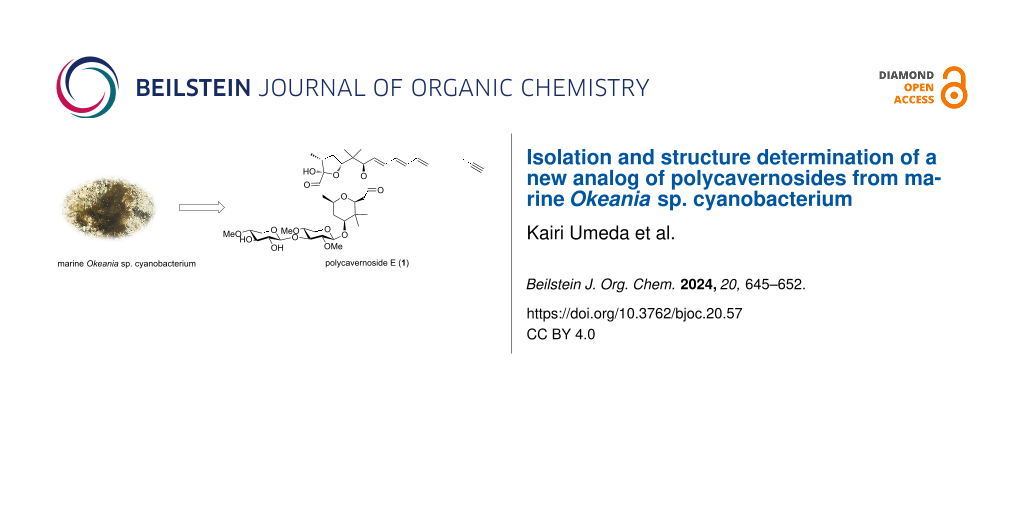
Abstract
Polycavernoside E (1), a new polycavernoside analog, was isolated from a marine Okeania sp. cyanobacterium. The relative configuration was elucidated primarily by analyzing the two dimensional nuclear magnetism resonance (2D NMR) data. The absolute configuration was clarified by comparing the electronic circular dichroism (ECD) data of 1 with those of known analogs. Polycavernoside E (1) exhibited moderate antitrypanosomal activity against Trypanosoma brucei rhodesiense. Furthermore, the isolation of polycavernoside E (1) from marine cyanobacteria provides additional evidence that marine cyanobacteria, and not red algae, are responsible for the biosynthesis of polycavernosides.

Graphical Abstract
Introduction
In 1991, an outbreak of food poisoning caused by a species of red algae known as ‘Polycavernosa tsudai’ occurred in Guam, which resulted in killing of three people. Two novel macrolide glycosides, polycavernosides A (2) and B (3), were reported as the causative compounds for the illness [1]. After that, the second fatal food poisoning incidents occurred in the Philippines caused by the ingestion of polycavernoside A (2)-contaminated red algae [2]. Subsequently, polycavernoside analogs such as polycavernoside C (4) were isolated from red algae [3,4]. In 2015, Navarro et al. isolated polycavernoside D (5) from a marine Okeania sp. cyanobacterium [5]. They suggested that polycavernosides were produced by marine cyanobacteria based on their high content and structural similarity to other cyanobacterial metabolites. In this study, polycavernoside E (1), a new polycavernoside analog, was isolated from a marine Okeania sp. cyanobacterium obtained from Okinawa Prefecture, Japan (Figure 1). This finding provides additional evidence that polycavernosides are secondary metabolites derived from marine Okeania sp. cyanobacteria.
![[1860-5397-20-57-1]](/bjoc/content/figures/1860-5397-20-57-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of compounds 1, 2, and 5.
Figure 1: Structures of compounds 1, 2, and 5.
Results and Discussion
The EtOH extract of marine Okeania sp. cyanobacterium (340 g, wet weight) collected from Akuna Beach, Okinawa, Japan, was partitioned between EtOAc and H2O. The EtOAc fraction was further partitioned into 90% aqueous MeOH and hexane. The aqueous MeOH portion was purified by reversed-phase column chromatography (ODS silica gel, MeOH/H2O), automated flash chromatography (hexane/EtOAc), and repeated reversed-phase HPLC to give polycavernoside E (1, 0.5 mg as a colorless oil). The isolation of compound 1 was directed by its characteristic UV absorption around 270 nm.
