Abstract
A series of 2,3-dihalo-1,1,1,4,4,4-hexafluorobutanes and 2-halo-1,1,1,4,4,4-hexafluorobut-2-enes were prepared from commercially available hydrofluoroolefins 1,1,1,4,4,4-hexafluorobut-2-enes and their 1H, 19F and 13C chemical shifts measured. Some reactions of synthesized 2-halo-1,1,1,4,4,4-hexafluorobut-2-enes have been investigated. A simple, one-pot procedure for the preparation of a new allene (1,1,4,4,4-pentafluorobuta-1,2-diene) and some of its transformations is presented.

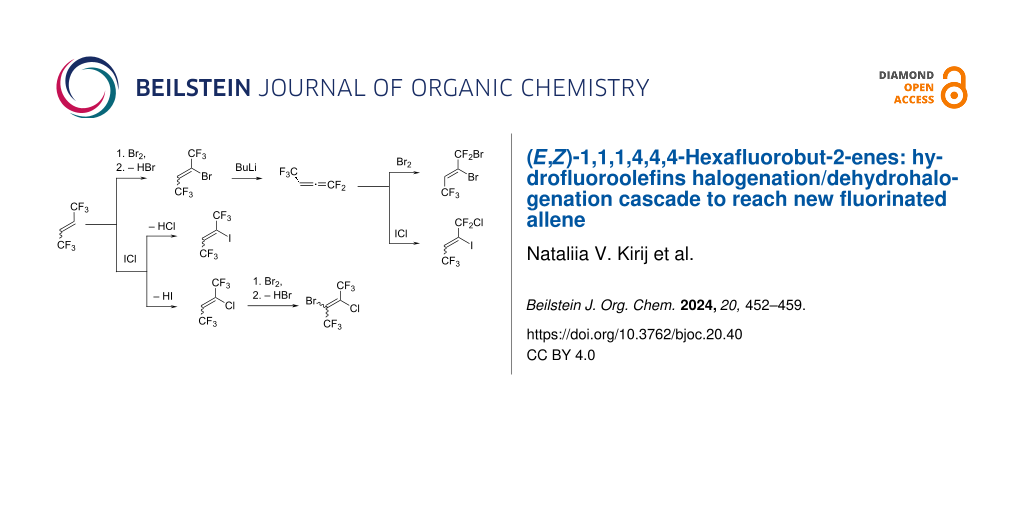
Graphical Abstract
Introduction
The first publication using (E)-1,1,1,4,4,4-hexafluorobut-2-ene (1a) was published in the middle of the 20th century [1]. Despite this, the real opportunity to study the properties of 1,1,1,4,4,4-hexafluorobut-2-ene appeared only after (Z)-1,1,1,4,4,4-hexafluorobut-2-ene (1b) began to be produced on an industrial scale [2]. These hydrofluoroolefins belong to the newest 4th generation of fluorocarbon refrigerants and are promising compounds and starting materials. Due to this, interest in the use of (E)- and (Z)-butenes 1a,b as synthons in various organic transformations has recently grown significantly. One of the new and budding directions in recent years is the stereoselective olefin metathesis processes based on catalysis by complexes of molybdenum, tungsten and ruthenium [3-5]. The first publications have recently appeared that molybdenum complexes can catalyze cross-metathesis of butene 1b. Wherein various alkyl and aryl olefins, including those that contain Lewis basic esters, carbamates and amines or α-branched moieties, may be used in efficient and exceptionally Z-selective cross-metathesis reactions [6-8]. A few years ago, some publications devoted to the cleavage of the C–F bond in butenes 1a,b have been presented in the literature. First, Crimmin et al. investigated the reaction of an aluminum(I) complex with fluoroalkenes. Unlike all the presented fluoroolefins, the reaction of the Al(I) complex with (Z)-butene 1b did not allow isolating the intermediate organoaluminum compound, but led to the elimination of two fluorine atoms with the formation of 1,1,4,4-tetrafluorobuta-1,3-diene [9]. It was also shown that the reactions of the boron reagent (CAACMe)BH2Li(thf)3 with hydrofluoroolefins, including (Z)-1,1,1,4,4,4-hexafluorobut-2-ene, results in defluoroborylation to form the corresponding =CF2 containing products [10]. In addition to complexes of aluminum and boron, several magnesium and lithium silyl reagents were prepared and proved to be good nucleophiles in reactions with (Z)-1,1,1,4,4,4-hexafluorobut-2-ene, as a result of which the corresponding defluorosilylation product was obtained [11]. In a related study of the hydrosilylation reaction of olefins 1a,b, it was shown that, depending on the catalyst used, platinum or rhodium compounds, along with the products of the addition of silane to the double bond, the elimination of the fluorine atom occurs with the formation of the corresponding olefin [12]. Another area of application of olefins 1a,b is based on C=C double bond addition reactions. As early as 1968, Atherton and Fields showed that (Z)- and (E)-butenes 1a,b reacted with diazotrifluoroethane to give 3,4,5-tris(trifluoromethyl)pyrazoline [13]. A few decades later, several new publications in this direction appeared in the literature. In a Chemours’ patent, it has been shown that the oxidation of (E)-butene 1a with sodium hypochlorite in the presence of tetrabutylphosphonium bromide leads to the formation of a bistrifluoromethyl containing oxirane [14]. A related study has demonstrated that (E)-butene 1a reacts with potassium persulfate to form 4,5-bistrifluoromethyl)-1,3,2-dioxathiolane 2,2-dioxide [15]. In 2021 Petrov published an article on the interaction of fluorinated olefins with fluorinated thioketones. In this publication it was demonstrated that 1,1,1,4,4,4-hexafluorobut-2-ene reacts with dithietane, sulfur and KF with the formation of the corresponding 1,3-dithiole [16]. Also, a recent patent presents a method for the preparation of 5,6-bis(trifluoromethyl)-1,2,4-triazine-3-carboxylic acid ethyl ester starting from 1,1,1,4,4,4-hexafluorobut-2-ene and an oxalamide hydrazone [17].
