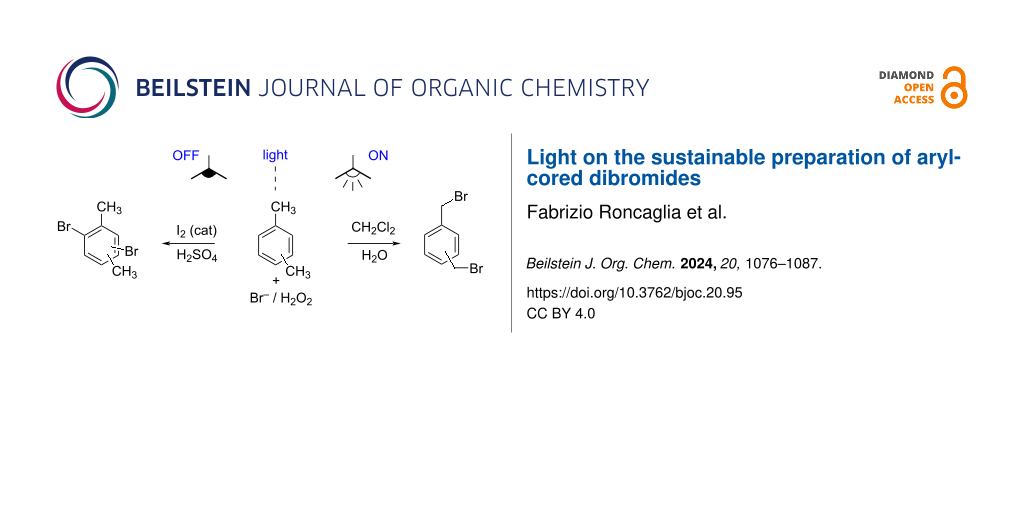
Abstract
Both aryl and benzyl polybromides have gained significant importance as reactive building blocks in polymer and materials chemistry. Their preparation primarily relies on established synthetic methods using molecular bromine or N-bromosuccinimide, known for their reliability and effectiveness. However, from a sustainability perspective, these methods suffer from the generation of stoichiometric amounts of byproducts and often encounter selectivity troubles. To mitigate these issues, we extended the greener peroxide-bromide halogenation method, initially developed for monobromides, to afford aryl-cored polybromides in high yields. The same method can be employed in two variants modulated by light irradiation. This external switch can be used to selectively trigger side-chain or core halogenation.

Graphical Abstract
Introduction
Activation through halogens has become a key strategy in achieving desired reactivity. Substitution of a hydrogen atom with a halogen atom within an organic skeleton significantly increases the electrophilicity of the linked carbon centre, enhancing concerted (SN2) as well as carbenium ion-mediated (SN1) substitutions, common – for instance – on benzylic positions. Useful alkenyl functional groups can also be obtained by means of elimination processes, if proper alkyl side chains are present. Additional opportunities offered by C(sp3)–Hal bonds arise from the ease of their homolytic cleavage, leading to the formation of reactive carbon-centred radicals. C(sp2)–Hal bonds of aryl halides also exhibit high reactivity, particularly towards transition-metal-mediated cross-coupling processes or Ar-SN reactions.
Benzyl and aryl halides, collectively referred to as 'aryl-cored halides', have found extensive applications across various fields, including synthesis [1], photonics [2], and diagnostics [3]. Their popularity stems from their structural rigidity, potential conjugation with the remaining structure, and the capability to form additional π–π-stacking interactions. Dihalides, in particular, are highly preferred in such applications, enabling the formation of molecular wires or extended structures – either linear or cyclic – with enhanced functionality. Consequently, they play a crucial role in the fabrication of polymers [4-7], cyclophanes [8], photoactive materials [9-11], membranes [12], and other architectures.
The synthetic potential of aryl-cored halides can be broadened by converting C–Hal functions into different functional groups. For example, aldehyde and amine functionalities can be readily derived from C(sp3)–Hal functions through hydrolysis–oxidation [13] or substitution [14], respectively. This is of significant interest in the context of covalent organic frameworks (COFs) and metal-organic frameworks (MOFs), frequently assembled through imine linkages.
While C–H activation through halogens presents clear technical advantages, it also brings forth concerns about the toxicity of halo compounds to both human health and the environment [15]. Nevertheless, in line with the European Union's 'green new deal' guidelines [16], addressing two pivotal issues could facilitate the environmentally conscious utilisation of halogenated compounds as intermediates in chemical processes:
- The development of more sustainable production methods for halo compounds, potentially involving the use of eco-friendly halogenation reagents.
- The development of methods for the efficient removal and recycle of halogens, advocating principles of a circular economy.
It is noteworthy that both aspects are influenced by the type of halogen employed. When considering the most atom-economical options, namely chlorine and bromine, the latter typically exhibits some advantages over the former. These include: (i) better regioselectivity in radical processes, attributed to the lower bond enthalpy of H–Br (88 kcal/mol) compared to H–Cl (103 kcal/mol) [17], (ii) greater electrophilicity of the halo compound due to better leaving group ability of the halide ion, (iii) reduced toxicity, presumably due to faster hydrolysis [18], and (iv) easier oxidation of the halide to molecular halogen (E0 = 1.087 V (SHE) for Br2/Br−; E0 = 1.358 V (SHE) for Cl2/Cl−) [19] resulting in easier recyclability.
