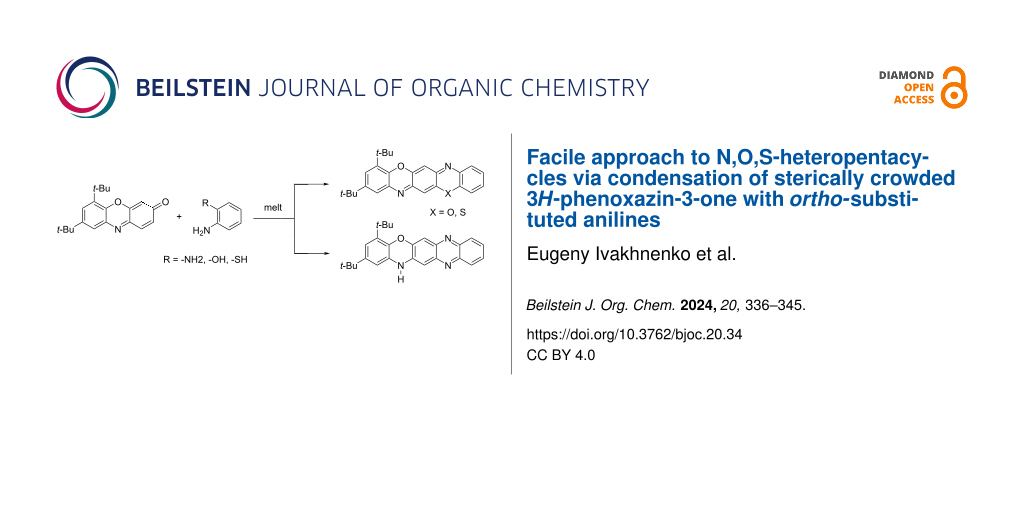
Abstract
A convenient method for the synthesis of a series of 2-(arylamino)-3H-phenoxazin-3-ones based on the nucleophilic substitution reaction between sterically crowded 3H-phenoxazin-3-one and arylamines performed by short-term heating of the melted reactants at 220–250 °C is described, and the compounds were characterized by means of single-crystal X-ray crystallography, NMR, UV–vis, and IR spectroscopy, as well as cyclic voltammetry. The reaction with o-amino-, o-hydroxy-, and o-mercapto-substituted arylamines widened the scope and provided an access to derivatives of N,O- and N,S-heteropentacyclic quinoxalinophenoxazine, triphenodioxazine and oxazinophenothiazine systems.

Graphical Abstract
Introduction
3H-Phenoxazin-3-one and its derivatives are widely distributed in nature in microorganisms and fungi, and they represent the key structural units of many important drugs with antibacterial, antifungal, anticancer, anti-inflammatory, and antiviral activities [1,2]. Due to the presence of several reactive centers in the structure, 3H-phenoxazin-3-ones can easily be accessed through oxidative couplings of o-aminophenols [3,4] or N-aryl-o-benzoquinone imines [5,6]. Further, they can serve as efficient precursors of pentacyclic N,O-heterocyclic compounds that possess promising properties for application in fluorescent probes, organic light-emitting diodes, and organic solar cells [2,7-9]. The principal way for the preparation of these heterocycles involves the coupling of 3H-phenoxazin-3-ones with variously functionalized aromatic amines. This is followed by the cyclization of the initially formed adducts [10-12]. At the first stage, this reaction follows one of three possible reaction pathways, including Schiff base formation (attack at the C(3) center), Michael addition at C(1), or nucleophilic substitution (SNH) at the C(2) center [13-15]. Most readily used is the pathway involving carbonyl–amine condensation and Schiff base formation, which is then cyclized [12,16]. The reaction of 1 with arylamines 2a is performed in toluene solution in the presence of a catalytic amount of p-toluenesulfonic acid. This readily affords 6,8-di-tert-butyl-N-aryl-3H-phenoxazin-3-imines 3 but proceeds smoothly only with highly basic amines (Scheme 1) [6]. The choice for one of the other two possible reaction pathways (nucleophilic additions to either the C(1) or C(2) center) critically depends on the electrophilicity. Figure 1 shows the distribution of electronic density in 6,8-di-tert-butyl-3H-phenoxazin-3-one (1). This is also the basic compound used in the transformations that are studied in this work due to the high kinetic stability and good solubility ensured by the tert-butyl groups. The largest positive charge of the C(1)–C(2)–C(3) segment is concentrated at the C(2) atom. The charge at the other electrophilic center C(1) of 1 is much lower.
![[1860-5397-20-34-i1]](/bjoc/content/inline/1860-5397-20-34-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 6,8-di-tert-butyl-N-aryl-3H-phenoxazin-3-imines 3 [6] and 6,8-di-tert-butyl-2-(arylamino)-3H-phenoxazin-3-ones 4.
Scheme 1: Synthesis of 6,8-di-tert-butyl-N-aryl-3H-phenoxazin-3-imines 3 [6] and 6,8-di-tert-butyl-2-(arylamino)...
![[1860-5397-20-34-1]](/bjoc/content/figures/1860-5397-20-34-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: DFT-calculated molecular geometry (B3LYP/6-311++G(d,p) level) and distribution of electronic density in 6,8-di-tert-butyl-3H-phenoxazin-3-one (1): Mulliken charges and molecular electrostatic potential (MEP, isovalue = 0.004).
Figure 1: DFT-calculated molecular geometry (B3LYP/6-311++G(d,p) level) and distribution of electronic densit...