In the present study, we investigated the reactions of commercially available butenes 1a,b with halogens, as well as subsequent transformations of the resulting compounds.
Results and Discussion
In 1952 Haszeldine found that the reaction of bromine with (E)-1,1,1,4,4,4-hexafluorobut-2-ene (1a) under UV irradiation leads to the formation of 2,3-dibromo-1,1,1,4,4,4-hexafluorobutane (2) [1]. Subsequent dehydrobromination of compound 2 by treatment with alcoholic potassium hydroxide formed a mixture of isomers 2-bromo-1,1,1,4,4,4-hexafluorobut-2-ene (3a,b) with a yield in two stages of 48%. It should be noted, that only boiling points and elemental analysis data were given for the obtained substances. Ten years later Knunyants and co-workers also synthesized compound 2 and density, refractive index and mass spectra were added to the already available data [18]. However, until now there was no information about the structure and spectral characteristics of the obtained compounds. We have now synthesized these compounds, fully characterized them, and studied some of their transformations.
We found that not only (E)-butene 1a but also (Z)-butene 1b reacted with bromine in the same manner under the influence of ultraviolet irradiation or sunlight with the formation of 2,3-dibromo-1,1,1,4,4,4-hexafluorobutane (2) in 95% yield (Scheme 1).
![[1860-5397-20-40-i1]](/bjoc/content/inline/1860-5397-20-40-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 2,3-dibromo-1,1,1,4,4,4-hexafluorobutane (2).
Scheme 1: Synthesis of 2,3-dibromo-1,1,1,4,4,4-hexafluorobutane (2).
The only difference was that under UV irradiation, the reaction proceeded faster. In both cases, product 2 represented a mixture of stereoisomers in 2:1 ratio. After isolation by distillation, 2,3-dibromo-1,1,1,4,4,4-hexafluorobutane (2) was characterized by 1H, 19F, 13C NMR and mass spectra.
We studied the reaction of dibromoalkane 2 with various bases such as DBU, Hünig’s base (iPr2NEt), and potassium hydroxide (Table 1).
Table 1: The reaction of 2,3-dibromo-1,1,1,4,4,4-hexafluorobutane (2) with bases.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-40-i15.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent | Base (equiv) | Conditions (°C/h) | Conversion (%) |
| 1 | Et2O | iPr2NEt (1) | 20/18 | 60 |
| 2 | Et2O | iPr2NEt (1) | 20/18 | 95 |
| 3 | Et2O | DBU (1) | 20/1 | 95 |
| 4 | diglyme | DBU (1) | 20/18 | 60 |
| 5 | diglyme | DBU (2) | 20/1 | 100 |
| 6 | H2O | KOH (1) | 20/18 | 55 |
| 7 | H2O | KOH (1.3) | 20/2 | 100 |
In all cases, except the reaction in diglyme (Table 1, entry 5), a mixture of (E)- and (Z)-2-bromo-1,1,1,4,4,4-hexafluorobut-2-enes (3a,b) in a ratio of 2:1 was formed. The configuration of the isomers was determined by the 5JFFcis coupling constant in the 19F{H} NMR spectrum (ca. 0 Hz for (Z)-isomer and ca. 11 Hz for (E)-isomer). The best results were obtained in Et2O with Hünig’s and DBU bases (Table 1, entries 2 and 3), but unfortunately in these cases the product olefins could not be separated from Et2O. Therefore, we decided to use high-boiling diglyme instead of ether. The reaction of a butane 2 with one equivalent of DBU (Table 1, entry 4) led to the same results as for the Hünig’s base (Table 1, entry 1). The use of two equivalents of DBU (Table 1, entry 6) led to the complete conversion of the initial substrate, but the selectivity of the reaction was significantly reduced in this case. The reaction mixture gave a complex mixture, in which (E)-butene 3a was identified as a major component. Product 3a was removed from bulk diglyme in vacuum and after subsequent distillation it was isolated with a yield of 23%. As the isolation of the 2-dehydrobromination products from organic bases was complex, we focused our attention on carrying out the reaction with KOH. The best result was obtained by treatment of 1 equivalent of butane 2 with 1.3 equivalents of KOH in the presence of 5 mol % of Bu4NBr as an interfacial carrier in water at room temperature for 2 h (Table 1, entry 7). After completion of the reaction the mixture of isomers 3a,b was separated from the water phase and distilled at 55 °C.
