 (d, J = 3.9 Hz, 1H), 7.24 (d, J = 6.9 Hz, 2H), 7.28–7.38 (m, 3H), 7.43 (d, J = 3.9 Hz, 1H), 8.42 (br. s, 1H), 9.15 (br. s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 48.5, 112.6, 121.2, 127.2, 127.6, 128.5, 135.7, 138.8, 143.6, 153.5, 174.5; IR (ATR, KBr, cm−1): ν 3400, 3359, 3255, 1625, 1602, 1563, 1554, 1508, 1495, 1483, 1455, 1436, 1425, 1402, 1385, 1356, 1332, 1319, 1303, 1283, 1257, 1215, 1151, 1095, 1067, 1053, 1035, 1011; HRMS–ESI-TOF (m/z): [M + H]+ calcd for C13H14N7S+, 300.1026; found, 300.1031.
X-ray structure determination
3g: Crystal data for C15H22N8O2 (M = 346.40 g/mol): monoclinic, space group P21/c (no. 14), a = 14.734(3) Å, b = 9.000(2) Å, c = 13.125(4) Å, β = 104.29°, V = 1686.6(8)Å3, Z = 4, T = 295(2) K, μ(Mo Kα) = 0.097 mm−1, Dcalc = 1.364 g/cm3, 6415 reflections measured (5.4° ≤ 2Θ ≤ 59°), 6415 unique (R(sigma) = 0.0978) which were used in all calculations. The final R1 was 0.0693 (I > 2σ(I)) and wR2 was 0.1849 (all data). The refined twin ratio was 0.6748(18):0.3252(18).
The experiment was accomplished on the automated X-ray diffractometer «Xcalibur Ruby» with CCD detector following standard procedures (Mo Kα irradiation, graphite monochromator, ω-scans with 1° step at T = 295(2) K). Empirical absorption correction was applied. The structure was solved using the intrinsic phases in ShelXT program [24] and refined by ShelXL [25] using full-matrix least-squared method for non-hydrogen atoms. The H-atoms atoms bound to carbon were placed in the calculated positions and were refined in isotropic approximation. The hydrogen atoms of NH2 groups were refined independently with isotropic displacement parameters. The solution and refinement of the structures were accomplished with the Olex2 program package [26]. The structure was refined using HKLF5 format file as twin with two components.
CCDC 2298850 (3g) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; Email: deposit@ccdc.cam.ac.uk)
Funding
Design and elaboration of a novel tandem process, synthesis of starting building blocks and 21-st target compounds was made by chemistry team and founded by Russian Science Foundation, project number 23-13-00248. The study of cytotoxic effect of 7 compounds was financially supported by the Ministry of Science and Higher Education of the Russian Federation within the framework of the Program of the Development of the Ural Federal University named after the first President of Russia B.N. Yeltsin under the Federal Academic Leadership Program “Priority 2030”.
Data Availability Statement
Data generated and analyzed during this study is openly available in CCDC at https://doi.org/10.5517/ccdc.csd.cc2h54g9.
References
-
Sauer, B.; Skinner-Adams, T. S.; Bouchut, A.; Chua, M. J.; Pierrot, C.; Erdmann, F.; Robaa, D.; Schmidt, M.; Khalife, J.; Andrews, K. T.; Sippl, W. Eur. J. Med. Chem. 2017, 127, 22–40. doi:10.1016/j.ejmech.2016.12.041
Return to citation in text: [1] -
Cindrić, M.; Sović, I.; Martin-Kleiner, I.; Kralj, M.; Mašek, T.; Hranjec, M.; Starčević, K. Med. Chem. Res. 2017, 26, 2024–2037. doi:10.1007/s00044-017-1912-z
Return to citation in text: [1] -
Racané, L.; Cindrić, M.; Perin, N.; Roškarić, P.; Starčević, K.; Mašek, T.; Maurić, M.; Dogan, J.; Karminski-Zamola, G. Croat. Chem. Acta 2017, 90, 187–195. doi:10.5562/cca3146
Return to citation in text: [1] -
Beryozkina, T.; Bakulev, V.; Dianova, L.; Berseneva, V.; Slepukhin, P.; Leban, J.; Kalaba, P.; Aher, N. Y.; Ilic, M.; Sitte, H. H.; Lubec, G. Synthesis 2016, 48, 1046–1054. doi:10.1055/s-0035-1561350