The molecular formula of 1 was determined to be C44H66O15 based on the HRESIMS data. The NMR data for 1 are summarized in Table 1. The 1H NMR spectrum of compound 1 was similar to those of known polycavernosides but matched none of them, suggesting that 1 was a new analog of polycavernosides [1,3-5]. A detailed analysis of the NMR data revealed the planar structure of 1, as shown in Figure 2. COSY and HMQC spectral analyses revealed several partial structures, indicated by the bold bonds in Figure 2. Four HMBC were observed from singlet methyl signals: δH 0.85 (H-28)/δC 19.4 (C-29), δH 0.86 (H-29)/δC 17.8 (C-28), δH 0.94 (H-30)/δC 13.9 (C-31), and δH 0.90 (H-31)/δC 22.2 (C-30). These correlations elucidated the presence of two gem-dimethyl groups. Moreover, three HMBC, δH 4.03 (H-5a’)/δC 106.1 (C-1’), δH 3.61 (H-6’)/δC 83.8 (C-2’), and δH 3.45 (H-7’)/δC 78.5 (C-4’), revealed the presence of a 2,4-di-O-methylpyranose substructure. Furthermore, an HMBC, δH 3.48 (H-6”)/δC 78.7 (C-4”), along with typical chemical shifts and coupling constants from C-1” to C-5” obtained in CD3OD (Table 2), indicated the presence of a 4-O-methylpyranose substructure. The HMBC, δH 3.64 (H-3’)/δC 103.0 (C-1”), indicated that these two sugar structures were connected through a glycosidic bond.
Table 1: NMR data for polycavernoside E (1) in CDCl3.
| position | δC, typea | δHb (J in Hz) | COSY | selected HMBC |
| 1 | 171.9, C | |||
| 2 | 35.6, CH2 | 2.29, m | 3 | 1 |
| 3 | 82.0, CH | 3.43, m | 2 | |
| 4 | 38.3, C | |||
| 5 | 85.3, CH | 3.32, m | 6a, 6b | |
| 6a | 37.7, CH2 | 1.95, m | 5, 6b, 7 | |
| 6b | 1.61, m | 5, 6a, 7 | ||
| 7 | 83.8, CH | 3.07, m | 6a, 6b, 8a, 8b | |
| 8a | 42.1, CH2 | 3.08, m | 7, 8b | 9 |
| 8b | 2.00, m | 7, 8a | ||
| 9 | 206.9, C | |||
| 10 | 103.0, C | |||
| 11 | 39.7, CH | 2.74, m | 12a, 12b, 27 | |
| 12a | 33.6, CH2 | 2.01, m | 11, 12b, 13 | |
| 12b | 1.70, m | 11, 12a | ||
| 13 | 83.5, CH | 4.18, dd (11.3, 5.0) | 12a, 12b | |
| 14 | 39.8, C | |||
| 15 | 78.4, CH | 5.17, d (8.2) | 16 | 1 |
| 16 | 127.4, CH | 5.55, dd (8.2, 15.0) | 15, 17 | |
| 17 | 135.4, CH | 6.26, m | 16, 18 | |
| 18 | 130.1, CH | 6.09, m | 17, 19 | |
| 19 | 133.9, CH | 6.13, m | 18, 20 | |
| 20 | 131.2, CH | 6.08, m | 19, 21 | |
| 21 | 134.6, CH | 5.67, dt (15.0, 7.3) | 20, 22 | |
| 22 | 31.8, CH2 | 2.19, m | 21, 23 | |
| 23 | 28.1, CH2 | 1.62, m | 22, 24 | 25 |
| 24 | 17.9, CH2 | 2.18, m | 23, 26 | 25, 26 |
| 25 | 84.6, C | |||
| 26 | 68.6, CH | 1.95, t (2.7) | 24 | |
| 27 | 13.3, CH3 | 0.99, d (6.8) | 11 | 10 |
| 28 | 17.8, CH3 | 0.85, s | 13, 14, 15, 29 | |
| 29 | 19.4, CH3 | 0.86, s | 13, 14, 15, 28 | |
| 30 | 22.2, CH3 | 0.94, s | 3, 4, 5, 31 | |
| 31 | 13.9, CH3 | 0.90, s | 3, 4, 5, 30 | |
| 32 | OH | 4.47, s | 9, 10, 11 | |
| 1’ | 106.1, CH | 4.27, d (7.7) | 2’ | 5 |
| 2’ | 83.8, CH | 3.07, m | 1’, 3’ | |
| 3’ | 79.9, CH | 3.64, m | 2’, 4’ | 1” |
| 4’ | 78.5, CH | 3.27, m | 3’, 5a’, 5b’ | |
| 5a’ | 63.2, CH2 | 4.03, dd (11.3, 5.0) | 4’, 5b’ | 1’ |
| 5b’ | 3.12, m | 4’,5a’ | ||
| 6’ | 61.1, CH3 | 3.61, s | 2’ | |
| 7’ | 58.8, CH3 | 3.45, s | 4’ | |
| 1” | 103.0, CH | 4.87, d (4.5) | 2” | |
| 2” | 71.7, CH | 3.53, m | 1”, 3” | |
| 3” | 71.0, CH | 3.75, m | 2”, 4” | |
| 4” | 78.7, CH | 3.34, m | 3”, 5a”, 5b” | |
| 5a” | 60.1, CH2 | 4.23, dd (12.2, 3.2) | 5b”, 4” | |
| 5b” | 3.46, m | 5a”, 4” | ||
| 6” | 58.1, CH3 | 3.48, s | 4” | |
| 7” | - | OH | ||
| 8” | - | OH | ||
aMeasured at 400 MHz. bMeasured at 100 MHz.