Further increase in the amount of KOH led to the elimination of the second mole of HBr with the formation of hexafluorobut-2-yne (4). By controlling the course of the reaction by the 19F NMR method it was possible to achieve complete dehydrobromination of (Z)-isomer 3b and isolation of (E)-isomer 3a in pure form in 35% yield (Scheme 2).
![[1860-5397-20-40-i2]](/bjoc/content/inline/1860-5397-20-40-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Unexpectedly the (E)-isomer 3a, upon long-term storage, transforms into the (Z)-isomer 3b. Therefore, we studied the isomerization of olefin 3a in the presence of SbCl5, AlCl3, Bu4NOH/MeOH and under UV irradiation. We found that under UV irradiation for several hours, the (E)-isomer 3a completely transformed into (Z)-isomer 3b in quantitative yield (Scheme 3).
![[1860-5397-20-40-i3]](/bjoc/content/inline/1860-5397-20-40-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Isomerization reaction of (E)-butene 3a to (Z)-butene 3b.
Scheme 3: Isomerization reaction of (E)-butene 3a to (Z)-butene 3b.
Next, our attention was directed toward the reaction of (E)- and (Z)-butenes 1a,b with iodine monochloride (ICl). We found that olefins 1a and 1b reacts with ICl under the influence of sunlight to form previously unknown 2-chloro-3-iodo-1,1,1,4,4,4-hexafluorobutane (5, Scheme 4).
![[1860-5397-20-40-i4]](/bjoc/content/inline/1860-5397-20-40-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 2-chloro-3-iodo-1,1,1,4,4,4-hexafluorobutane (5).
Scheme 4: Synthesis of 2-chloro-3-iodo-1,1,1,4,4,4-hexafluorobutane (5).
Pure final product 5 was isolated by distillation at 112 °C in 85% yield. NMR analysis as in the case of bromo derivative 2 showed a mixture of stereoisomers with a 2:1 ratio.
The dehydrohalogenation reaction of 2-chloro-3-iodo-1,1,1,4,4,4-hexafluorobutane (5) was studied. Like the dehydrobromination of alkane 2, the reaction of compound 5 with 1.3 equivalents of KOH in water in the presence of 5 mol % of Bu4NBr was carried out. In this case, a mixture of 2-chloro-1,1,1,4,4,4-hexafluorobut-2-enes (6a,b) and 2-iodo-1,1,1,4,4,4-hexafluorobut-2-enes (7a,b) in a ratio of 3:2 was formed from the concurrent elimination reactions of hydrogen iodide and hydrogen chloride (Scheme 5).
![[1860-5397-20-40-i5]](/bjoc/content/inline/1860-5397-20-40-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Dehydrohalogenation reaction of 2-chloro-3-iodo-1,1,1,4,4,4-hexafluorobutane (5).
Scheme 5: Dehydrohalogenation reaction of 2-chloro-3-iodo-1,1,1,4,4,4-hexafluorobutane (5).
The chloro- and iodobutenes were separated by distillation in 52% yield for 6a,b and 34% yield for 7a,b. NMR analysis showed the mixtures of (E)- and (Z)-isomers in both cases with the ratio E/Z = ≈4:5 for 6a,b and E/Z = ≈1:2 for 7a,b. The configuration of the isomers was determined by the 5JFFcis coupling constant in the 19F{H} NMR spectrum (ca. 0 Hz for (Z)-isomer and ≈11 Hz for (E)-isomer). The 2-chloro-1,1,1,4,4,4-hexafluorobut-2-enes were first described by Haszeldine in 1952 [1] and only then the (Z)-isomer. We report here the isolation and complete characterization of both (E)- and (Z)-isomers. The spectral characteristics of product 6b obtained by us fully correspond to the literature data [19,20]. The 1H and 19F NMR spectra of compounds 7a,b also corresponded to the data given in the literature [21,22]. We present here the spectral data for isomer 6a, as well as the missing data of 13C NMR spectra for iodoolefins 7a,b.
It should be noted that the reaction of alkane 5 with DBU or Hünig’s base in Et2O or dimethoxyethane (DME) as a solvent, resulted only in the formation of 2-chloro-1,1,1,4,4,4-hexafluorobut-2-enes 6a,b (Scheme 5). Unfortunately, due to difficulties in separation from solvent, olefins 6a,b in this case were not isolated in a pure state.
For iodoolefin 7a, we found an alternative route for its synthesis. We have previously shown that hydrosilylation reaction of hexafluorobut-2-yne with triethylsilanes gave (E)-1,1,1,4,4,4-hexafluoro-2-triethylsilylbut-2-ene (8) [23]. Going back to the study of the obtained silane reactivity we performed the reaction with iodine. All experiments were carried out in THF, N-methylpyrrolidone and sulfolane with iodine in the presence of a source of fluoride ion. The best result was observed when the reaction was carried out in dry sulfolane with a two-fold excess of iodine and 1.5-fold excess of anhydrous KF (Scheme 6).
![[1860-5397-20-40-i6]](/bjoc/content/inline/1860-5397-20-40-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: The reaction of silane 8 with I2/KF.