It comes, therefore, with no surprise that the interaction of arylamines and the 5-hydroxy and 5-acetoxy derivatives of 3H-phenoxazin-3-one is directed toward the C(2) reaction center to yield 2-amino-3H-phenoxazin-3-ones as the final products under aerobic conditions. The reactions proceed readily in refluxing acidified (pKa = 1–5) ethanol solutions of the amine hydrochlorides to give 2-monosubstituted derivatives of 3H-phenoxazin-3-ones in a moderate yield [10,17]. In the present work, we intended to explore the reaction of 3H-phenoxazin-3-ones with aromatic amines, the direction of which is controlled by the large positive charge at the C(2) center of the p-quinone imine moiety of the heterocycle. With this in mind, we turned our attention to solid-state organic reactions. Numerous examples of nucleophilic substitutions at carbon centers are discussed in comprehensive reviews [18,19], but none is directly related to aromatic SNH reactions. The developed procedure was applied to the synthesis of compounds 4 and extended to arylamines with o-amino, o-hydroxy, and o-mercapto substituents, providing access to N-, O-, and S-containing heteropolycyclic structures.
Results and Discussion
We found that a convenient way toward 6,8-di-tert-butyl-2-(arylamino)-3H-phenoxazin-3-ones 4 involves the short-term heating (30 min) of a molten mixture of 1 and an arylamine at 250 °C, followed by purification of the products by column chromatography. No preliminary grinding of the crystalline samples, which is otherwise typical for solid-state reaction, was employed in this case. As seen in Scheme 2, the nucleophilic substitution reaction occured in good yield and with no restrictions in terms of amine basicity.
![[1860-5397-20-34-i2]](/bjoc/content/inline/1860-5397-20-34-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: 6,8-Di-tert-butyl-2-(arylamino)-3H-phenoxazin-3-ones 4 prepared by the one-pot reaction between 6,8-di-tert-butyl-3H-phenoxazin-3-one (1) and aromatic amines 2b (the yield is given in parentheses).
Scheme 2: 6,8-Di-tert-butyl-2-(arylamino)-3H-phenoxazin-3-ones 4 prepared by the one-pot reaction between 6,8...
The molecular structures of compounds 4c,d,f were determined by X-ray crystallography and are shown in Figure 2 (i.e., 4f) and Figures S1 and S2, Supporting Information File 1 (i.e., 4c,d). The geometry of the phenoxazine-3-one fragment of 4c,d,f coincides with that found for 6,8-di-tert-butyl-3H-phenoxazin-3-one (1) [6]. A strong hydrogen bridge, N(15)–H···O(31), is formed between the nitro and imino groups of the N-aryl ring.
![[1860-5397-20-34-2]](/bjoc/content/figures/1860-5397-20-34-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of 6,8-di-tert-butyl-2-(o-nitrophenylamino)-3H-phenoxazin-3-one (4f). a) Selected bond distances (Å) and angles: N(1)–C(11) 1.3061(19), N(1)–C(14) 1.3836(18), O(23)–C(4) 1.2314(19), N(15)–C(3) 1.3765(19), N(15)–C(16) 1.383(2), C(11)–N(1)–C(14) 117.55(12), C(3)–N(15)–C(16) 131.12(14). b) Crystal packing of 4f. Important crystallographic parameters and bond distances are given in Tables S2 and S5, Supporting Information File 1. Thermal ellipsoids are drawn at the 50% probability level.
Figure 2: Molecular structure of 6,8-di-tert-butyl-2-(o-nitrophenylamino)-3H-phenoxazin-3-one (4f). a) Select...
The compounds 4a–h intensely absorb light in the spectral range of 400–550 nm, with maxima at 439–459 nm, ε = 20600–37100 M−1⋅cm−1 (Figure 3 and Table 1). The introduction of an amino group into the p-position of the N-phenyl fragment gave rise to the appearance of an additional long-wavelength absorption band with λmax = 520 nm and ε = 9200 M−1⋅сm−1.
![[1860-5397-20-34-3]](/bjoc/content/figures/1860-5397-20-34-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis spectra of 6,8-di-tert-butyl-2-(arylamino)-3H-phenoxazin-3-ones 4a–h (toluene, c = 2⋅10−5 M, l = 1 cm, T = 293 K).
Figure 3: UV–vis spectra of 6,8-di-tert-butyl-2-(arylamino)-3H-phenoxazin-3-ones 4a–h (toluene, c = 2⋅10−5 M, ...
Table 1: UV–vis absorption data of 6,8-di-tert-butyl-2-(arylamino)-3H-phenoxazin-3-ones 4a–h in toluene.
| compound | λmax, nm (ε, 104 M−1·cm−1) |
| 4a | 445 (2.75)a, 458 (2.80) |
| 4b | 447 (3.04) |
| 4c | 444 (2.83)a, 459 (2.88) |
| 4d | 449 (2.99) |
| 4e | 357 (1.26), 457 (3.19) |
| 4f | 459 (3.71) |
| 4g | 439 (2.07), 459 (2.06), 520 (0.92) |
| 4h | 309 (2.35), 454 (3.20) |
aShoulder.
Subjecting o-phenylenediamines 2с to the reaction with 3H-phenoxazin-3-one makes the simultaneous activation of two principle reaction pathways (SNH and Schiff base formation) possible. By using a similar procedure to that applied to the synthesis of compounds 4, we succeeded in the preparation of 14Н-quinoxaline[2,3-b]phenoxazine derivatives 5 (Scheme 3).
![[1860-5397-20-34-i3]](/bjoc/content/inline/1860-5397-20-34-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 14H-quinoxaline[2,3-b]phenoxazines 5 and 6.
Scheme 3: Synthesis of 14H-quinoxaline[2,3-b]phenoxazines 5 and 6.
The nitrogen atoms in the oxazine and pyrazine rings of 5, N(7), N(12), and N(14), offer three possible positions for the N–H proton. Therefore, three tautomeric forms are possible for 5 (Scheme 4), one of which, the 7H-tautomer 7b, inevitably adopts a bipolar or biradical structure. According to the data from the DFT calculations performed at the B3LYP/6-311++G(d,p) approximation (Figure S6, Supporting Information File 1), the energetically preferred tautomer is the 14H-form 7a. The least stable 7H-isomer 7b conforms to a minimum on the corresponding potential energy surface. However, the stable wave function of 7b corresponds to an electronic state with a broken symmetry [20], indicating the presence of two unpaired electrons and the singlet biradical form.