Light irradiation often significantly influences the selectivity of halogenation processes. Photolytic cleavage of molecular halogens gives rise to radicals that are known to favour benzylic functionalisation [17]. Conversely, the same molecular halogens exhibit prominent functionalisation on the aromatic ring when used in the dark [20]. A classic example is the bromination of toluene with molecular bromine. When the system is exposed to light (right side of Figure 1), a radical mechanism is initiated by Br• coming from Br2 homolysis. Propagation involves the reversible abstraction of a benzylic hydrogen atom from the substrate by Br•, to give HBr and a structure-stabilised carbon-centred radical, which may react with Br2 to give the brominated product, thus regenerating Br• that is able to sustain the chain process. In the absence of light (left side of Figure 1), the reaction follows a different mechanism, producing the ortho and para-bromoarenes through Ar-SE, that involves cationic intermediates. In this case, a catalytic amount of iodine [21,22] or FeCl3 [23] is added to enhance the electrophilicity of bromine.
![[1860-5397-20-95-1]](/bjoc/content/figures/1860-5397-20-95-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Comparison between the light-initiated radical halogenation of toluene (right), and the Ar-SE bromination in the dark (left).
Figure 1: Comparison between the light-initiated radical halogenation of toluene (right), and the Ar-SE bromi...
While widely employed and capable of producing reliable results on various substrates, the direct use of molecular bromine poses sustainability challenges. Its innate reactivity requires stringent safety protocols during transportation, storage, and handling [24]. A related concern involves the stability of the diluting media, often requiring the use of toxic CCl4 to prevent undesired solvent degradation. Furthermore, the direct use of Br2, even if used in low concentration, exhibits limited selectivity towards benzylic bromination, primarily due to co-bromination occurring on the aromatic ring. This side process produces awkward halogenated byproducts that can complicate product separation, and will require disposal. Lastly, only one half of the halogen load is incorporated into the product, the other half being lost as bromide ion.
A solution for some of the aforementioned problems is the in situ generation of bromine: firstly, the handling of Br2 is no longer an issue, since it is formed inside the reaction vessel, secondly this approach allows enhanced selectivity of the bromination, as the amount and timing of the chemical generation can be modulated. In addition, in situ regeneration of bromine from bromide byproducts improves the atom economy of the overall process. A pertinent example of this technique is represented by the Wohl–Ziegler halogenation protocol, which is based on the stable and easily handled N-bromosuccinimide (NBS), which is able to slowly deliver molecular bromine through its interaction with bromide ions [17,25]. This method found prevalent application in the bromination of side-chain positions (right side of Figure 2) [26,27]. However, the addition of molecular iodine in catalytic amounts makes it suitable for aromatic bromination “in the dark” (left side of Figure 2). This gives rise to a radical-initiated Ar-SE mechanism, which is reported to proceed through the generation of a mixed molecular halogen [28]. As an alternative to iodine, the trityl cation [29] is reported too. Additional benefits of the method include its ability to work in neutral conditions, and the potential quantitative incorporation of the halogen into the product (no bromide byproducts are generated).
![[1860-5397-20-95-2]](/bjoc/content/figures/1860-5397-20-95-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Toluene halogenation mediated by NBS in absence (left) or exposed to light (right).
Figure 2: Toluene halogenation mediated by NBS in absence (left) or exposed to light (right).
Unfortunately, the safety and selectivity advantages of using NBS are counterbalanced by other issues. Specifically, light irradiation is often ineffective as an initiator for NBS [26,30], introducing the necessity to add a radical initiator, such as azobisisobutyronitrile (AIBN) [31-34] or benzoyl peroxide [35,36]; substances affected by safety concerns on transportation, storage, and use. Moreover, the regeneration of the halogen comes at a cost: the release of stoichiometric amounts of succinimide, whose recovery requires additional chemical operations, and reconversion to NBS is typically accomplished with stoichiometric Br2 [37,38]. Recently, certain bromine carriers, potentially offering improvements over NBS, have been proposed [24], demonstrating notable success in specific halogenation reactions. Nevertheless, additional studies are required to broaden the scope of most alternatives and to evaluate options for carrier recovery/regeneration.
Regardless of the employed method, the concept of (re-)generating molecular bromine from “spent” bromide ions is a crucial idea to improve the atom economy of the process. Therefore, various forms of oxidative halogenations have been reported over the years. For example, Ishii described the use of the NaBrO3–NaHSO3 couple to slowly generate Br2 through an aqueous redox equilibrium [39]. Few years later, Adimurthy et al. proposed an improved variant based on the redox couple NaBr–NaBrO3 in acidic media [40,41]. Other variations include the system KBr–Oxone® [42].