![[1860-5397-20-57-2]](/bjoc/content/figures/1860-5397-20-57-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Planar structure of polycavernoside E (1) based on 2D NMR analysis.
Figure 2: Planar structure of polycavernoside E (1) based on 2D NMR analysis.
The geometries of the two olefins at C-16 and C-20 were determined to be trans based on the large coupling constants, 3JH-16/H-17 15.0 Hz and 3JH-20/H-21 15.0 Hz, respectively. The geometry of the remaining double bond at C-18 was established to be trans by comparing the 13C NMR chemical shifts at C-16 and C-21 between 1 and polycavernoside D (5) (Table S1 in Supporting Information File 1) [5]. In addition, a 4J long-range coupling between δH 1.95 (H-26) and δH 2.18 (H-24) and three HMBC δH 1.62 (H-23)/δC 84.6 (C-25), δH 2.18 (H-24)/δC 84.6 (C-25), and δH 2.18 (H-24)/δC 68.6 (C-26) revealed a terminal alkyne structure. Additionally, COSY correlations shown in Figure 2 revealed the side chain structure of 1 containing a terminal alkyne and a conjugated trans triene (C-15 to C-26).
We then focused on the macrolide structure of 1. Six HMBC, δH 0.94 (H-30)/δC 82.0 (C-3), δH 0.94 (H-30)/δC 38.3 (C-4), δH 0.94 (H-30)/δC 85.3 (C-5), δH 0.90 (H-31)/δC 82.0 (C-3), δH 0.90 (H-31)/δC 38.3 (C-4), and δH 0.90 (H-31)/δC 85.3 (C-5), along with COSY correlations shown in Figure 2, revealed a chain structure from C-2 to C-8. In addition, eight HMBC, δH 5.17 (H-15)/δC 171.9 (C-1), δH 2.29 (H-2)/δC 171.9 (C-1), δH 0.85 (H-28)/δC 83.5 (C-13), δH 0.85 (H-28)/δC 39.8 (C-14), δH 0.85 (H-28)/δC 78.4 (C-15), δH 0.86 (H-29)/δC 83.5 (C-13), δH 0.86 (H-29)/δC 39.8 (C-14), and δH 0.86 (H-29)/δC 78.4 (C-15), and COSY correlations shown in Figure 2, clarified the connection of C-1 to C-8 and C-15 to C-11(-C27) through an ester bond. Furthermore, five HMBC, δH 0.99 (H-27)/δC 103.0 (C-10), δH 4.47 (H-32)/δC 206.9 (C-9), δH 4.47 (H-32)/δC 103.0 (C-10), δH 4.47 (H-32)/δC 39.7 (C-11), and δH 3.08 (H-8a)/δC 206.9 (C-9) connected C-11 and C-8 through a ketone carbonyl carbon at C-9 and hemiacetal carbon at C-10, revealing the 16-membered macrolide structure of 1. The HMBC, δH 4.27 (H-1’)/δC 85.3 (C-5), revealed that the disaccharide moiety was connected to C-5. Finally, considering the molecular formula of 1 and the chemical shifts of known polycavernosides, we established the presence of a THP ring containing C-3 to C-7 and a THF ring containing C-10 to C-13 in the macrolide structure. Consequently, we established the planar structure of 1, as shown in Figure 2.