![[1860-5397-20-34-i4]](/bjoc/content/inline/1860-5397-20-34-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Relative stability of the tautomers 7 and 7a,b of quinoxaline[2,3-b]phenoxazine calculated at the DFT B3LYP/6-311++G(d,p) level.
Scheme 4: Relative stability of the tautomers 7 and 7a,b of quinoxaline[2,3-b]phenoxazine calculated at the D...
In previous studies on the coupling of 3H-phenoxazin-3-one derivatives 8 and 9 with o-phenylenediamine [10,11], the preference was given to the 12Н-quinoxaline[2,3-b]phenoxazine form 7 (Scheme 5). A series of N-aryl derivatives of this form was also obtained via treatment of 6,8-di-tert-butyl-N-aryl-3H-phenoxazin-3-imines with various arylamines in the presence of an excess of trifluoroacetic acid [9].
![[1860-5397-20-34-i5]](/bjoc/content/inline/1860-5397-20-34-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparation of quinoxaline[2,3-b]phenoxazine (7) from 2-amino-3H-phenoxazin-3-one (8) [10] and 2-ethoxy-3H-phenoxazin-3-one (9) [11], respectively.
Scheme 5: Preparation of quinoxaline[2,3-b]phenoxazine (7) from 2-amino-3H-phenoxazin-3-one (8) [10] and 2-ethoxy...
The structure of the newly synthesized compounds 5, which are derivatives of a previously unknown 14Н-quinoxaline[2,3-b]phenoxazine system 7a, was unambiguously established based on COSY, HSQC, and HMBC NMR-spectroscopic data. Further, the 15N NMR spectrum of 5a confirmed the typical pyrrole-like character of the N(12) atom as well as the pyridine-like character of the N(7) and N(14) atoms (Figure S31, Supporting Information File 1) [21,22]. The molecular structure of 5c was also determined using X-ray crystallography (Figure 4).
![[1860-5397-20-34-4]](/bjoc/content/figures/1860-5397-20-34-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Molecular structure of ethyl 2,4-di-tert-butyl-14H-quinoxalino[2,3-b]phenoxazine-10-carboxylate (5c), with atom numbering scheme. Selected bond distances (Å) and angles: N(1)–C(5) 1.390(2), N(1)–C(6) 1.365(3), N(2)–C(14) 1.363(3), N(2)–C(15) 1.336(3), N(3)–C(8) 1.338(3), N(3)–C(9) 1.356(3), C(6)–N(1)–C(5) 122.13(17), C(15)–N(2)–C(14) 116.88(17), C(8)–N(3)–C(9) 116.66(17). All bond lengths, angles, and important crystallographic parameters are given in Tables S6 and S7, Supporting Information File 1. Hydrogen atoms are omitted for clarity.
Figure 4: Molecular structure of ethyl 2,4-di-tert-butyl-14H-quinoxalino[2,3-b]phenoxazine-10-carboxylate (5c...
We assumed that the scope of the reaction shown in Scheme 3 could be expanded via replacement of one of the amino groups of o-phenylenediamine by another strong nucleophilic center. It was earlier found [23] that condensation of 3H-phenoxazin-3-one (1) with various o-aminophenols (in refluxing DMF for 8–10 h), upon formation of the corresponding imine intermediate, affords benzo[5,6][1,4]oxazino[2,3-b]phenoxazines derivatives 10a,b (triphenodioxazines). As shown in the present work, this reaction can also be performed successfully under the conditions applied to the preparation of 14H-quinoxaline[2,3-b]phenoxazines 5. The reaction proceeds readily with o-mercaptoaniline to produce the benzo[5,6][1,4]oxazino[2,3-b]phenothiazine derivative 10c (Scheme 6).
![[1860-5397-20-34-i6]](/bjoc/content/inline/1860-5397-20-34-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Triphenodioxazine and oxazinophenothiazine derivatives 10 via condensation of 3H-phenoxazin-3-one 1 with o-aminophenol and o-mercaptoaniline derivatives 2d.
Scheme 6: Triphenodioxazine and oxazinophenothiazine derivatives 10 via condensation of 3H-phenoxazin-3-one 1...
Electronic absorption spectra of the prepared 14Н-quinoxaline[2,3-b]phenoxazines 5 and 6 (Table 2, Figures 5–7, and Figures S7–S11, Supporting Information File 1) exhibit broad and high-intensity absorption maxima in the range of 450–550 nm, which encompass the strongest emissive part of the solar spectrum. In contrast to nonfluorescent 6,8-di-tert-butyl-2-(arylamino)-3H-phenoxazin-3-ones 4, compounds 5 and 6 display intense fluorescence in solution at room temperature (Figure 5). The excitation spectra of the compounds (Figures S7–S11, Supporting Information File 1) correspond to the longest-wavelength absorption bands. The absorption and emission spectra of benzo[5,6][1,4]oxazino[2,3-b]phenothiazine 10c (Figure 7) were bathochromically shifted by about 50 nm relative to those of the quinoxaline[2,3-b]phenoxazines 6.
Table 2: UV–vis absorption and fluorescence emission data of compounds 5a–c, 6a,b, and 10c in toluene.
| compound | absorption λmax, nm (ε, 104 M−1⋅cm−1) | emission λfl, nm | Φfla |
| 5a | 311 (1.13), 327b (0.83), 380b (0.46), 400 (0.59), 466 (2.36), 486b (2.18) | 526, 550b | 0.19 |
| 5b | 307 (1.07), 326b (0.72), 379b (0.45), 401b (0.60), 462 (2.23), 482 (2.12) | 519, 542b | 0.20 |
| 5c | 325 (1.32), 339 (1.22), 389b (0.59), 408 (1.47), 484 (2.52), 505b (2.32) | 550, 573b | 0.13 |
| 6a | 310 (1.04), 326b (0.78), 379b (0.41), 399 (0.49), 471 (2.29), 486b (2.10) | 531, 555b | 0.19 |
| 6b | 314 (1.11), 327b (0.93), 380b (0.46), 400 (0.55), 473 (2.64), 497 (2.50) | 531, 555b | 0.19 |
| 10c | 474b (2.13), 502 (4.61), 538 (6.56) | 556, 590b | 0.35 |
aFluorescence quantum yield. bShoulder.