However, based on a literature review, we concluded that unparalleled efficiency and sustainability can be achieved through the well-established redox equilibria between hydrogen peroxide and halide ion in aqueous acidic media (Equation 1) [43]. Its practical implementation is surprisingly simple as standard aqueous HBr and H2O2 solutions are effective. The generated bromine can be involved in different chemical mechanisms, such as an Ar-SE (Equation 2) [44] or a radical substitution on activated positions (Equation 3). In the context of the radical process, it is noteworthy that a standard household LED lamp can serve as an efficient initiator, capable of triggering bromine photolysis (Equation 3, top) [45]. Hence, no oxidant-derived residues apart from water are formed and, at the end of the process, the residual halogen (if any) can decompose hydrogen peroxide into molecular oxygen (Equation 4) [45].
![[1860-5397-20-95-i1]](/bjoc/content/inline/1860-5397-20-95-i1.svg?max-width=590&scale=1.18182)
![[1860-5397-20-95-i2]](/bjoc/content/inline/1860-5397-20-95-i2.svg?max-width=590&scale=1.18182)
![[1860-5397-20-95-i3]](/bjoc/content/inline/1860-5397-20-95-i3.svg?max-width=590&scale=1.18182)
![[1860-5397-20-95-i4]](/bjoc/content/inline/1860-5397-20-95-i4.svg?max-width=590&scale=1.18182)
Over the years, various peroxide-bromide processes have been developed based on this chemistry. In particular, significant attention has been given to the preparation of benzyl (mono)bromides [46-49], which can be produced with high efficiency. The peroxide-bromide method appears to be especially suitable for obtaining aryl-cored dibromides, too. However, to the best of our knowledge, this option has been scarcely exploited, except for a few reports on some activated arenes [50,51].
Based on a SciFinder® survey, we came to realise that a small number of aryl-cored dibromides, which can be derived from xylenes (1, 2, 3) or mesitylene (4, Figure 3), play a predominant role as building blocks for the construction of functional materials and/or polymeric architectures. Some aryl/benzyl polybromides are also well known. Looking at this subject, the number of found reaction hits (role = reagent) for the structure “as drawn” provides a quantitative measurement of the synthetic usefulness of these halides.
![[1860-5397-20-95-3]](/bjoc/content/figures/1860-5397-20-95-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Scifinder® reaction hits for the structure “as drawn” (January 2024).
Figure 3: Scifinder® reaction hits for the structure “as drawn” (January 2024).
This work focuses on the application of the peroxide-bromide method to the synthesis of aryl-cored polybromides obtainable from xylenes or mesitylene. The selected targets are those of heightened interest to the scientific community, identified as those with a score of at least 100 reaction hits in the Scifinder® search (Figure 3).
Results and Discussion
As p- and o-xylenes represent a primary source for aryl-cored halides, our investigation started with the conversion of 3 into 3a, taking as “zero-point” conditions those reported for monobromides [48,49], with the only change of a double amount of brominating agent (HBr·2H2O2). A household white LED lamp was used as the activator, and hydrogen peroxide was slowly added, by means of a syringe pump. As the bromine colour generated after each H2O2 drop disappeared quickly, the reaction time was reduced from 24 h to 8 h. In these conditions, the process exhibited complete conversion, but strong selectivity in favour of the monohalide 3am (entry 1, Table 1).
Table 1: Light-mediated bromination of p-xylene with in situ-generated Br2.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-95-i5.svg?max-width=637&scale=1.0)
|
|||||||
| entry | H2O2 dropping time (h) | post- dropping time (h) | H2O (mL) | CH2Cl2 (mL) | 3am (%) | 3a (%) | 3at (%) |
| 1 | 8.0 | – | 4.0 | – | 78 | 22 | traces |
| 2 | 8.0 | – | 1.0 | – | 18 | 26 | 56 |
| 3 | 3.0 | 1.0 | 1.0 | – | 63 | 37 | 1 |
| 4 | 3.0 | 1.0 | 2.0 | – | 52 | 46 | 2 |
| 5 | 3.0 | 1.0 | 2.0 | 1.0 | 20 | 74 | 6 |
| 6 | 3.0 | 1.0 | 2.0 | 0.5 | 21 | 74 | 5 |
| 7 | 2.0 | 1.5 | 2.0 | 0.5 | 9 | 86 | 5 |
| 8 | 2.0 | 1.5 | 1.5 | 1.0 | 1 | 90 | 9 |
aReaction conditions: p-xylene (531 mg, 5 mmol), H2O (see Table), HBr (48 wt % aqueous solution, 1.25 mL, 11 mmol), white LED lamp, H2O2 (35 wt % aqueous solution, 2.00 mL, 23.3 mmol) slowly dropped by a syringe pump (see Table), CH2Cl2 (see Table). Yields are reported as molar % obtained from 1H NMR spectra.