The relative configuration of compound 1 was determined based on the NMR data obtained in CD3OD and CDCl3 (Table 1 and Table 2). The relative configuration of the THP ring and the disaccharide moiety of 1 was determined by analyzing the proton coupling constants and NOESY correlations (Figure 3). The two coupling constants in CD3OD, 3JH-5/H-6b (11.9 Hz) and 3JH-6b/H-7 (11.9 Hz), indicated that H-5, H-6b, and H-7 were in the axial position. The two NOESY correlations in CD3OD, δH 1.80 (H-6b)/δH 0.91 (H-31) and δH 2.36 (H-2)/δH 0.91 (H-31), revealed that H-6b, C-31, and C-2 were located in the same face of the THP ring as shown in Figure 3. Consequently, the relative configuration of the THP ring was determined to be 3S*,5S*,7S*. For the 2,4-di-O-methylpyranose moiety, the two large coupling constants in CD3OD, 3JH-1’/H-2’ (7.8 Hz) and 3JH-2’/H-3’ (9.0 Hz), indicated that H-1’, H-2’, and H-3’ were in the axial position. The two NOESY correlations in CDCl3, δH 3.45 (H-7’)/δH 4.87 (H-1”) and δH 3.45 (H-7’)/δH 4.23 (H-5a”), revealed that the methoxy group at C-4’ was in the equatorial position and H-4’ was in the axial position. The 2,4-di-O-methylpyranose moiety was identified as 2,4-di-O-methylxylose. For the 4-O-methylpyranose moiety, the two large coupling constants in CD3OD, 3JH-1”/H-2” (7.3 Hz) and 3JH-2”/H-3” (9.1 Hz), indicated that H-1”, H-2”, and H-3” were in the axial position. NOESY correlations in CDCl3, δH 3.75 (H-3”)/δH 3.48 (H-6”), revealed that the methoxy group at C-4” was in the equatorial position and H-4” was in the axial position. The 4-O-methylpyranose moiety was identified as 4-O-methylxylose. The relationship between the relative configuration of the 2,4-di-O-methylxylose moiety and 4-O-methylxylose moiety was identified using three NOESY correlations in CDCl3, δH 3.45 (H-7’)/δH 4.87 (H-1”), δH 3.45 (H-7’)/δH 4.23 (H-5a”), and δH 3.64 (H-3’)/δH 4.87 (H-1”), as shown in Figure 3. Furthermore, the relationship of the relative configuration between the disaccharide moiety and the THP ring was revealed by two NOESY correlations in CDCl3, δH 4.27 (H-1’)/δH 3.32 (H-5) and δH 4.27 (H-1’)/δH 0.94 (H-30), shown in Figure 3. The validity of the relative configurations shown in Figure 3 is further substantiated by the good agreement with the corresponding chemical shifts of polycavernoside D (5), both of which possess the same disaccharide moiety attached to a THP ring (Tables S1 and S2 in Supporting Information File 1) [5].
Table 2: NMR data for polycavernoside E (1) in CD3OD.
| position | δC, typea | δHb (J in Hz) | COSY | selected HMBC |
| 1 | 174.3, C | |||
| 2 | 36.5, CH2 | 2.36, d (7.7) | 3 | 1 |
| 3 | 83.2, CH | 3.40, m | 2 | |
| 4 | 39.4, C | |||
| 5 | 86.1, CH | 3.40, m | 6a, 6b | |
| 6a | 38.0, CH2 | 1.91, m | 5, 6b, 7 | |
| 6b | 1.80, ddd (11.9, 11.9, 11.9) | 5, 6a, 7 | ||
| 7 | 76.6, CH | 3.65, m | 6a, 6b, 8a, 8b | |
| 8a | 42.3, CH2 | 2.85, m | 7, 8b | 9 |
| 8b | 2.37, m | 7, 8a | ||
| 9 | 207.4, C | |||
| 10 | 104.8, C | |||
| 11 | 39.7, CH | 2.82, m | 12a, 12b, 27 | |
| 12a | 34.6, CH2 | 1.98, m | 11, 12b, 13 | |
| 12b | 1.62, m | 11, 12a | ||
| 13 | 83.8, CH | 4.12, dd (11.6, 4.7) | 12a, 12b | |
| 14 | 40.6, C | |||
| 15 | 80.3, CH | 5.10, d (8.1) | 16 | 1 |
| 16 | 128.5, CH | 5.61, dd (8.1, 15.1) | 15, 17 | 18 |
| 17 | 136.4, CH | 6.21, m | 16, 18 | |
| 18 | 131.1, CH | 6.13, m | 17 | |
| 19 | 135.0, CH | 6.19, m | ||
| 20 | 132.4, CH | 6.12, m | 21 | |
| 21 | 135.6, CH | 5.72, dt (15.3, 7.1) | 20, 22 | 19 |
| 22 | 32.7, CH2 | 2.22, m | 21, 23 | |