![[1860-5397-20-34-5]](/bjoc/content/figures/1860-5397-20-34-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: a) UV–vis (solid lines) and fluorescence emission (λex = 365 nm, dashed) spectra of compounds 5a–c (toluene, c = 2 10−5 M, l = 1 cm). b) Solutions of compounds 5a–c in toluene before irradiation (no emission) and c) during irradiation (photoluminescence, λex = 365 nm) at room temperature.
Figure 5: a) UV–vis (solid lines) and fluorescence emission (λex = 365 nm, dashed) spectra of compounds 5a–c ...
![[1860-5397-20-34-6]](/bjoc/content/figures/1860-5397-20-34-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: UV–vis (solid lines) and fluorescence emission (dashed, λex = 365 nm) spectra of compounds 6a,b in toluene (c = 2 10−5 M, l = 1 cm) at room temperature.
Figure 6: UV–vis (solid lines) and fluorescence emission (dashed, λex = 365 nm) spectra of compounds 6a,b in ...
![[1860-5397-20-34-7]](/bjoc/content/figures/1860-5397-20-34-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: UV–vis, fluorescence emission (λex = 500 nm), and fluorescence excitation (λobs = 590 nm) spectra of benzo[5,6][1,4]oxazino[2,3-b]phenothiazine 10c in toluene (c = 2 10−5 M (UV–vis) or c = 2 10−6 M (fluorescence), l = 1 cm) at room temperature.
Figure 7: UV–vis, fluorescence emission (λex = 500 nm), and fluorescence excitation (λobs = 590 nm) spectra o...
The electrochemical behavior of compounds 4a–h, 5a–c, 6a,b, and 10c was studied using cyclic voltammetry (CV). As exemplified by the CV curves (Figure S12, Supporting Information File 1), 2-(arylamino)-3H-phenoxazin-3-ones 4a–h manifest two reduction waves at Е1/2RED1 = −1.36 ± 1.69 V and Е1/2RED2 = −1.85 ± 2.12 V. Oxidation of 4a–f,h occurs as an irreversible process at Е1/2OX = 0.81–1.07 V. For 4g bearing an amino group, the oxidation potential is shifted to Е1/2OX = 0.25 V. The irreversible two-wave reduction (Е1/2RED1 = −1.40 ± 1.60 V and Е1/2RED2 = −1.92 ± 2.45 V) is also characteristic of 14H-quinoxaline[2,3-b]phenoxazines 5a–c and 6a,b. In contrast to 5a–c and 6a,b, benzo[5,6][1,4]oxazino[2,3-b]phenothiazine 10c is reversibly reduced at Е1/2RED1 = −1.39 V to a radical anion and then undergoes irreversible reduction at Е1/2RED2 = −1.91 V and irreversible oxidation at Е1/2OX = 0.48 V. These CV parameters are close to those recorded for triphenodioxazines [23]. The energy of the HOMO and LUMO orbitals assessed on the basis of the CV and electronic absorption spectral data are given in Table 3.
Table 3: CV parameters and calculated energy levels of 4a–h, 5a–c, 6a,b, and 10c.
| compound | CV (vs Fc+/Fc) | UV–vis | ||||||
| E1/2OX, V | E1/2RED1, V | E1/2RED2, V | HOMO, eV | LUMO, eV | ∆E, eV | ∆E, eV | LUMO, eV | |
| 4a | 0.89 | −1.41 | −1.97 | −5.69 | −3.39 | 2.30 | 2.71 | −2.98 |
| 4b | 0.91 | −1.36 | −1.90 | −5.71 | −3.44 | 2.27 | 2.77 | −2.94 |
| 4c | 0.83 | −1.42 | −1.92 | −5.63 | −3.38 | 2.25 | 2.70 | −2.93 |
| 4d | 0.81 | −1.36 | −1.94 | −5.61 | −3.44 | 2.17 | 2.76 | −2.85 |
| 4e | 1.07 | −1.59 | −1.91 | −5.87 | −3.21 | 2.66 | 2.71 | −3.16 |
| 4f | 1.05 | −1.69 | −2.12 | −5.85 | −3.11 | 2.74 | 2.70 | −3.15 |
| 4g | 0.25 | −1.54 | −1.96 | −5.05 | −3.26 | 1.79 | 2.38 | −2.67 |
| 4h | 0.92 | −1.43 | −1.85 | −5.72 | −3.37 | 2.35 | 2.73 | −2.99 |
| 5a | 0.86 | −1.40 | −2.32 | −5.66 | −3.40 | 2.26 | 2.55 | −3.11 |
| 5b | 0.95 | −1.60 | −2.45 | −5.75 | −3.20 | 2.55 | 2.57 | −3.18 |
| 5c | 0.82 | −1.40 | −2.15 | −5.62 | −3.40 | 2.22 | 2.46 | −3.16 |
| 6a | 1.10 | −1.45 | −1.92 | −5.9 | −3.35 | 2.55 | 2.55 | −3.35 |
| 6b | 1.15 | −1.48 | −2.02 | −.95 | −3.32 | 2.63 | 2.49 | −3.46 |
| 10c | 0.48 | −1.39 | −1.91 | −.28 | −3.41 | 1.87 | 2.30 | −2.98 |
Conclusion
The diverse reactions of 3H-phenoxazin-3-one derivatives with nucleophilic reagents are primarily directed toward the p-quinone imine fragment [5,13,15]. In the presence of protonic acids, the reaction with amines proceeds through Schiff base formation [6]. In turn, without an acidic catalyst, it is driven by the distribution of the electron density (Figure 1), such that the nucleophilic attack occurs at the most electrophilic C(2) center. With this in mind, we presented a convenient procedure for the SNH reaction of aromatic amines with sterically crowded 6,8-di-tert-butyl-3H-phenoxazin-3-one (1) that afforded a series of 6,8-di-tert-butyl-2-(arylamino)-3H-phenoxazin-3-ones 4 prepared in 68–93% yield (Scheme 2). Using o-amino-, o-hydroxy- and o-mercapto-substituted anilines in this process allowed to pursue both principal reaction pathways (Schiff base formation and SNH), which led to the formation of derivatives of the previously unknown 14Н-quinoxaline[2,3-b]phenoxazine system 5 (Scheme 3) as well as N,O- and N,S-heteropentacyclic triphenodioxazines and oxazinophenothiazine 10a–c. The structural assignment [10,11] of the N,O-containing reaction products as 12H-quinoxaline[2,3-b]phenoxazines was confirmed through DFT calculations, X-ray crystallography, and NMR spectroscopy.