The reduction of the volume of the aqueous phase (Table 1, entry 2) resulted in a small shift towards the desired product 3a but also in the predominant overreaction towards tribromide 3at. Formation of 3at was substantially controlled by reducing the reaction time (4 h overall, entries 3 and 4 in Table 1). In these last experiments a whitish solid appeared during addition of the oxidant, initially observed in the one with lower water amount (Table 1, entry 3). The 1H NMR confirmation of the nature of the solid as 3a, makes clear that phase separation was an important issue, resulting in a limitation of reagents’ diffusion. The addition of a small amount of CH2Cl2 was thus considered (Table 1, entries 5 and 6) and resulted in a substantial selectivity improvement. The achievement of a better homogeneity, let it possible to further reduce the reaction time (Table 1, entry 7), giving total conversion and excellent selectivity toward 3a. Water is an essential constituent of the system, as its polar nature favours the extraction of HBr from the organic phase, resulting in a useful shift of the reversible hydrogen abstraction from the substrate (Figure 1 and Equation 3, middle) [45]. This two-phase system is likely to benefit from efficient stirring as well as from a proper volume ratio between aqueous and organic phases, implemented in entry 8, which resulted in further enhancement of selectivity toward 3a.
Different wavelengths were also evaluated, through the use of different types of lamps, particularly those with partial UV emission. In a specific implementation, in consideration of the glass filter’s capabilities, some UV emitting diodes were inserted inside the vessel. Anyway, no significant modulation was observed, probably due to the negligible adsorption of Br2 at wavelengths shorter than 380 nm [52].
We finally evaluated a variation in the H2O2/HBr molar ratio to determine the optimal amount of H2O2, typically used in a twofold molar ratio relative to HBr [49]. These experiments raised on the observation of gas evolution (presumably O2) starting when the added moles of H2O2 reached that of HBr. In order to confirm that this phenomenon was not attributable exclusively to the undesired peroxide decomposition described in Equation 4, we repeated the same reaction on p-xylene as in entry 8 of Table 1, but with different H2O2/HBr molar ratios. Our data confirmed that using reduced amounts of peroxide (in relation to H2O2/HBr = 2.0) leads to proportionally reduced conversion with remaining unreacted 3 (Supporting Information File 1, Table S1, entries 1, 2 and 3). The reason can be ascribed to the partial physiological peroxide decomposition due to the presence of both bromine and bromide in acid media [43]. Besides, an increased amount of peroxide did not show any noticeable benefits in selectivity toward 3a, but it led to increased formation of over-halogenated products (Table S1, entries 4 and 5 in Supporting Information File 1).
The developed method was finally successfully scaled up to 10 g scale, giving 3a in excellent isolated yield (87%), with only small amounts of polybrominated byproducts as contaminants (<2%). The pure product was obtained through (unoptimised) crystallisation from toluene, giving 3a in 80% overall yield, and undetectable 1H NMR impurities.
The application of the same protocol on o-xylene (1, Figure 3) cleanly gave dibromide 1a in almost quantitative yield. Some issues emerged during the isolation step, because of the high lacrimatory activity of 1a [53,54], while 3a lacks the same effect.
Higher nucleophilic aromatic cores are affected by minor, yet inevitable, bromination on the ring. This is the case of dialkyl-substituted arenes having o,p-activated positions (ortho with respect to one alkyl substituent and para with respect to another alkyl substituent). For instance, bromination of m-xylene (2, Figure 3) resulted in lower selectivity for the benzyl α,α’-dibromide (2a) compared to 1a or 3a, due to the partial ring bromination [55]. The structure of mesitylene (4), featuring three o,p-activated positions, makes the selective halogenation of benzylic positions even more challenging. The conventional bromination with NBS in CCl4 yields no more than 30% of the desired 1,3,5-tris(bromomethyl)benzene (4a) [56], due to the concurrent ring bromination [57]. A modified method working in refluxing benzene and benzoyl peroxide initiator was claimed to provide a clean conversion to 4a with very high yields [36,58]. However, in our hands, this procedure resulted in significant amounts of ring bromination too. Using the peroxide-bromide conditions developed for p-xylene, 4a was obtained with a 42% yield. In this case, two chromatographic separations in sequence were necessary to remove most of the core brominated byproducts, that were still detected in small amounts through 1H NMR (see the image in the Supporting Information File 1).
Ring bromination of xylenes is commonly carried out using molecular bromine in the absence of light, along with the aforementioned catalytic promoters (FeBr3 or I2) [59,60]. Some reports of ring bromination by means of NBS or the peroxide-bromide method are also known, but mainly for highly activated arenes (phenols or anisoles, for instance) or for the synthesis of monobromides [49,50,61]. Continuing our investigation on the subject, the ring bromination of p-xylene (3, Figure 3) towards 3b was considered. The starting conditions were taken from procedures established for monobromides [49,62], adapting the molar amount of the brominating agent (HBr·2H2O2) and including a small amount of CH2Cl2, according to the previous discussion. A catalytic amount of iodine (≈1 mol %) was also included. Under these conditions, a first assessment of the reaction time was performed (entries 1, 2, 3 of Table 2), resulting in the predominant ring monobromination, even after 48 h. Considering the continuous conversion of HBr into Br2 by means of the peroxide, we speculated on the decreasing acidity throughout the reaction progress. Since acidity was claimed to have a crucial role [63,64], we contemplated the addition of a small amount of sulphuric acid (Table 2, entry 4).