| 23 | 29.3, CH2 | 1.60, quint (7.2) | 22, 24 | 25 |
| 24 | 18.5, CH2 | 2.17, m | 23 | 25, 26 |
| 25 | 84.7, C | |||
| 26 | 69.8, CH | 2.21, m | ||
| 27 | 13.6, CH3 | 0.98, d (6.8) | 11 | 10 |
| 28 | 18.0, CH3 | 0.868, s | 13, 14, 15, 29 | |
| 29 | 19.5, CH3 | 0.870, s | 13, 14, 15, 28 | |
| 30 | 22.3, CH3 | 0.98, s | 3, 4, 5, 31 | |
| 31 | 14.1, CH3 | 0.91, s | 3, 4, 5, 30 | |
| 1’ | 107.1, CH | 4.33, d (7.8) | 2’ | 5 |
| 2’ | 85.5, CH | 3.08, dd (7.8, 9.0) | 1’, 3’ | |
| 3’ | 81.9, CH | 3.63, m | 2’, 4’ | 1” |
| 4’ | 79.4, CH | 3.27, m | 3’, 5a’, 5b’ | |
| 5a’ | 64.3, CH2 | 3.96, dd (11.5, 5.1) | 4’, 5b’ | 1’ |
| 5b’ | 3.16, m | 4’, 5a’ | ||
| 6’ | 61.3, CH3 | 3.61, s | 2’ | |
| 7’ | 59.5, CH3 | 3.46, s | 4’ | |
| 1” | 105.0, CH | 4.62, d (7.3) | 2” | |
| 2” | 75.3, CH | 3.21, dd (7.3, 9.1) | 1”, 3” | |
| 3” | 76.5, CH | 3.39, m | 2”, 4” | |
| 4” | 81.0, CH | 3.18, m | 3”, 5a”, 5b” | |
| 5a” | 64.1, CH2 | 4.07, dd (11.0, 4.4) | 5b”, 4” | |
| 5b” | 3.13, m | 5a”, 4” | ||
| 6” | 58.9, CH3 | 3.47, s | 4” | |
aMeasured at 400 MHz. bMeasured at 100 MHz.
![[1860-5397-20-57-3]](/bjoc/content/figures/1860-5397-20-57-3.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 3: Relative configuration of the THP ring and disaccharide moiety of 1.
Figure 3: Relative configuration of the THP ring and disaccharide moiety of 1.
The remaining relative configuration of 1 was determined by a comparison of the carbon chemical shifts between 1 and 5 in CDCl3 [5]. As shown in Table S1 (Supporting Information File 1), the two sets of data were in good agreement, indicating that the relative configurations of compounds 1 and 5 were identical.
To reveal the absolute configuration, we recorded the ECD spectrum of 1 (Figure 4) and compared it with those of 2 and 5 reported in previous papers [5,6]. We detected a Cotton effect of negative sign at around 280 nm corresponding to the n–π* transition of a ketone group as same as the literature data for 2 and 5. As a result, the absolute configuration of polycavernoside E was determined to be 1.
![[1860-5397-20-57-4]](/bjoc/content/figures/1860-5397-20-57-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Next, we examined the antitrypanosomal activity of 1 against the bloodstream form of Trypanosoma brucei rhodesiense IL-1501 (the causative organism of African trypanosomiasis) (Table 3). As a result, 1 showed moderate growth-inhibitory activities against Trypanosoma brucei rhodesiense (IC50: 9.9 μM). In addition, we examined the growth-inhibitory activity of 1 on normal human fibroblasts WI-38 (Table 3). In summary, 1 showed a moderate activity against Trypanosoma brucei rhodesiense.
Conclusion
In conclusion, we isolated a new polycavernoside analog, named polycavernoside E (1), from a marine Okeania sp. cyanobacterium. The relative configuration was elucidated mainly by analyzing the 2D NMR data. The absolute configuration was determined based on a comparison of the ECD data for 1 and its known analogs. Polycavernoside E (1) showed selective antitrypanosomal activity against Trypanosoma brucei rhodesiense with an IC50 value of 9.9 μM. This discovery provides additional evidence that polycavernosides, previously thought to be derived from red algae, are produced by the marine Okeania sp. cyanobacterium. In the field, this type of cyanobacterium that produces the analog of human lethal toxin is often observed with macroalgae and shells, and, therefore, it can be a potential risk for food poisoning of fishery resources.