Electronic absorption spectra (Table 2 and Figures 5–7) and electrochemical properties (Table 3) of the heteropentacyclic compounds 4a–h, 5a–c, 6a,b, and 10c revealed potential for testing as potential donors for organic solar cells or as dye sensitizers for dye-sensitized solar cells [24,25].
Experimental
All reagents and solvents were purchased from commercial sources (Aldrich) and used without additional purification. The compounds were characterized by 1H, 13C, and 15N NMR spectroscopy (NMR spectra of compounds 4a–h, 5a–c, 6a,b, and 10c are given in Figures S13–S43, Supporting Information File 1), mass spectrometry (Figures S44–S56, Supporting Information File 1), IR and UV–vis spectroscopy, as well as elemental analysis. The NMR spectra were recorded on the spectrometers Varian UNITY-300 (300 MHz for 1H) and Bruker AVANCE-600 (600 MHz for 1H, 151 MHz for 13C, and 60 MHz for 15N) in CDCl3 solutions. Chemical shifts are reported in ppm using the residual solvent peaks as reference (7.24 ppm for 1H, 77.0 ppm for 13C, and 384 ppm for 15N using nitromethane). Chemical shifts were measured with a precision of 0.01 ppm, and 0.1 Hz for spin–spin coupling constants J. The assignment of resonance peaks was carried out using COSY, HSQC, and 1H,13C as well as 1H,15N HMBC. Melting points were determined using a PTP (M) apparatus and were left uncorrected. IR spectra were recorded on a Varian Excalibur 3100 FTIR instrument using the attenuated total internal reflection technique (ZnSe crystal). UV–vis spectra were recorded at c = 2⋅10−5 M in toluene solutions with a Varian Cary 100 spectrophotometer. Photoluminescent spectra were recorded at c = 2⋅10−5 M (compounds 5a–c and 6a,b) and c = 2⋅10−6 M (compound 10c) in toluene solutions with a Varian Cary Eclipse fluorescence spectrophotometer. UV–vis and fluorescence spectra were recorded using standard 1 cm quartz cells at room temperature. Toluene of spectroscopic grade (Aldrich) was used to prepare the solutions. The fluorescence quantum yield was determined relative to quinine bisulfate in 0.05 M H2SO4 as standard (ΦF = 0.52, excitation at 365 nm for 5a–c and 6a,b) [26] and cresyl violet perchlorate in ethanol (ΦF = 0.54, excitation at 510 nm for 10c) [27]. Mass spectrometric analysis was performed on a Bruker UHR-TOF Maxis™ Impact (resolving power (FWHM) of 40,000 at m/z 1222, electrospray ionization). The cyclic voltammograms of 4a–h, 5a–c, 6a,b, and 10c were measured with the use of three-electrode configuration (glassy carbon working electrode, Pt counter electrode, Ag/Ag+ reference electrode using 0.01 M AgNO3 in CH3CN) in CH2Cl2 (4a–h), CH3CN (5a–c, 6a,b, and 10c) and potentiostat–galvanostat Elins P-45X. X-ray data collection was performed on an Agilent SuperNova diffractometer using a microfocus X-ray source with copper anode (Cu Kα radiation, λ = 1.54184 Å) and Atlas S2 CCD detector. The diffraction data of 4c,d,f, 5c, and 10b were obtained at 100 K. Crystals of 5c were obtained in the form of a solvate with molecules of isopropanol and water present. The protons attached to heteroatoms were localized by difference Fourier synthesis and refined with isotropic thermal parameters. The collection of reflexes as well as the determination and refinement of unit cell parameters were performed by using the specialized CrysAlisPro 1.171.38.41 software suite [28]. The structures were solved by using the SHELXT program [29]. Structural refinement was performed with the SHELXL program [30]. Molecular graphics were rendered and prepared for publication with the Olex2 version 1.3.0 software suite [31]. The complete X-ray diffraction datasets were deposited in the Cambridge Crystallographic Data Center (CCDC numbers 2292841, 2292840, 2292847, 2308520, and 2292848, Tables S2–S9, Supporting Information File 1). The DFT calculations [32] were performed using the Gaussian 16 program package [33] with the B3LYP functional [34] and the 6-311++G(d,p) basis set. The structures corresponding to minima on the potential energy surface and states with broken symmetry [20] were found through complete optimization of the geometry without imposing symmetry restrictions, followed by analyzing the stability of the DFT wave function. The images of the molecular structures in Figure 1 and Figure S6, Supporting Information File 1, were obtained using the Chemcraft program [35].
Supporting Information
| Supporting Information File 1: Synthetic details, compound characterization and additional analytic data, including copies of spectra and Cartesian coordinates. | ||
| Format: PDF | Size: 5.5 MB | Download |
Funding
This work was financially supported by the Russian Science Foundation (Project No. 19-13-00022, https://rscf.ru/project/19-13-00022/).