Table 2: p-Xylene bromination with in situ-generated Br2 in absence of light.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-95-i6.svg?max-width=637&scale=1.0)
|
||||||
| entry | CH2Cl2 (mL) | T (°C) | t (h) | H2SO4 (mmol) | 3bm (%) | 3b (%) |
| 1 | 1.5 | rt | 16 | – | 90 | 10 |
| 2 | 1.5 | rt | 24 | – | 75 | 25 |
| 3 | 1.5 | rt | 48 | – | 73 | 27 |
| 4 | 1.5 | rt | 48 | 0.5 | 50 | 50 |
| 5 | 1.5 | rt | 168 | 0.5 | 43 | 57 |
| 6 | 1.5 | 60 | 72 | 0.5 | 40 | 60 |
| 7 | – | 60 | 72 | 0.5 | 78 | 22 |
| 8b | 1.5 | 60 | 48 | 11.5 | 14 | 86 |
| 9b,c | 1.5 | 60 | 48 | 11.5 | 0 | 100 |
aReaction conditions: 3 (531 mg, 0.62 mL, 5 mmol), I2 (15 mg, 0.07 mmol), HBr (48 wt % aqueous solution, 1.25 mL, 11 mmol), H2SO4 (98 wt %, see Table), H2O2 (35 wt % aqueous solution, 2.00 mL, 20.6 mmol) dropped in 2 h by a syringe pump, CH2Cl2 (see Table). Selectivity is reported as molar % obtained from 1H NMR spectra. bNaBr (11 mmol) and H2SO4 (98 wt %, 11.5 mmol) instead of HBr (48 wt % aqueous solution). cChanged reagent order: NaBr (11 mmol) dissolved in H2O2 (35 wt % aqueous solution, 2.00 mL, 20.6 mmol) dropped in 15 min to a mixture of 3 (5 mmol, solution in CH2Cl2 (1.5 mL) and H2SO4 (98 wt %, 11.5 mmol).
This adjustment led to a significant enhancement towards 3b, resulting in a respectable 57 mol % selectivity, after prolonged reaction time (Table 2, entry 5). Furthermore, a slight improvement was observed at higher temperatures (Table 2, entry 6), while the removal of CH2Cl2, possibly limiting the temperature inside the flask, had a negative impact (entry 7). Nevertheless, yields and selectivity seemed to plateau around 60%.
Data available in the literature suggested a beneficial effect coming from the reduction of the water amount [62]. As all the existing water originates from the employed reagents like aqueous HBr and H2O2, the combination of NaBr and H2SO4 was explored as a potential source of anhydrous HBr. A notable increase in selectivity towards 3b was suddenly observed (Table 2, entry 8). However, this method led to the undesired, uncontrolled generation of Br2 when neat H2SO4 was mixed with NaBr. This issue was addressed through an alternative reagent introduction scheme, where NaBr was dissolved in aqueous hydrogen peroxide and was gradually added to the reaction mixture, containing the remaining chemicals. The addition time of this aqueous reagent was also shortened to 15 minutes, to counteract the slow decomposition of H2O2 caused by NaBr. This modification ultimately resulted in nearly complete conversion of 3 into 3b (Table 2, entry 9).
The method was successfully scaled up to 10 g of substrate, giving 1,4-dibromo-2,5-dimethylbenzene (3b) in quantitative isolated yield. The method was promptly extended to the other xylenes and to mesitylene, giving bromides 1b, 2b, and 4b in excellent isolated yields.
An evaluation of the E factors involved in the here developed benzylic bromination towards 3a (Figure S1 in Supporting Information File 1) and ring bromination towards 1b (Figure S2) was done, based on the 10-gram scale procedures (see Supporting Information File 1). The obtained values, both inferior to 4.5 (i.e. less than 4.5 kg of wastes to get 1 kg of product), denotes a good sustainability. Moreover, more than 2 kg of wastes come from losses in the recycle of CH2Cl2. Therefore, these results can be easily improved by increasing the efficiency of solvent recovery, here set at 85%.
The aryl-cored bromides featuring both core and side chain halogens (1c, 3c, and 4c of Figure 3) are also of interest for the chemical community. The preparation of these polybromides can be ideally approached with two synthetic strategies: first-benzylic-then-ring or first-ring-then-benzylic halogenation. Of the two alternatives, the latter is more advantageous, due to three favourable features [65,66]: (i) Optimal control as each ring halogenation inhibits further ring halogenations, (ii) ring halogenation do not negatively affect benzylic bromination, and (iii) aryl bromides often display better solubility in organic solvents than benzyl bromides, reducing precipitation issues. Applying now the first-ring-then-benzylic halogenation strategy to p-xylene, dibromide 3b was successfully converted into 3c, by means of the method herein developed for the benzylic bromination. The process was then telescoped, resulting in a one-pot, two-step synthesis of 3c from 3, with improved operativity and yield (see experimental part). The same strategies were followed for the preparation of 1c (from 1b or from 1), as well as for 4c (from 4b or from 4). The preparation of the tetrabromo compound 2c was instead not investigated, due to low SciFinder® score (Figure 3).
The nucleophilicity of 4 provides useful support for its ring bromination towards 4b, resulting in an excellent isolated yield of 89%. Moreover, the intermediacy of 4b is advantageous for obtaining 4c, thus avoiding the selectivity issues observed during the benzylic bromination of 4. As a result, 4c was obtained from 4 (in a one-pot, two-step process) with an 85% isolated yield. The particular cleanliness of the conversion of 4b into 4c, featuring negligible formation of gem dihalides, could arise from the useful steric hindrance given by the halogen bound to the core.