Experimental
General experimental procedures
Optical rotations were measured with a JASCO DIP-1000 polarimeter. UV spectra were recorded on a UV-3600. ECD spectra were measured with JASCO J-1100. IR spectra were recorded on a Bruker ALPHA instrument. All NMR data were recorded on a JEOL ECX-400/ECS-400 spectrometer for 1H (400 MHz) and 13C (100 MHz). 1H NMR chemical shifts (referenced to the residual solvent signal of CHD2OD: δ 3.31, CHCl3: δ 7.26) were assigned using a combination of data from COSY and HMQC experiments. Similarly, 13C NMR chemical shifts (referenced to the solvent signal CD3OD: δ 49.0, CDCl3: δ 77.16) were assigned based on HMBC and HMQC experiments. HRESIMS spectra were obtained on a Bruker timsTOF mass spectrometer. For reversed-phase column chromatography, ODS silica gel Cosmosil 75C18-OPN (Nacalai Tesque) was used. For medium pressure column chromatography, AFCS (Smart Flash AKROS, Yamazen) consisting of a pump and a UV detector was used. HPLC analysis was conducted using a pump (model PU-2080, Jasco) and a UV detector (model UV-2075, Jasco). All chemicals and solvents used in this study were the best grade available and obtained from a commercial source (Nacalai Tesque).
Collection and identification of the sample
The marine cyanobacterium producing polycavernoside E (1) was collected in March 2022 at the coast in Akuna beach, Yonashiromiyagi, Uruma city, Okinawa, Japan. It was classified into Okeania sp. based on the phylogenetic analysis as described in the previous paper (accession no. LC771053) [7].
Isolation of polycavernoside E (1)
In a manner analogous to [7], the collected cyanobacterium (340 g) was extracted with EtOH (0.5 L) for 10 days at room temperature (rt). The extract was filtered, and the residue was homogenized with a blender and re-extracted with EtOH (0.5 L) at room temperature for one day. The extract was filtered, and the combined filtrates were concentrated. The residue was partitioned between EtOAc (3 × 300 mL) and H2O (300 mL). The combined organic layers were concentrated, and the residue was partitioned between 90% aqueous MeOH (300 mL) and hexane (3 × 300 mL). The aqueous MeOH layer was concentrated, and the obtained residue (673 mg) was separated by column chromatography on ODS (7 g) eluted with 35 mL of 40%, 60%, 80%, and 90% aqueous MeOH, followed by 35 mL of MeOH and 70 mL of CHCl3/MeOH 1:1. The fraction (244.4 mg) eluted with 80% MeOH was subjected to AFCS [Ø 11 × 300 mm; flow rate 5 mL/min; detection at 254 nm; solvent gradient condition, hexane/EtOAc 28:72 → 7:93] to give a fraction that contained compound 1 (17.5 mg, tR = 32.0 min). The fraction that contained 1 was further purified by HPLC [Cosmosil 5C18-MS-II (Ø 20 mm × 250 mm); solvent MeOH/H2O 85:15; flow rate 5 mL/min; detection UV 215 nm] to give a fraction that contained 1 (8.6 mg, last collected fraction). The fraction that contained 1 was further separated by HPLC [Cosmosil Cholester (Ø 20 mm × 250 mm); solvent MeCN/H2O 75:25; flow rate 5 mL/min; detection UV 254 nm] to give a fraction that contained 1 (0.6 mg, tR = 32.8 min). The fraction that contained 1 was further separated by HPLC [Cosmosil 5PE-MS (Ø 20 mm × 250 mm); solvent MeOH/H2O 85:15; flow rate 5 mL/min; detection UV 270 nm] to give 1 (0.5 mg, tR = 37.3 min).
Polycavernoside E (1): colorless oil; [α]D26 −19 (c 0.04, MeOH); UV (MeOH) λmax, nm (log ε): 281 (2.27), 270 (2.96), 260 (2.40); ECD (100 μg/mL; MeOH), λmax, nm (Δε): 226 (−0.31), 274 (−0.76), 282 (−0.96); IR (neat): 3443, 2965, 2925, 2896, 1646, 1457, 1086 cm−1; HRESIMS (m/z): [M + Na]+ calcd for C44H66O15Na+, 857.4294; found, 857.4294.
In vitro antitrypansomal assay
The bloodstream form of Trypanosoma brucei rhodesiense strain IL-1501 was cultured at 37 °C under a humidified 5% CO2 atmosphere in HMI-9 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) [8,9]. For in vitro studies, compounds were dissolved in DMSO and diluted in culture medium prior to being assayed. The maximum DMSO concentration in the in vitro assays was 1%. The compounds were tested in an AlamarBlue serial drug dilution assay to determine the 50% inhibitory concentrations (IC50) [10]. Serial drug dilutions were prepared in 96-well microtiter plates, containing 50 μL of culture medium. Subsequently, 50 μL of a parasite suspension with a concentration of 4.0 × 104 cells/mL was introduced into each well. Cultures were incubated for 69 h at 37 °C under a humidified 5% CO2 atmosphere. After this time, 10 μL of resazurin (12.5 mg resazurin (Sigma) dissolved in 100 mL phosphate-buffered saline) was added to each well. The plates were incubated for an additional 3 h. The plates were read in a SpectraMax Gemini XS microplate fluorescence scanner (Molecular Devices) using an excitation wavelength of 536 nm and an emission wavelength of 588 nm.