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Zorrilla, J. G.; Rial, C.; Cabrera, D.; Molinillo, J. M. G.; Varela, R. M.; Macías, F. A. Molecules 2021, 26, No. 3453. doi:10.3390/molecules26113453
Return to citation in text: [1] -
Sadhu, C.; Mitra, A. K. Mol. Diversity 2023, 10619. doi:10.1007/s11030-023-10619-5
Return to citation in text: [1] [2] -
Diepolder, E. Ber. Dtsch. Chem. Ges. 1902, 35, 2816–2822. doi:10.1002/cber.19020350360
Return to citation in text: [1] -
Podder, N.; Mandal, S. New J. Chem. 2020, 44, 12793–12805. doi:10.1039/d0nj02558e
Return to citation in text: [1] -
Abakumov, G. A.; Druzhkov, N. O.; Kurskii, Y. A.; Abakumova, L. G.; Shavyrin, A. S.; Fukin, G. K.; Poddel’skii, A. I.; Cherkasov, V. K.; Okhlopkova, L. S. Russ. Chem. Bull. 2005, 54, 2571–2577. doi:10.1007/s11172-006-0157-7
Return to citation in text: [1] [2] -
Ivakhnenko, E. P.; Knyazev, P. A.; Kovalenko, A. A.; Romanenko, G. V.; Revinskii, Y. V.; Starikov, A. G.; Minkin, V. I. Tetrahedron Lett. 2020, 61, No. 151429. doi:10.1016/j.tetlet.2019.151429
Return to citation in text: [1] [2] [3] [4] [5] -
Kulszewicz-Bajer, I.; Guzauskas, M.; Makowska-Janusik, M.; Zagórska, M.; Mahmoudi, M.; Grazulevicius, J. V.; Proń, A.; Volyniuk, D. J. Mater. Chem. C 2022, 10, 12377–12391. doi:10.1039/d2tc02270b
Return to citation in text: [1] -
Lv, L.; Luo, W.; Diao, Q. Spectrochim. Acta, Part A 2021, 246, No. 118959. doi:10.1016/j.saa.2020.118959
Return to citation in text: [1] -
Ivakhnenko, E. P.; Knyazev, P. A.; Omelichkin, N. I.; Makarova, N. I.; Starikov, A. G.; Aleksandrov, A. E.; Ezhov, A. V.; Tameev, A. R.; Demidov, O. P.; Minkin, V. I. Dyes Pigm. 2022, 197, No. 109848. doi:10.1016/j.dyepig.2021.109848
Return to citation in text: [1] [2] -
Martinek, M.; Kotouček, M.; Ružička, E. Monatsh. Chem. 1967, 98, 1532–1536. doi:10.1007/bf00909022
Return to citation in text: [1] [2] [3] [4] [5] -
Afanas'eva, G. B.; Postovskii, I. Y.; Viktorova, T. S. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1978, 14, 966–968. doi:10.1007/bf00509550
Return to citation in text: [1] [2] [3] [4] -
Ivakhnenko, E. P.; Romanenko, G. V.; Kovalenko, A. A.; Revinskii, Y. V.; Knyazev, P. A.; Kuzmin, V. A.; Minkin, V. I. Dyes Pigm. 2018, 150, 97–104. doi:10.1016/j.dyepig.2017.11.009
Return to citation in text: [1] [2] -
Yang, J.; Cohen Stuart, M. A.; Kamperman, M. Chem. Soc. Rev. 2014, 43, 8271–8298. doi:10.1039/c4cs00185k
Return to citation in text: [1] [2] -
Vasu, D.; Leitch, J. A.; Dixon, D. J. Tetrahedron 2019, 75, No. 130726. doi:10.1016/j.tet.2019.130726
Return to citation in text: [1] -
Kutyrev, A. A. Tetrahedron 1991, 47, 8043–8065. doi:10.1016/s0040-4020(01)91002-6
Return to citation in text: [1] [2] -
Ivakhnenko, E.; Malay, V.; Demidov, O.; Knyazev, P.; Makarova, N.; Minkin, V. Org. Biomol. Chem. 2023, 21, 621–631. doi:10.1039/d2ob02165j
Return to citation in text: [1] -
Viktorova, T. S.; Afanas'eva, G. B.; Postovskii, I. Y.; Ivanova, L. V. Chem. Heterocycl. Compd. 1974, 10, 1038–1041. doi:10.1007/bf00472117
Return to citation in text: [1] -
Kaupp, G. Organic Solid-State Reactions. In Encyclopedia of Physical Organic Chemistry; Wang, Z., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, U.S.A., 2016. doi:10.1002/9781118468586.epoc2005
Return to citation in text: [1] -
Kaupp, G. Organic Solid-State Reactions with 100% Yield. In Organic Solid State Reactions; Toda, F., Ed.; Topics in Current Chemistry, Vol. 254; Springer: Berlin, Germany, 2005; pp 95–183. doi:10.1007/b100997
Return to citation in text: [1] -
Noodleman, L. J. Chem. Phys. 1981, 74, 5737–5743. doi:10.1063/1.440939
Return to citation in text: [1] [2] -
Świderski, G.; Wojtulewski, S.; Kalinowska, M.; Świsłocka, R.; Lewandowski, W. J. Mol. Struct. 2011, 993, 448–458. doi:10.1016/j.molstruc.2011.01.026
Return to citation in text: [1] -
Pearse, G. A.; Raithby, P. R.; Lewis, J. Polyhedron 1989, 8, 301–304. doi:10.1016/s0277-5387(00)80418-0
Return to citation in text: [1] -
Ivakhnenko, E. P.; Romanenko, G. V.; Makarova, N. I.; Kovalenko, A. A.; Knyazev, P. A.; Rostovtseva, I. A.; Starikov, A. G.; Minkin, V. I. Dyes Pigm. 2020, 176, No. 108174. doi:10.1016/j.dyepig.2019.108174
Return to citation in text: [1] [2] -
Yahya, M.; Bouziani, A.; Ocak, C.; Seferoğlu, Z.; Sillanpää, M. Dyes Pigm. 2021, 192, No. 109227. doi:10.1016/j.dyepig.2021.109227
Return to citation in text: [1] -
Li, Y.; Huang, W.; Zhao, D.; Wang, L.; Jiao, Z.; Huang, Q.; Wang, P.; Sun, M.; Yuan, G. Molecules 2022, 27, No. 1800. doi:10.3390/molecules27061800
Return to citation in text: [1] -
Meech, S. R.; Phillips, D. J. Photochem. 1983, 23, 193–217. doi:10.1016/0047-2670(83)80061-6
Return to citation in text: [1] -
Magde, D.; Brannon, J. H.; Cremers, T. L.; Olmsted, J. J. Phys. Chem. 1979, 83, 696–699. doi:10.1021/j100469a012
Return to citation in text: [1] -
CrysAlisPro 1.171.38.41. Rigaku, 2015; https://www.rigaku.com/en/products/smc/chrysalis (accessed Nov 24, 2023).