Scheme 6: The reaction of silane 8 with I2/KF.
The mixture was stirred at 30 °C for several days until none of the starting 8 was detected in 19F NMR spectra. The desired iodoolefin 7a together with byproduct triethylfluorosilane (Et3SiF) were removed from sulfolane under vacuum and after double distillation with column 7a was isolated in 67% yield. The 19F{1H} NMR spectra of 7a, showed the coupling constant of CF3 and CF3 to be 11.3 Hz, suggesting strongly that product 7a like the original silane 8 has (E)-configuration.
Although some olefins presented above were obtained several decades ago, there was almost no information about their reactivity in the literature. Therefore, we became interested in exploring the synthetic potential of halo-bistrifluoromethyl-containing olefins 3, 6 and 7, which are readily available and can be synthesized in the laboratory in appreciable amounts.
Fluorinated organic compounds are important synthons, which are widely used in agrochemicals, pharmaceuticals and other fields [24-26]. Fluoroorganic lithium and Grignard reagents have been obtained by the metalation reactions of organofluorine compounds containing bromine and iodine atoms with alkyllithium and Grignard reagents.
Although olefin 3 has been available for many decades, only one paper describes its lithiation with methyllithium and the subsequent reaction of the lithium compound with trifluoroacetophenone [27]. We began our research on the reactivity of the bromobutenes 3a,b with isopropylmagnesium chloride (iPrMgCl) and butyllithium (BuLi), as well as the reactions of the resulting organometallic compounds.
1.2 Equivalents of a solution of iPrMgCl in THF were added to bromoolefin 3a in Et2O or THF at −78 °C and then after 1 h at the same temperature 1 equivalent of 4-fluorobenzaldehyde (9) was added. After completion of the reaction and subsequent treatment of the reaction mixture with 2 N hydrochloric acid, 2,3-bis(trifluoromethyl)-1-(4-fluorophenyl)prop-2-ene-1-ol (10) was detected in the 19F NMR spectrum. It should be noted that, in addition to the unreacted starting aldehyde 9, the formation of olefin 1b and previously unknown product 11 were also recorded in the reaction mixture (Scheme 7). The 19F NMR spectrum of compound 11 showed two signals at −65.6 and −99.1 ppm in a ratio of 3:2 and in the 1H NMR spectrum, a multiplet at 6.5 ppm was detected. Based on the received data, we assumed that product 11 had an allene structure. It was also important to note that the reaction proceeded more selectively in ether, which significantly reduced the amount of byproducts.
![[1860-5397-20-40-i7]](/bjoc/content/inline/1860-5397-20-40-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: The reaction of 3a with iPrMgCl and 4-fluorobenzaldehyde (9).
Scheme 7: The reaction of 3a with iPrMgCl and 4-fluorobenzaldehyde (9).
Pure final alcohol 10 was isolated by column chromatography on SiO2 in 46% yield and 1H, 19F and 13C NMR spectra were in full agreement with the published data [23].
The most interesting outcome from our point of view was the formation of 1,1,4,4,4-pentafluorobuta-1,2-diene (11). 1,1-Difluoroallenes are building blocks for a great number of valuable transformations [28]. Therefore, the synthesis of new fluorinated allenes continues to be relevant. One of the methods for the synthesis of allenes was based on the interaction of bromoolefins with organolithium compounds, followed by the elimination of lithium fluoride [29-31]. It was logical to assume that in our case a similar reaction of the Grignard reagent 12 with aldehyde 9, elimination of MgBrF results in the formation of allene 11. To confirm our hypothesis, we studied the reaction of haloolefins 3 and 7 with iPrMgCl and BuLi.
Thus, olefin 3a in Et2O reacted with iPrMgCl solution in THF at −80 °C to form Grignard reagent 12 and by heating the reaction mixture to room temperature MgBrF was produced together with allene 11 (Scheme 8). In addition to the allene, the formation of olefin 1b was also recorded in the 19F NMR spectrum.
![[1860-5397-20-40-i8]](/bjoc/content/inline/1860-5397-20-40-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: The reaction of olefin 3a with iPrMgCl.
Scheme 8: The reaction of olefin 3a with iPrMgCl.
Unfortunately, due to difficulties in separating from olefin 1b and diethyl ether, allene 11 was not isolated in a pure state. Therefore, we turned to study the reaction of adduct 3a with butyllithium.
Unlike the reaction of bromobutene 3a with iPrMgCl, the reaction mixture with BuLi in hexane solution did not contain olefin 1b and only desired allene 11 was identified with 95% purity in the 19F NMR spectra (Scheme 9).
![[1860-5397-20-40-i9]](/bjoc/content/inline/1860-5397-20-40-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: The reaction of (E)-butene 3a with BuLi.
Scheme 9: The reaction of (E)-butene 3a with BuLi.
The volatile product 11 was removed from the reaction mixture at 20 °C for 2 hours with a slow flow of argon through the system. Unfortunately, we failed to completely separate it from the hexane and allene 11 was produced with 80–85% purity. We managed to partially solve this problem by replacing hexane with the higher-boiling heptane. In this case, the desired product was isolated by double distillation at 7 °C in 50% yield and >90% purity and was fully characterized by 1H, 19F, 13C NMR and IR spectra. The presence of a characteristic band at 2038 cm−1 in the IR spectrum confirmed the allene structure of product 11. As expected, allene 11 is extremely reactive and therefore it decomposes rather quickly during storage both in solvent and in the individual state.