Return to citation in text: [1] -
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5
Return to citation in text: [1] -
Moses, J. E.; Moorhouse, A. D. Chem. Soc. Rev. 2007, 36, 1249–1262. doi:10.1039/b613014n
Return to citation in text: [1] -
Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j
Return to citation in text: [1] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4
Return to citation in text: [1] -
Akter, M.; Rupa, K.; Anbarasan, P. Chem. Rev. 2022, 122, 13108–13205. doi:10.1021/acs.chemrev.1c00991
Return to citation in text: [1] -
Vala, D. P.; Vala, R. M.; Patel, H. M. ACS Omega 2022, 7, 36945–36987. doi:10.1021/acsomega.2c04883
Return to citation in text: [1] -
Dheer, D.; Singh, V.; Shankar, R. Bioorg. Chem. 2017, 71, 30–54. doi:10.1016/j.bioorg.2017.01.010
Return to citation in text: [1] -
Dehaen, W.; Bakulev, V. A., Eds. Chemistry of 1,2,3-triazoles; Topics in Heterocyclic Chemistry, Vol. 40; Springer International Publishing: Cham, Switzerland, 2015. doi:10.1007/978-3-319-07962-2
Return to citation in text: [1] -
Bakulev, V. A.; Beryozkina, T.; Thomas, J.; Dehaen, W. Eur. J. Org. Chem. 2018, 262–294. doi:10.1002/ejoc.201701031
Return to citation in text: [1] -
Belyaev, N. A.; Beryozkina, T. V.; Lubec, G.; Dehaen, W.; Bakulev, V. A. Chem. Heterocycl. Compd. 2018, 54, 1050–1055. doi:10.1007/s10593-018-2390-z
Return to citation in text: [1] -
Filimonov, V. O.; Dianova, L. N.; Beryozkina, T. V.; Mazur, D.; Beliaev, N. A.; Volkova, N. N.; Ilkin, V. G.; Dehaen, W.; Lebedev, A. T.; Bakulev, V. A. J. Org. Chem. 2019, 84, 13430–13446. doi:10.1021/acs.joc.9b01599
Return to citation in text: [1] -
Bakulev, V. A.; Shafran, Yu. M.; Beliaev, N. A.; Beryozkina, T. V.; Volkova, N. N.; Joy, M. N.; Fan, Z. Russ. Chem. Rev. 2022, 91, RCR5042. doi:10.1070/rcr5042
Return to citation in text: [1] -
Silaichev, P. S.; Beryozkina, T. V.; Ilkin, V.; Novikov, M. S.; Dehaen, W.; Bakulev, V. A. J. Org. Chem. 2023, 88, 8163–8174. doi:10.1021/acs.joc.3c00151
Return to citation in text: [1] [2] [3] -
Bhuyan, P. J.; Borah, H. N.; Sandhu, J. S. J. Chem. Soc., Perkin Trans. 1 1999, 3083–3084. doi:10.1039/a906222j
Return to citation in text: [1] -
Sako, M.; Ohara, S.; Hirota, K.; Kano, K.; Maki, Y.; Taylor, E. C. J. Org. Chem. 1991, 56, 6302–6306. doi:10.1021/jo00022a017
Return to citation in text: [1] -
Pagoti, S.; Surana, S.; Chauhan, A.; Parasar, B.; Dash, J. Catal. Sci. Technol. 2013, 3, 584–588. doi:10.1039/c2cy20776a
Return to citation in text: [1] -
Beliaev, N. A.; Shafikov, M. Z.; Efimov, I. V.; Beryozkina, T. V.; Lubec, G.; Dehaen, W.; Bakulev, V. A. New J. Chem. 2018, 42, 7049–7059. doi:10.1039/c7nj04243d
Return to citation in text: [1] -
Chen, X.; Jin, Q.; Wu, L.; Tung, C.; Tang, X. Angew. Chem., Int. Ed. 2014, 53, 12542–12547. doi:10.1002/anie.201408422
Return to citation in text: [1] -
Saito, T.; Ohkubo, T.; Kuboki, H.; Maeda, M.; Tsuda, K.; Karakasa, T.; Satsumabayashi, S. J. Chem. Soc., Perkin Trans. 1 1998, 3065–3080. doi:10.1039/a805574b
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1]
| 1. | Sauer, B.; Skinner-Adams, T. S.; Bouchut, A.; Chua, M. J.; Pierrot, C.; Erdmann, F.; Robaa, D.; Schmidt, M.; Khalife, J.; Andrews, K. T.; Sippl, W. Eur. J. Med. Chem. 2017, 127, 22–40. doi:10.1016/j.ejmech.2016.12.041 |
| 2. | Cindrić, M.; Sović, I.; Martin-Kleiner, I.; Kralj, M.; Mašek, T.; Hranjec, M.; Starčević, K. Med. Chem. Res. 2017, 26, 2024–2037. doi:10.1007/s00044-017-1912-z |