Figure 4 collects the results obtained in the preparation of aryl-cored halides of Figure 3, by means of the peroxide-bromide process. For products featuring bromine on both the aromatic core and the side chain, the “direct” one-pot two-stage and the “indirect” first-ring-then-benzylic halogenation methods are compared.
![[1860-5397-20-95-4]](/bjoc/content/figures/1860-5397-20-95-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Yields obtained in the preparation of aryl-cored halides.
Figure 4: Yields obtained in the preparation of aryl-cored halides.
Conclusion
In summary, the peroxide-bromide halogenation method, originally developed for the monobromination of benzenoid structures, has been extended to synthesize aryl-cored polybromides with high yield and high atom economy. This method, when used with light irradiation as halogen initiator, is capable of selectively convert xylene isomers and mesitylene into their corresponding benzyl bromides, regardless the presence of halogen atoms on the core. Moreover, under dark conditions, a modification of the same method allows the preparation of aryl polybromides through ring halogenation. The two variants, tested on a 10-gram scale, can be telescoped to achieve polybromo derivatives that feature both core and side chain substitution, in high yield. The application of this method enables the effective preparation of high-interest building blocks for polymer and materials chemists, offering improved sustainability compared to standard methodologies.
Experimental
General
Solvents and reagents were commercial grade and used as received. 1H NMR spectra were acquired with a Bruker Avance 400 spectrometer (Billerica, MA, USA). The lighting was achieved by means of a standard household white LED bulb (OSRAM 6.5 W, 2700 K, 806 lm). The H2O2 solution was dispensed by means of a syringe pump, mod. SyringeOne NE-300, from NewEra Instruments.
All the aryl-cored halides here prepared are known, and thoroughly characterized in [6] (compounds 1a, 2a, 3a), [67] (compounds 1b, 3b, 4b), [60] (compounds 2b, 3b, 3c), [68] (compound 1c), [56] (compound 4a), and [69] (compound 4c).
General procedure for benzylic bromination
In a 15 mL Schlenk tube with screw cap, equipped with a magnetic stirring bar, the substrate (5 mmol, 1.0 equiv), solvent (either CH2Cl2 or chlorobenzene, 1.0–4.0 mL), H2O (1.5 mL), and HBr (48 wt % aqueous solution, d = 1.49 g/mL, 1.25 mL, 11 mmol, 2.2 equiv) were inserted. The mixture was kept under stirring at rt and irradiated with a LED lightbulb placed at 10 cm from the side of the reaction tube. Aqueous H2O2 (35 wt % solution, d = 1.13 g/mL, 2.00 mL, 23.3 mmol, 4.7 equiv) was added over 2 h using a syringe pump, through a small PTFE tube inserted through the side arm of the Schlenk tube. After the addition was complete, the mixture was left under stirring for 1 h and 30 min. Once the mixture was neutralised with solid NaHCO3, the product was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were concentrated to dryness to give the crude product.
General procedure for ring bromination
In a 25 mL round-bottomed flask, equipped with a magnetic stirring bar, the substrate (12 mmol, 1.0 equiv), H2SO4 (96 wt %, 3.00 g, 30 mmol, 2.5 equiv), I2 (0.04 g, 0.16 mmol, 0.01 equiv), and CH2Cl2 (1.50 mL) were inserted. The flask, kept under stirring at room temperature (rt), was light-shielded with aluminium foil and a water-cooled condenser was installed on its top. A solution of NaBr (2.88 g, 28 mmol, 2.33 equiv) in H2O2 (35 wt % aqueous solution, 4.8 mL, 56 mmol, 4.67 equiv) was added to the mixture over 15 min with a syringe pump, through a PTFE tube inserted in the top of the condenser. The system was then refluxed for 48 h. Once cooled to rt, the reaction mixture was neutralised with solid NaHCO3, and the product was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were concentrated to dryness to give the crude product.
General procedure for one-pot first-ring-then-benzylic bromination
In a 25 mL round-bottomed flask, equipped with a magnetic stirring bar, the substrate (12 mmol, 1.0 equiv), H2SO4 (96 wt %, 3.00 g, 30 mmol, 2.5 equiv), I2 (0.04 g, 0.16 mmol, 0.01 equiv), and chlorobenzene (8.0 mL) were inserted at rt. The flask was shielded from light by means of an aluminium foil and a water-cooled condenser was installed on its top. Once the stirring was started, a solution of NaBr (2.88 g, 28 mmol, 2.33 equiv) in H2O2 (35 wt % aqueous solution, 4.8 mL, 56 mmol, 4.67 equiv) was added over 15 min through a PTFE tube inserted at the top of the condenser, by means of a syringe pump. The flask was then heated to 50 °C for 48 h, cooled to rt, and unwrapped.