Since we were able to obtain both isomers 3a and 3b in the individual state, we investigated the reaction of (Z)-isomer 3b with BuLi. Thus, 3b also reacted with butyllithium to form allene 11. However, when studying the reaction of each of the isomers under the same conditions, it turned out that the (Z)-isomer reacts faster than the (E)-isomer. Therefore, in the case of the (E)-isomer, to achieve its complete conversion, it was necessary to increase the lithiation reaction time. Based on the results obtained, it was logical to assume that the reaction can be carried out with a mixture of 3a,b. We found that the reaction of 3a,b with 1.2 equivalents of BuLi in heptane at −80 °C for 1 hour, followed by warming to room temperature, led to the complete conversion of the original olefins and the formation of allene 11.
Thus, we have developed a method for the synthesis of the previously unknown allene 11. In addition, the possibility of using a mixture of olefins 3a,b made the allene 11 a more accessible synthon for studying its further transformations.
We started studying the reactivity of allene 11 with the bromination reaction. The reaction was carried out without a solvent at a reagent ratio of 1:1. Bromine was added dropwise to allene 11 at −30 °C and the reaction mixture was stirred at the same temperature until colorless. The bromination of allene resulted in the formation of (Z)-1,2-dibromo-1,1,4,4,4-pentafluorobut-2-ene (13) in 92% yield, which was isolated as a colorless liquid and fully characterized by NMR spectroscopy (Scheme 10).
![[1860-5397-20-40-i10]](/bjoc/content/inline/1860-5397-20-40-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: The reaction of allene 11 with bromine.
Scheme 10: The reaction of allene 11 with bromine.
The 19F NMR spectrum showed two signals with the integrated intensity ratio 3:2, a doublet for a CF3 group at δ −57.7 ppm with the coupling constant 3JFH = 6.8 Hz and the characteristic signal of a CF2Br group as a singlet at δ −50.1 ppm. In the 1H NMR spectrum, there was a signal attributed to the CH proton at 6.9 ppm as a quartet with the coupling constant of 3JHF = 6.8 Hz. The (Z)-configuration of product 13 was determined by the absence of the F–F constant in the 19F NMR spectra. It should be noted that the bromination reaction could also be carried out in diethyl ether. In this case, the bromo derivative 13 could be isolated in pure form only after several distillations, which significantly reduces its yield.
Continuing to study the reactivity of allene 11, we investigated its reaction with ICl. The reaction was carried out at a reagent ratio of 1:1. ICl was added to a solution of allene in pentane at −20 °C and then the reaction mixture was stirred at rt until the color disappeared. In contrast to bromination, the reaction with ICl is less selective and leads to the formation of addition products at both double bonds. At the same time, the reaction proceeded predominantly at the terminal double bond with the formation of a mixture of isomers (Z)-14a and (E)-14b. The content of the addition product 15 at the second double bond in the reaction mixture was about 15% (Scheme 11).
![[1860-5397-20-40-i11]](/bjoc/content/inline/1860-5397-20-40-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: The reaction of allene 11 with ICl.
Scheme 11: The reaction of allene 11 with ICl.
It should be noted that we failed to isolate the pure product 15 from reaction mixtures because of the small difference in boiling points and volatiles between isomers 14a,b and product 15. Nevertheless, it was tentatively identified in the mixture, based on 1H, 19F and 13C NMR data. The 19F NMR spectrum showed three signals with the integrated intensity ratio 3:1:1, a doublet for a CF3 group at δ −71.5 ppm with a coupling constant of 3JFH = 6 Hz and two highly characteristic signals of geminal fluorine atoms of the CF2 group as doublets of multiplets at δ −66.7 and −70.3 ppm with a coupling constant of 2JFF = 15 Hz. In the 1H NMR spectrum, there was a signal attributed to CH proton at 4.9 ppm as a quartet of multiplets with a coupling constant of 3JHF = 6 Hz.
Thus, during the study of reactions of allene 11 with Br2 and ICl, we obtained new synthons, which due to the presence of several reaction centers, could be of particular interest in various kinds of transformations. Therefore, we decided to synthesize a bistrifluoromethyl containing olefin with bromine and chlorine atoms and explore the possibility of using it to obtain another allene.
We found that 2-chloro-1,1,1,4,4,4-hexafluorobut-2-enes (6a,b) react with bromine under the influence of sunlight with the formation of 2,3-dibromo-2-chloro-1,1,1,4,4,4-hexafluorobutane 16 in 84% yield (Scheme 12).
![[1860-5397-20-40-i12]](/bjoc/content/inline/1860-5397-20-40-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Synthesis of 2,3-dibromo-2-chloro-1,1,1,4,4,4-hexafluorobutane (16).
Scheme 12: Synthesis of 2,3-dibromo-2-chloro-1,1,1,4,4,4-hexafluorobutane (16).