WI-38 cells assay
In a manner analogous to [7], WI-38 cells were cultured at 37 °C with 5% CO2 in DMEM (Nissui) supplemented with 10% heat-inactivated FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, 0.25 μg/mL amphotericin, 300 μg/mL ʟ-glutamine, and 2.25 mg/mL NaHCO3. Cells were seeded at 4 × 103 cells/well in 96-well plates (Iwaki) and cultured overnight. Various concentrations of compounds were then added, and cells were incubated for 72 h. Cell proliferation was measured by the MTT assay.
Supporting Information
| Supporting Information File 1: NMR data for polycavernoside E (1). | ||
| Format: PDF | Size: 3.0 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Yotsu-Yamashita, M.; Haddock, R. L.; Yasumoto, T. J. Am. Chem. Soc. 1993, 115, 1147–1148. doi:10.1021/ja00056a048
Return to citation in text: [1] [2] -
Yotsu-Yamashita, M.; Yasumoto, T.; Yamada, S.; Bajarias, F. F. A.; Formeloza, M. A.; Romero, M. L.; Fukuyo, Y. Chem. Res. Toxicol. 2004, 17, 1265–1271. doi:10.1021/tx0498556
Return to citation in text: [1] -
Yotsu-Yamashita, M.; Seki, T.; Paul, V. J.; Naoki, H.; Yasumoto, T. Tetrahedron Lett. 1995, 36, 5563–5566. doi:10.1016/0040-4039(95)01052-j
Return to citation in text: [1] [2] -
Yotsu-Yamashita, M.; Abe, K.; Seki, T.; Fujiwara, K.; Yasumoto, T. Tetrahedron Lett. 2007, 48, 2255–2259. doi:10.1016/j.tetlet.2007.02.003
Return to citation in text: [1] [2] -
Navarro, G.; Cummings, M. E.; Lee, J.; Moss, N.; Glukhov, E.; Valeriote, F. A.; Gerwick, L.; Gerwick, W. H. Environ. Sci. Technol. Lett. 2015, 2, 166–170. doi:10.1021/acs.estlett.5b00116
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Blakemore, P. R.; Browder, C. C.; Hong, J.; Lincoln, C. M.; Nagornyy, P. A.; Robarge, L. A.; Wardrop, D. J.; White, J. D. J. Org. Chem. 2005, 70, 5449–5460. doi:10.1021/jo0503862
Return to citation in text: [1] -
Umeda, K.; Iwasaki, A.; Taguchi, R.; Kurisawa, N.; Jeelani, G.; Nozaki, T.; Suenaga, K. J. Nat. Prod. 2023, 86, 2529–2538. doi:10.1021/acs.jnatprod.3c00742
Return to citation in text: [1] [2] [3] -
Kuboki, N.; Inoue, N.; Sakurai, T.; Di Cello, F.; Grab, D. J.; Suzuki, H.; Sugimoto, C.; Igarashi, I. J. Clin. Microbiol. 2003, 41, 5517–5524. doi:10.1128/jcm.41.12.5517-5524.2003
Return to citation in text: [1] -
Räz, B.; Iten, M.; Grether-Bühler, Y.; Kaminsky, R.; Brun, R. Acta Trop. 1997, 68, 139–147. doi:10.1016/s0001-706x(97)00079-x
Return to citation in text: [1] -
Huber, W.; Koella, J. C. Acta Trop. 1993, 55, 257–261. doi:10.1016/0001-706x(93)90083-n
Return to citation in text: [1]
| 1. | Yotsu-Yamashita, M.; Haddock, R. L.; Yasumoto, T. J. Am. Chem. Soc. 1993, 115, 1147–1148. doi:10.1021/ja00056a048 |
| 1. | Yotsu-Yamashita, M.; Haddock, R. L.; Yasumoto, T. J. Am. Chem. Soc. 1993, 115, 1147–1148. doi:10.1021/ja00056a048 |
| 3. | Yotsu-Yamashita, M.; Seki, T.; Paul, V. J.; Naoki, H.; Yasumoto, T. Tetrahedron Lett. 1995, 36, 5563–5566. doi:10.1016/0040-4039(95)01052-j |