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1] -
Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, No. A1133. doi:10.1103/physrev.140.a1133
Return to citation in text: [1] -
Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, 2016.
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Chemcraft 1.7. 2013; http://www.chemcraftprog.com (accessed Nov 24, 2023).
Return to citation in text: [1]
| 29. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930 |
| 30. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 31. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 1. | Zorrilla, J. G.; Rial, C.; Cabrera, D.; Molinillo, J. M. G.; Varela, R. M.; Macías, F. A. Molecules 2021, 26, No. 3453. doi:10.3390/molecules26113453 |
| 2. | Sadhu, C.; Mitra, A. K. Mol. Diversity 2023, 10619. doi:10.1007/s11030-023-10619-5 |
| 10. | Martinek, M.; Kotouček, M.; Ružička, E. Monatsh. Chem. 1967, 98, 1532–1536. doi:10.1007/bf00909022 |
| 11. | Afanas'eva, G. B.; Postovskii, I. Y.; Viktorova, T. S. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1978, 14, 966–968. doi:10.1007/bf00509550 |
| 12. | Ivakhnenko, E. P.; Romanenko, G. V.; Kovalenko, A. A.; Revinskii, Y. V.; Knyazev, P. A.; Kuzmin, V. A.; Minkin, V. I. Dyes Pigm. 2018, 150, 97–104. doi:10.1016/j.dyepig.2017.11.009 |
| 9. | Ivakhnenko, E. P.; Knyazev, P. A.; Omelichkin, N. I.; Makarova, N. I.; Starikov, A. G.; Aleksandrov, A. E.; Ezhov, A. V.; Tameev, A. R.; Demidov, O. P.; Minkin, V. I. Dyes Pigm. 2022, 197, No. 109848. doi:10.1016/j.dyepig.2021.109848 |
| 2. | Sadhu, C.; Mitra, A. K. Mol. Diversity 2023, 10619. doi:10.1007/s11030-023-10619-5 |
| 7. | Kulszewicz-Bajer, I.; Guzauskas, M.; Makowska-Janusik, M.; Zagórska, M.; Mahmoudi, M.; Grazulevicius, J. V.; Proń, A.; Volyniuk, D. J. Mater. Chem. C 2022, 10, 12377–12391. doi:10.1039/d2tc02270b |
| 8. | Lv, L.; Luo, W.; Diao, Q. Spectrochim. Acta, Part A 2021, 246, No. 118959. doi:10.1016/j.saa.2020.118959 |
| 9. | Ivakhnenko, E. P.; Knyazev, P. A.; Omelichkin, N. I.; Makarova, N. I.; Starikov, A. G.; Aleksandrov, A. E.; Ezhov, A. V.; Tameev, A. R.; Demidov, O. P.; Minkin, V. I. Dyes Pigm. 2022, 197, No. 109848. doi:10.1016/j.dyepig.2021.109848 |
| 10. | Martinek, M.; Kotouček, M.; Ružička, E. Monatsh. Chem. 1967, 98, 1532–1536. doi:10.1007/bf00909022 |
| 5. | Abakumov, G. A.; Druzhkov, N. O.; Kurskii, Y. A.; Abakumova, L. G.; Shavyrin, A. S.; Fukin, G. K.; Poddel’skii, A. I.; Cherkasov, V. K.; Okhlopkova, L. S. Russ. Chem. Bull. 2005, 54, 2571–2577. doi:10.1007/s11172-006-0157-7 |
| 6. | Ivakhnenko, E. P.; Knyazev, P. A.; Kovalenko, A. A.; Romanenko, G. V.; Revinskii, Y. V.; Starikov, A. G.; Minkin, V. I. Tetrahedron Lett. 2020, 61, No. 151429. doi:10.1016/j.tetlet.2019.151429 |
| 3. | Diepolder, E. Ber. Dtsch. Chem. Ges. 1902, 35, 2816–2822. doi:10.1002/cber.19020350360 |
| 4. | Podder, N.; Mandal, S. New J. Chem. 2020, 44, 12793–12805. doi:10.1039/d0nj02558e |
| 10. | Martinek, M.; Kotouček, M.; Ružička, E. Monatsh. Chem. 1967, 98, 1532–1536. doi:10.1007/bf00909022 |
| 11. | Afanas'eva, G. B.; Postovskii, I. Y.; Viktorova, T. S. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1978, 14, 966–968. doi:10.1007/bf00509550 |
| 6. | Ivakhnenko, E. P.; Knyazev, P. A.; Kovalenko, A. A.; Romanenko, G. V.; Revinskii, Y. V.; Starikov, A. G.; Minkin, V. I. Tetrahedron Lett. 2020, 61, No. 151429. doi:10.1016/j.tetlet.2019.151429 |
| 18. | Kaupp, G. Organic Solid-State Reactions. In Encyclopedia of Physical Organic Chemistry; Wang, Z., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, U.S.A., 2016. doi:10.1002/9781118468586.epoc2005 |
| 19. | Kaupp, G. Organic Solid-State Reactions with 100% Yield. In Organic Solid State Reactions; Toda, F., Ed.; Topics in Current Chemistry, Vol. 254; Springer: Berlin, Germany, 2005; pp 95–183. doi:10.1007/b100997 |