As in the case of alkanes 2 and 5, product 16 represented a mixture of stereoisomers in 2:1 ratio. After isolation by distillation butane 16 was characterized by 1H, 19F, 13C NMR and mass spectra.
The reaction of alkane 16 with DBU in pentane as a solvent led exclusively to dehydrobromination with the formation of a mixture of (Z)- and (E)-2-bromo-3-chloro-1,1,1,4,4,4-hexafluorobut-2-enes (17a,b) in a ratio of 2:1 (Scheme 13).
![[1860-5397-20-40-i13]](/bjoc/content/inline/1860-5397-20-40-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Synthesis of (Z, E)-2-bromo-3-chloro-1,1,1,4,4,4-hexafluorobut-2-enes (17a,b).
Scheme 13: Synthesis of (Z, E)-2-bromo-3-chloro-1,1,1,4,4,4-hexafluorobut-2-enes (17a,b).
After completion of the reaction the mixture of isomers 17a,b was isolated by distillation at 79 °C and fully characterized. The configuration of the isomers was determined by the 5JFFcis coupling constant in the 19F{H} NMR spectrum (ca. 0 Hz for (Z)-isomer and ca. 13 Hz for (E)-isomer).
Like the reaction of bromobutenes 3a,b with BuLi, the reaction of olefins 17a,b with BuLi was carried out. Unfortunately, we were unable to detect the formation of allene 18 in this reaction. In the 19F NMR spectrum, along with the unreacted starting olefins 17a,b, the main reaction product is hexafluorobut-2-yne (4, Scheme 14).
![[1860-5397-20-40-i14]](/bjoc/content/inline/1860-5397-20-40-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: The reaction of olefins 17a,b with BuLi.
Scheme 14: The reaction of olefins 17a,b with BuLi.
Although attempts to obtain allene 18 were unsuccessful, olefins 17a,b may be of great interest as synthons due to the presence of several reaction centers in the molecule.
Conclusion
In conclusion, we synthesized a series of synthons from available industrial starting compounds – (E,Z)-1,1,1,4,4,4-hexafluorobut-2-enes (1a,b) using simple procedures. We demonstrated the halogenation reactions of butenes 1a,b with bromine and iodine monochloride to form 2,3-dihalo-1,1,1,4,4,4-hexafluorobutanes. Dehydrohalogenation of the obtained butanes leads to the formation of a number of bistrifluoromethyl-containing haloolefins, which are widely used in subsequent transformations. Based on bromoolefins 3a,b, a new polyfluoro-containing allene 11 was synthesized and its reactions with bromine and iodine monochloride were also studied.
Supporting Information
| Supporting Information File 1: Experimental part and copies of NMR spectra. | ||
| Format: PDF | Size: 6.2 MB | Download |
References
-
Haszeldine, R. N. J. Chem. Soc. 1952, 2504–2513. doi:10.1039/jr9520002504
Return to citation in text: [1] [2] [3] -
Sicard, A. J.; Baker, R. T. Chem. Rev. 2020, 120, 9164–9303. doi:10.1021/acs.chemrev.9b00719
Return to citation in text: [1] -
Malcolmson, S. J.; Meek, S. J.; Sattely, E. S.; Schrock, R. R.; Hoveyda, A. H. Nature 2008, 456, 933–937. doi:10.1038/nature07594
Return to citation in text: [1] -
Yu, M.; Wang, C.; Kyle, A. F.; Jakubec, P.; Dixon, D. J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 479, 88–93. doi:10.1038/nature10563
Return to citation in text: [1] -
Herbert, M. B.; Grubbs, R. H. Angew. Chem., Int. Ed. 2015, 54, 5018–5024. doi:10.1002/anie.201411588
Return to citation in text: [1] -
Koh, M. J.; Nguyen, T. T.; Lam, J. K.; Torker, S.; Hyvl, J.; Schrock, R. R.; Hoveyda, A. H. Nature 2017, 542, 80–85. doi:10.1038/nature21043
Return to citation in text: [1] -
Mu, Y.; Nguyen, T. T.; van der Mei, F. W.; Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2019, 58, 5365–5370. doi:10.1002/anie.201901132
Return to citation in text: [1] -
Hoveyda, A. H.; Ming, J.; Nauyen, T. T.; Schrock, R. R.; Hyvl, J. Halogen-containing metathesis catalysts and methods thereof. WO Patent WO2018/013943, Jan 18, 2018.
Return to citation in text: [1] -
Bakewell, C.; White, A. J. P.; Crimmin, M. R. Angew. Chem., Int. Ed. 2018, 57, 6638–6642. doi:10.1002/anie.201802321
Return to citation in text: [1] -
Phillips, N. A.; Coates, G. J.; White, A. J. P.; Crimmin, M. R. Chem. – Eur. J. 2020, 26, 5365–5368. doi:10.1002/chem.202000636
Return to citation in text: [1] -
Coates, G.; Tan, H. Y.; Kalff, C.; White, A. J. P.; Crimmin, M. R. Angew. Chem., Int. Ed. 2019, 58, 12514–12518. doi:10.1002/anie.201906825
Return to citation in text: [1] -
Pavlenko, N. V.; Peng, S.; Petrov, V.; Jackson, A.; Sun, X.; Sprague, L.; Yagupolskii, Y. L. Eur. J. Org. Chem. 2020, 5425–5435. doi:10.1002/ejoc.202000853
Return to citation in text: [1] -
Atherton, J. H.; Fields, R. J. Chem. Soc. C 1968, 1507–1513. doi:10.1039/j39680001507
Return to citation in text: [1] -
Petrov, V. A.; Brandt, D. R.; Minor, B. H.; Kontomaris, K.; Robin, M. L.; Wysong, E. B.; Musyimi, H. K.; Glatt, C. M. Uses of fluorinated epoxides and novel mixtures thereof. WO Patent WO2018/165623, Sept 13, 2018.