Under stirring at rt, H2O (3.6 mL) and HBr (48 wt % aqueous solution, 3.00 mL, 26.5 mmol, 2.2 equiv) were inserted. The mixture was then irradiated with a LED lightbulb, placed at 10 cm from the side of the flask and aqueous H2O2 (35 wt % solution, 4.8 mL, 56 mmol, 4.7 equiv) was added over 2 h using a syringe pump, through a PTFE tube inserted at the top of the condenser. After the addition was complete, the mixture was left under stirring for 1 h and 30 min. The reaction mixture was then neutralised with solid NaHCO3 and the product was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were concentrated to dryness to give the crude product.
1,2-Bis(bromomethyl)benzene (1a): white solid, yield: 98%. 1H NMR (400 MHz, 298 K, CDCl3) δ 7.37 (m, 2H), 7.31 (m, 2H), 4.67 (s, 4H) ppm.
1,2-Dibromo-4,5-dimethylbenzene (1b): For the 10-gram scale procedure, see Figure S2 (Supporting Information File 1), brown solid, yield: 93%. Recrystallisation from hot petroleum ether gave the pure product. 1H NMR (400 MHz, 298 K, CDCl3) δ 7.37 (s, 2H), 2.19 (s, 6H) ppm.
1,2-Dibromo-4,5-bis(bromomethyl)benzene (1c): From 1b: chlorobenzene (4.0 mL) was used as the solvent instead of CH2Cl2 (1 mL). Brown solid, yield: 86%. From 1 (two-step, one pot): Brown solid, yield: 82%. Recrystallisation from hot hexane gave the pure product. White solid, 1H NMR (400 MHz, 298 K, CDCl3) δ 7.62 (m, 2H), 4.53 (s, 4H) ppm.
1,3-Bis(bromomethyl)benzene (2a): Petroleum ether (1.5 mL) was used as the solvent instead of CH2Cl2. The reaction mixture was kept at 5 °C instead of rt and time was extended by 15% (2 h 20 min for H2O2 dropping and 2 h after the addition). The title product was obtained as pale-yellow solid, yield: 73%. Recrystallisation from hot petroleum ether gave the pure product. White solid, 1H NMR (400 MHz, 298 K, CDCl3) δ 7.42 (m, 1H), 7.33 (m, 1H), 7.32 (m, 2H), 4.48 (s, 4H) ppm.
1,5-Dibromo-2,4-dimethylbenzene (2b): Brown solid, yield: 87%. Recrystallisation from hot ethanol gave the pure product as pale-yellow solid. 1H NMR (400 MHz, 298 K, CDCl3) δ 7.68 (s, 1H), 7.10 (s, 1H), 2.31 (s, 6H) ppm.
1,4-Bis(bromomethyl)benzene (3a): For the 10-gram scale procedure, see Figure S1 (Supporting Information File 1), white-yellowish solid, yield: 87%. Recrystallisation from hot toluene gave the pure product as white solid. 1H NMR (400 MHz, 298 K, CDCl3) δ 7.37 (s, 4H), 4.48 (s, 4H) ppm.
1,4-Dibromo-2,5-dimethylbenzene (3b): pale orange solid, yield: quantitative. Recrystallisation from hot hexane gave the pure product as pale-yellow solid. 1H NMR (400 MHz, 298 K, CDCl3) δ 7.39 (s, 2H), 2.33 (s, 6H) ppm.
1,4-Dibromo-2,5-bis(bromomethyl)benzene (3c): From 3b: chlorobenzene (4.0 mL) was used as the solvent instead of CH2Cl2 (1.0 mL). Pale-yellow solid, yield: 94%. From 3 (two-step, one pot): yellow solid, yield 96%. Recrystallisation from hot petroleum ether gave the pure product as white solid. 1H NMR (400 MHz, 298 K, CDCl3) δ 7.66 (s, 2H), 4.51 (s, 4H) ppm.
1,3,5-Tris(bromomethyl)benzene (4a): The general procedure was adapted considering the three benzylic positions. In a 15 mL Schlenk tube with screw cap, equipped with a magnetic stirring bar, substrate 4 (5 mmol, 1.0 equiv), CH2Cl2 (2.0 mL), H2O (2.0 mL), and HBr (48 wt % aqueous solution, d = 1.49 g/mL, 1.87 mL, 16.5 mmol, 3.3 equiv) were inserted. The mixture was kept under stirring at rt and irradiated with a LED lightbulb placed at 10 cm from the side of the reaction tube. Aqueous H2O2 (35 wt % solution, 3.0 mL, 23.3 mmol, 4.7 equiv) was added over 2 h using a syringe pump, through a small PTFE tube inserted through the side arm of the Schlenk. After the addition was complete, the mixture was left under stirring for 1 h and 30 min. After the workup, operated as described previously, a brown slurry was obtained. Yield: 42%. The crude product was purified via column chromatography on silica gel 60, eluting with petroleum ether. The pure product appears as a white solid. 1H NMR (400 MHz, 298 K, CDCl3) δ 7.35 (s, 3H), 4.45 (s, 6H) ppm.
1,3,5-Tribromo-2,4,6-trimethylbenzene (4b): The general procedure was adapted considering the three ring positions to be halogenated (3.5 equiv of NaBr were used instead of 2.33 equiv). The raw product appears as pale-yellow solid, yield: 89%. Recrystallisation from hot chloroform gave the pure product as white solid. 1H NMR (400 MHz, 298 K, CDCl3) δ 2.65 (s, 9H) ppm.