| 4. | Yotsu-Yamashita, M.; Abe, K.; Seki, T.; Fujiwara, K.; Yasumoto, T. Tetrahedron Lett. 2007, 48, 2255–2259. doi:10.1016/j.tetlet.2007.02.003 |
| 5. | Navarro, G.; Cummings, M. E.; Lee, J.; Moss, N.; Glukhov, E.; Valeriote, F. A.; Gerwick, L.; Gerwick, W. H. Environ. Sci. Technol. Lett. 2015, 2, 166–170. doi:10.1021/acs.estlett.5b00116 |
| 5. | Navarro, G.; Cummings, M. E.; Lee, J.; Moss, N.; Glukhov, E.; Valeriote, F. A.; Gerwick, L.; Gerwick, W. H. Environ. Sci. Technol. Lett. 2015, 2, 166–170. doi:10.1021/acs.estlett.5b00116 |
| 3. | Yotsu-Yamashita, M.; Seki, T.; Paul, V. J.; Naoki, H.; Yasumoto, T. Tetrahedron Lett. 1995, 36, 5563–5566. doi:10.1016/0040-4039(95)01052-j |
| 4. | Yotsu-Yamashita, M.; Abe, K.; Seki, T.; Fujiwara, K.; Yasumoto, T. Tetrahedron Lett. 2007, 48, 2255–2259. doi:10.1016/j.tetlet.2007.02.003 |
| 10. | Huber, W.; Koella, J. C. Acta Trop. 1993, 55, 257–261. doi:10.1016/0001-706x(93)90083-n |
| 2. | Yotsu-Yamashita, M.; Yasumoto, T.; Yamada, S.; Bajarias, F. F. A.; Formeloza, M. A.; Romero, M. L.; Fukuyo, Y. Chem. Res. Toxicol. 2004, 17, 1265–1271. doi:10.1021/tx0498556 |
| 7. | Umeda, K.; Iwasaki, A.; Taguchi, R.; Kurisawa, N.; Jeelani, G.; Nozaki, T.; Suenaga, K. J. Nat. Prod. 2023, 86, 2529–2538. doi:10.1021/acs.jnatprod.3c00742 |
| 5. | Navarro, G.; Cummings, M. E.; Lee, J.; Moss, N.; Glukhov, E.; Valeriote, F. A.; Gerwick, L.; Gerwick, W. H. Environ. Sci. Technol. Lett. 2015, 2, 166–170. doi:10.1021/acs.estlett.5b00116 |
| 6. | Blakemore, P. R.; Browder, C. C.; Hong, J.; Lincoln, C. M.; Nagornyy, P. A.; Robarge, L. A.; Wardrop, D. J.; White, J. D. J. Org. Chem. 2005, 70, 5449–5460. doi:10.1021/jo0503862 |
| 7. | Umeda, K.; Iwasaki, A.; Taguchi, R.; Kurisawa, N.; Jeelani, G.; Nozaki, T.; Suenaga, K. J. Nat. Prod. 2023, 86, 2529–2538. doi:10.1021/acs.jnatprod.3c00742 |
| 5. | Navarro, G.; Cummings, M. E.; Lee, J.; Moss, N.; Glukhov, E.; Valeriote, F. A.; Gerwick, L.; Gerwick, W. H. Environ. Sci. Technol. Lett. 2015, 2, 166–170. doi:10.1021/acs.estlett.5b00116 |
| 8. | Kuboki, N.; Inoue, N.; Sakurai, T.; Di Cello, F.; Grab, D. J.; Suzuki, H.; Sugimoto, C.; Igarashi, I. J. Clin. Microbiol. 2003, 41, 5517–5524. doi:10.1128/jcm.41.12.5517-5524.2003 |
| 9. | Räz, B.; Iten, M.; Grether-Bühler, Y.; Kaminsky, R.; Brun, R. Acta Trop. 1997, 68, 139–147. doi:10.1016/s0001-706x(97)00079-x |
| 5. | Navarro, G.; Cummings, M. E.; Lee, J.; Moss, N.; Glukhov, E.; Valeriote, F. A.; Gerwick, L.; Gerwick, W. H. Environ. Sci. Technol. Lett. 2015, 2, 166–170. doi:10.1021/acs.estlett.5b00116 |
| 5. | Navarro, G.; Cummings, M. E.; Lee, J.; Moss, N.; Glukhov, E.; Valeriote, F. A.; Gerwick, L.; Gerwick, W. H. Environ. Sci. Technol. Lett. 2015, 2, 166–170. doi:10.1021/acs.estlett.5b00116 |
| 7. | Umeda, K.; Iwasaki, A.; Taguchi, R.; Kurisawa, N.; Jeelani, G.; Nozaki, T.; Suenaga, K. J. Nat. Prod. 2023, 86, 2529–2538. doi:10.1021/acs.jnatprod.3c00742 |
© 2024 Umeda et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.