Return to citation in text: [1] -
Long, Z.; Xiaodong, G.; Xiaolong, W.; Zheng, L. Preparation method of ethylene sulfate and derivatives thereof. Chin. Patent CN114805291, July 29, 2022.
Return to citation in text: [1] -
Petrov, V. J. Fluorine Chem. 2021, 248, 109775. doi:10.1016/j.jfluchem.2021.109775
Return to citation in text: [1] -
Wissler, M.; Mueller, E.; Lorenz, P.; Kliem, C.; Fleischhacker, H. Process for the covalent coupling of two molecules by means of a Diels-Alder reaction with inverse electron requirement. U.S. Patent US2010/0016545, Jan 21, 2010.
Return to citation in text: [1] -
Knunyants, I. L.; German, L. S.; Rozhkov, I. N. Russ. Chem. Bull. 1962, 11, 1587–1588. doi:10.1007/bf00907244
Return to citation in text: [1] -
Bo, Z.; Jian, L.; Wei, Z.; Jijun, Z.; Sheng, H.; Zhiqiang, Y.; Xiaobo, T.; Zhijun, H.; Jianping, K.; Fengxian, L. Catalytic conversion method for hexachlorobutadiene. Chin. Patent CN110372471, Oct 25, 2019.
Return to citation in text: [1] -
Jian, L.; Bo, Z.; Wei, Z.; Jijun, Z.; Sheng, H.; Zhiqiang, Y.; Xiaobo, T.; Zhijun, H.; Jianping, K.; Fengxian, L. Synthesis method of 2-chloro-1,1,1,4,4,4-hexafluoro-2-butene. Chin. Patent CN110372472, Oct 25, 2019.
Return to citation in text: [1] -
Leedham, K.; Haszeldine, R. N. J. Chem. Soc. 1954, 1634–1638. doi:10.1039/jr9540001634
Return to citation in text: [1] -
El Soueni, A.; Tedder, J. M.; Walton, J. C. J. Chem. Soc., Faraday Trans. 1 1981, 77, 89–100. doi:10.1039/f19817700089
Return to citation in text: [1] -
Kirij, N. V.; Filatov, A. A.; Yagupolskii, Yu.. L.; Peng, S.; Jackson, A. J. Fluorine Chem. 2022, 253, 109922. doi:10.1016/j.jfluchem.2021.109922
Return to citation in text: [1] [2] -
Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity and Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393x
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Gassman, P. G.; Ray, J. A.; Wenthold, P. G.; Mickelson, J. W. J. Org. Chem. 1991, 56, 5143–5146. doi:10.1021/jo00017a029
Return to citation in text: [1] -
Fuchibe, K.; Ichikawa, J. Sci. Synth., Knowl. Updates 2014, 2, 217–231. doi:10.1055/sos-sd-124-00001
Return to citation in text: [1] -
Drakesmith, F. G.; Stewart, O. J.; Tarrant, P. J. Org. Chem. 1968, 33, 280–285. doi:10.1021/jo01265a055
Return to citation in text: [1] -
Zens, A. P.; Ellis, P. D.; Ditchfield, R. J. Am. Chem. Soc. 1974, 96, 1309–1312. doi:10.1021/ja00812a008
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Burkholder, C. R.; Piedrahita, C. A. J. Fluorine Chem. 1982, 20, 637–647. doi:10.1016/s0022-1139(00)82289-7
Return to citation in text: [1]
| 23. | Kirij, N. V.; Filatov, A. A.; Yagupolskii, Yu.. L.; Peng, S.; Jackson, A. J. Fluorine Chem. 2022, 253, 109922. doi:10.1016/j.jfluchem.2021.109922 |
| 19. | Bo, Z.; Jian, L.; Wei, Z.; Jijun, Z.; Sheng, H.; Zhiqiang, Y.; Xiaobo, T.; Zhijun, H.; Jianping, K.; Fengxian, L. Catalytic conversion method for hexachlorobutadiene. Chin. Patent CN110372471, Oct 25, 2019. |
| 20. | Jian, L.; Bo, Z.; Wei, Z.; Jijun, Z.; Sheng, H.; Zhiqiang, Y.; Xiaobo, T.; Zhijun, H.; Jianping, K.; Fengxian, L. Synthesis method of 2-chloro-1,1,1,4,4,4-hexafluoro-2-butene. Chin. Patent CN110372472, Oct 25, 2019. |
| 21. | Leedham, K.; Haszeldine, R. N. J. Chem. Soc. 1954, 1634–1638. doi:10.1039/jr9540001634 |
| 22. | El Soueni, A.; Tedder, J. M.; Walton, J. C. J. Chem. Soc., Faraday Trans. 1 1981, 77, 89–100. doi:10.1039/f19817700089 |
| 9. | Bakewell, C.; White, A. J. P.; Crimmin, M. R. Angew. Chem., Int. Ed. 2018, 57, 6638–6642. doi:10.1002/anie.201802321 |