| 6. | Ivakhnenko, E. P.; Knyazev, P. A.; Kovalenko, A. A.; Romanenko, G. V.; Revinskii, Y. V.; Starikov, A. G.; Minkin, V. I. Tetrahedron Lett. 2020, 61, No. 151429. doi:10.1016/j.tetlet.2019.151429 |
| 6. | Ivakhnenko, E. P.; Knyazev, P. A.; Kovalenko, A. A.; Romanenko, G. V.; Revinskii, Y. V.; Starikov, A. G.; Minkin, V. I. Tetrahedron Lett. 2020, 61, No. 151429. doi:10.1016/j.tetlet.2019.151429 |
| 12. | Ivakhnenko, E. P.; Romanenko, G. V.; Kovalenko, A. A.; Revinskii, Y. V.; Knyazev, P. A.; Kuzmin, V. A.; Minkin, V. I. Dyes Pigm. 2018, 150, 97–104. doi:10.1016/j.dyepig.2017.11.009 |
| 16. | Ivakhnenko, E.; Malay, V.; Demidov, O.; Knyazev, P.; Makarova, N.; Minkin, V. Org. Biomol. Chem. 2023, 21, 621–631. doi:10.1039/d2ob02165j |
| 32. | Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, No. A1133. doi:10.1103/physrev.140.a1133 |
| 13. | Yang, J.; Cohen Stuart, M. A.; Kamperman, M. Chem. Soc. Rev. 2014, 43, 8271–8298. doi:10.1039/c4cs00185k |
| 14. | Vasu, D.; Leitch, J. A.; Dixon, D. J. Tetrahedron 2019, 75, No. 130726. doi:10.1016/j.tet.2019.130726 |
| 15. | Kutyrev, A. A. Tetrahedron 1991, 47, 8043–8065. doi:10.1016/s0040-4020(01)91002-6 |
| 10. | Martinek, M.; Kotouček, M.; Ružička, E. Monatsh. Chem. 1967, 98, 1532–1536. doi:10.1007/bf00909022 |
| 17. | Viktorova, T. S.; Afanas'eva, G. B.; Postovskii, I. Y.; Ivanova, L. V. Chem. Heterocycl. Compd. 1974, 10, 1038–1041. doi:10.1007/bf00472117 |
| 23. | Ivakhnenko, E. P.; Romanenko, G. V.; Makarova, N. I.; Kovalenko, A. A.; Knyazev, P. A.; Rostovtseva, I. A.; Starikov, A. G.; Minkin, V. I. Dyes Pigm. 2020, 176, No. 108174. doi:10.1016/j.dyepig.2019.108174 |
| 11. | Afanas'eva, G. B.; Postovskii, I. Y.; Viktorova, T. S. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1978, 14, 966–968. doi:10.1007/bf00509550 |
| 21. | Świderski, G.; Wojtulewski, S.; Kalinowska, M.; Świsłocka, R.; Lewandowski, W. J. Mol. Struct. 2011, 993, 448–458. doi:10.1016/j.molstruc.2011.01.026 |
| 22. | Pearse, G. A.; Raithby, P. R.; Lewis, J. Polyhedron 1989, 8, 301–304. doi:10.1016/s0277-5387(00)80418-0 |
| 27. | Magde, D.; Brannon, J. H.; Cremers, T. L.; Olmsted, J. J. Phys. Chem. 1979, 83, 696–699. doi:10.1021/j100469a012 |
| 28. | CrysAlisPro 1.171.38.41. Rigaku, 2015; https://www.rigaku.com/en/products/smc/chrysalis (accessed Nov 24, 2023). |
| 24. | Yahya, M.; Bouziani, A.; Ocak, C.; Seferoğlu, Z.; Sillanpää, M. Dyes Pigm. 2021, 192, No. 109227. doi:10.1016/j.dyepig.2021.109227 |
| 25. | Li, Y.; Huang, W.; Zhao, D.; Wang, L.; Jiao, Z.; Huang, Q.; Wang, P.; Sun, M.; Yuan, G. Molecules 2022, 27, No. 1800. doi:10.3390/molecules27061800 |
| 26. | Meech, S. R.; Phillips, D. J. Photochem. 1983, 23, 193–217. doi:10.1016/0047-2670(83)80061-6 |
| 6. | Ivakhnenko, E. P.; Knyazev, P. A.; Kovalenko, A. A.; Romanenko, G. V.; Revinskii, Y. V.; Starikov, A. G.; Minkin, V. I. Tetrahedron Lett. 2020, 61, No. 151429. doi:10.1016/j.tetlet.2019.151429 |
| 10. | Martinek, M.; Kotouček, M.; Ružička, E. Monatsh. Chem. 1967, 98, 1532–1536. doi:10.1007/bf00909022 |
| 11. | Afanas'eva, G. B.; Postovskii, I. Y.; Viktorova, T. S. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1978, 14, 966–968. doi:10.1007/bf00509550 |
| 23. | Ivakhnenko, E. P.; Romanenko, G. V.; Makarova, N. I.; Kovalenko, A. A.; Knyazev, P. A.; Rostovtseva, I. A.; Starikov, A. G.; Minkin, V. I. Dyes Pigm. 2020, 176, No. 108174. doi:10.1016/j.dyepig.2019.108174 |
| 5. | Abakumov, G. A.; Druzhkov, N. O.; Kurskii, Y. A.; Abakumova, L. G.; Shavyrin, A. S.; Fukin, G. K.; Poddel’skii, A. I.; Cherkasov, V. K.; Okhlopkova, L. S. Russ. Chem. Bull. 2005, 54, 2571–2577. doi:10.1007/s11172-006-0157-7 |
| 13. | Yang, J.; Cohen Stuart, M. A.; Kamperman, M. Chem. Soc. Rev. 2014, 43, 8271–8298. doi:10.1039/c4cs00185k |
| 15. | Kutyrev, A. A. Tetrahedron 1991, 47, 8043–8065. doi:10.1016/s0040-4020(01)91002-6 |
© 2024 Ivakhnenko et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.