| 3. | Racané, L.; Cindrić, M.; Perin, N.; Roškarić, P.; Starčević, K.; Mašek, T.; Maurić, M.; Dogan, J.; Karminski-Zamola, G. Croat. Chem. Acta 2017, 90, 187–195. doi:10.5562/cca3146 |
| 4. | Beryozkina, T.; Bakulev, V.; Dianova, L.; Berseneva, V.; Slepukhin, P.; Leban, J.; Kalaba, P.; Aher, N. Y.; Ilic, M.; Sitte, H. H.; Lubec, G. Synthesis 2016, 48, 1046–1054. doi:10.1055/s-0035-1561350 |
| 13. | Bakulev, V. A.; Beryozkina, T.; Thomas, J.; Dehaen, W. Eur. J. Org. Chem. 2018, 262–294. doi:10.1002/ejoc.201701031 |
| 26. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 9. | Akter, M.; Rupa, K.; Anbarasan, P. Chem. Rev. 2022, 122, 13108–13205. doi:10.1021/acs.chemrev.1c00991 |
| 10. | Vala, D. P.; Vala, R. M.; Patel, H. M. ACS Omega 2022, 7, 36945–36987. doi:10.1021/acsomega.2c04883 |
| 11. | Dheer, D.; Singh, V.; Shankar, R. Bioorg. Chem. 2017, 71, 30–54. doi:10.1016/j.bioorg.2017.01.010 |
| 12. | Dehaen, W.; Bakulev, V. A., Eds. Chemistry of 1,2,3-triazoles; Topics in Heterocyclic Chemistry, Vol. 40; Springer International Publishing: Cham, Switzerland, 2015. doi:10.1007/978-3-319-07962-2 |
| 7. | Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j |
| 8. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4 |
| 24. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
| 5. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5 |
| 6. | Moses, J. E.; Moorhouse, A. D. Chem. Soc. Rev. 2007, 36, 1249–1262. doi:10.1039/b613014n |
| 25. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 17. | Silaichev, P. S.; Beryozkina, T. V.; Ilkin, V.; Novikov, M. S.; Dehaen, W.; Bakulev, V. A. J. Org. Chem. 2023, 88, 8163–8174. doi:10.1021/acs.joc.3c00151 |
| 18. | Bhuyan, P. J.; Borah, H. N.; Sandhu, J. S. J. Chem. Soc., Perkin Trans. 1 1999, 3083–3084. doi:10.1039/a906222j |
| 19. | Sako, M.; Ohara, S.; Hirota, K.; Kano, K.; Maki, Y.; Taylor, E. C. J. Org. Chem. 1991, 56, 6302–6306. doi:10.1021/jo00022a017 |
| 20. | Pagoti, S.; Surana, S.; Chauhan, A.; Parasar, B.; Dash, J. Catal. Sci. Technol. 2013, 3, 584–588. doi:10.1039/c2cy20776a |
| 21. | Beliaev, N. A.; Shafikov, M. Z.; Efimov, I. V.; Beryozkina, T. V.; Lubec, G.; Dehaen, W.; Bakulev, V. A. New J. Chem. 2018, 42, 7049–7059. doi:10.1039/c7nj04243d |
| 22. | Chen, X.; Jin, Q.; Wu, L.; Tung, C.; Tang, X. Angew. Chem., Int. Ed. 2014, 53, 12542–12547. doi:10.1002/anie.201408422 |
| 23. | Saito, T.; Ohkubo, T.; Kuboki, H.; Maeda, M.; Tsuda, K.; Karakasa, T.; Satsumabayashi, S. J. Chem. Soc., Perkin Trans. 1 1998, 3065–3080. doi:10.1039/a805574b |
| 16. | Bakulev, V. A.; Shafran, Yu. M.; Beliaev, N. A.; Beryozkina, T. V.; Volkova, N. N.; Joy, M. N.; Fan, Z. Russ. Chem. Rev. 2022, 91, RCR5042. doi:10.1070/rcr5042 |
| 17. | Silaichev, P. S.; Beryozkina, T. V.; Ilkin, V.; Novikov, M. S.; Dehaen, W.; Bakulev, V. A. J. Org. Chem. 2023, 88, 8163–8174. doi:10.1021/acs.joc.3c00151 |
| 15. | Filimonov, V. O.; Dianova, L. N.; Beryozkina, T. V.; Mazur, D.; Beliaev, N. A.; Volkova, N. N.; Ilkin, V. G.; Dehaen, W.; Lebedev, A. T.; Bakulev, V. A. J. Org. Chem. 2019, 84, 13430–13446. doi:10.1021/acs.joc.9b01599 |
| 14. | Belyaev, N. A.; Beryozkina, T. V.; Lubec, G.; Dehaen, W.; Bakulev, V. A. Chem. Heterocycl. Compd. 2018, 54, 1050–1055. doi:10.1007/s10593-018-2390-z |
| 17. | Silaichev, P. S.; Beryozkina, T. V.; Ilkin, V.; Novikov, M. S.; Dehaen, W.; Bakulev, V. A. J. Org. Chem. 2023, 88, 8163–8174. doi:10.1021/acs.joc.3c00151 |
© 2024 Silaichev et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.