| 18. | Knunyants, I. L.; German, L. S.; Rozhkov, I. N. Russ. Chem. Bull. 1962, 11, 1587–1588. doi:10.1007/bf00907244 |
| 6. | Koh, M. J.; Nguyen, T. T.; Lam, J. K.; Torker, S.; Hyvl, J.; Schrock, R. R.; Hoveyda, A. H. Nature 2017, 542, 80–85. doi:10.1038/nature21043 |
| 7. | Mu, Y.; Nguyen, T. T.; van der Mei, F. W.; Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2019, 58, 5365–5370. doi:10.1002/anie.201901132 |
| 8. | Hoveyda, A. H.; Ming, J.; Nauyen, T. T.; Schrock, R. R.; Hyvl, J. Halogen-containing metathesis catalysts and methods thereof. WO Patent WO2018/013943, Jan 18, 2018. |
| 3. | Malcolmson, S. J.; Meek, S. J.; Sattely, E. S.; Schrock, R. R.; Hoveyda, A. H. Nature 2008, 456, 933–937. doi:10.1038/nature07594 |
| 4. | Yu, M.; Wang, C.; Kyle, A. F.; Jakubec, P.; Dixon, D. J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 479, 88–93. doi:10.1038/nature10563 |
| 5. | Herbert, M. B.; Grubbs, R. H. Angew. Chem., Int. Ed. 2015, 54, 5018–5024. doi:10.1002/anie.201411588 |
| 17. | Wissler, M.; Mueller, E.; Lorenz, P.; Kliem, C.; Fleischhacker, H. Process for the covalent coupling of two molecules by means of a Diels-Alder reaction with inverse electron requirement. U.S. Patent US2010/0016545, Jan 21, 2010. |
| 29. | Drakesmith, F. G.; Stewart, O. J.; Tarrant, P. J. Org. Chem. 1968, 33, 280–285. doi:10.1021/jo01265a055 |
| 30. | Zens, A. P.; Ellis, P. D.; Ditchfield, R. J. Am. Chem. Soc. 1974, 96, 1309–1312. doi:10.1021/ja00812a008 |
| 31. | Dolbier, W. R., Jr.; Burkholder, C. R.; Piedrahita, C. A. J. Fluorine Chem. 1982, 20, 637–647. doi:10.1016/s0022-1139(00)82289-7 |
| 2. | Sicard, A. J.; Baker, R. T. Chem. Rev. 2020, 120, 9164–9303. doi:10.1021/acs.chemrev.9b00719 |
| 13. | Atherton, J. H.; Fields, R. J. Chem. Soc. C 1968, 1507–1513. doi:10.1039/j39680001507 |
| 15. | Long, Z.; Xiaodong, G.; Xiaolong, W.; Zheng, L. Preparation method of ethylene sulfate and derivatives thereof. Chin. Patent CN114805291, July 29, 2022. |
| 23. | Kirij, N. V.; Filatov, A. A.; Yagupolskii, Yu.. L.; Peng, S.; Jackson, A. J. Fluorine Chem. 2022, 253, 109922. doi:10.1016/j.jfluchem.2021.109922 |
| 12. | Pavlenko, N. V.; Peng, S.; Petrov, V.; Jackson, A.; Sun, X.; Sprague, L.; Yagupolskii, Y. L. Eur. J. Org. Chem. 2020, 5425–5435. doi:10.1002/ejoc.202000853 |
| 16. | Petrov, V. J. Fluorine Chem. 2021, 248, 109775. doi:10.1016/j.jfluchem.2021.109775 |
| 28. | Fuchibe, K.; Ichikawa, J. Sci. Synth., Knowl. Updates 2014, 2, 217–231. doi:10.1055/sos-sd-124-00001 |
| 11. | Coates, G.; Tan, H. Y.; Kalff, C.; White, A. J. P.; Crimmin, M. R. Angew. Chem., Int. Ed. 2019, 58, 12514–12518. doi:10.1002/anie.201906825 |
| 24. | Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity and Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393x |
| 25. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 26. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 10. | Phillips, N. A.; Coates, G. J.; White, A. J. P.; Crimmin, M. R. Chem. – Eur. J. 2020, 26, 5365–5368. doi:10.1002/chem.202000636 |
| 14. | Petrov, V. A.; Brandt, D. R.; Minor, B. H.; Kontomaris, K.; Robin, M. L.; Wysong, E. B.; Musyimi, H. K.; Glatt, C. M. Uses of fluorinated epoxides and novel mixtures thereof. WO Patent WO2018/165623, Sept 13, 2018. |
| 27. | Gassman, P. G.; Ray, J. A.; Wenthold, P. G.; Mickelson, J. W. J. Org. Chem. 1991, 56, 5143–5146. doi:10.1021/jo00017a029 |
© 2024 Kirij et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.