1,3,5-Tribromo-2,4,6-tris(bromomethyl)benzene (4c): The general procedure was adapted considering the three ring and the three benzylic positions. From 4b: White solid, yield: 93%. From 4 (two-step, one pot): Pale yellow solid, yield 85%. 1H NMR (400 MHz, 298 K, CDCl3) δ 4.92 (s, 6H) ppm.
Supporting Information
| Supporting Information File 1: Addtional figures and tables and copies of spectra. | ||
| Format: PDF | Size: 1.0 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
La Manna, P.; Soriente, A.; De Rosa, M.; Buonerba, A.; Talotta, C.; Gaeta, C.; Neri, P. ChemSusChem 2019, 12, 1673–1683. doi:10.1002/cssc.201900137
Return to citation in text: [1] -
Winter, A.; Friebe, C.; Hager, M. D.; Schubert, U. S. Eur. J. Org. Chem. 2009, 801–809. doi:10.1002/ejoc.200800857
Return to citation in text: [1] -
Nakamura, H.; Lee, J.-D.; Ueno, M.; Miyajima, Y.; Ban, H. S. NanoBiotechnology 2007, 3, 135–145. doi:10.1007/s12030-008-9000-6
Return to citation in text: [1] -
Al-Masri, M.; Kricheldorf, H. R.; Fritsch, D. Macromolecules 1999, 32, 7853–7858. doi:10.1021/ma9910742
Return to citation in text: [1] -
Yang, X.; Conrad, C. A.; Wan, W.; Bedford, M. S.; Hu, L.; Chumanov, G.; Smith, R. C. J. Mater. Chem. C 2015, 3, 4537–4544. doi:10.1039/c4tc02576h
Return to citation in text: [1] -
Liu, Z.; Wang, Q. Polymer 2016, 94, 14–18. doi:10.1016/j.polymer.2016.04.057
Return to citation in text: [1] [2] -
Liu, Z.; Wang, Q. Polymer 2016, 100, 56–59. doi:10.1016/j.polymer.2016.08.013
Return to citation in text: [1] -
Dale, E. J.; Ferris, D. P.; Vermeulen, N. A.; Henkelis, J. J.; Popovs, I.; Juríček, M.; Barnes, J. C.; Schneebeli, S. T.; Stoddart, J. F. J. Am. Chem. Soc. 2016, 138, 3667–3670. doi:10.1021/jacs.6b01368
Return to citation in text: [1] -
Park, Y.-I.; Lee, J. S.; Kim, B. J.; Kim, B.; Lee, J.; Kim, D. H.; Oh, S.-Y.; Cho, J. H.; Park, J.-W. Chem. Mater. 2011, 23, 4038–4044. doi:10.1021/cm2016824
Return to citation in text: [1] -
Yun, J. H.; Han, S. H.; Lee, J. Y. Dyes Pigm. 2020, 173, 107947. doi:10.1016/j.dyepig.2019.107947
Return to citation in text: [1] -
Broløs, L.; Kilde, M. D.; Hammerich, O.; Nielsen, M. B. J. Org. Chem. 2020, 85, 3277–3286. doi:10.1021/acs.joc.9b03118
Return to citation in text: [1] -
Zhu, T.; Shi, B.; Wu, H.; You, X.; Wang, X.; Fan, C.; Peng, Q.; Jiang, Z. Ind. Eng. Chem. Res. 2021, 60, 6337–6343. doi:10.1021/acs.iecr.1c00418
Return to citation in text: [1] -
Bratulescu, G. Synth. Commun. 2008, 38, 2748–2752. doi:10.1080/00397910802222795
Return to citation in text: [1] -
Kamali, E.; Mohammadkhani, A.; Pazoki, F.; Heydari, A. ChemistrySelect 2023, 8, e202204642. doi:10.1002/slct.202204642
Return to citation in text: [1] -
Sasaki, Y. F.; Saga, A.; Akasaka, M.; Ishibashi, S.; Yoshida, K.; Su, Y. Q.; Matsusaka, N.; Tsuda, S. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 1998, 419, 13–20. doi:10.1016/s1383-5718(98)00114-4
Return to citation in text: [1] -
European Commission; Directorate-General for Communication; European green deal – delivering on our targets. Publications Office of the European Union: 2021. https://data.europa.eu/doi/10.2775/373022.
Return to citation in text: [1] -
Bruckner, R. Radical Halogenation of Hydrocarbons. In Organic Mechanisms Reactions Stereochemistry and Synthesis; Harmata, M., Ed.; Springer: Berlin, Heidelberg, 2010; pp 21–35.
Return to citation in text: [1] [2] [3] -
Compare, for example, the SDS of benzyl chloride with that of benzyl bromide. Source: Sigma-Aldrich® site. SDS are up to date at 05/12/2023. https://www.sigmaaldrich.com/IT/ru/sds/aldrich/185558?userType=undefined, https://www.sigmaaldrich.com/IT/en/sds/aldrich/b17905?userType=undefined.
Return to citation in text: [1] -
Sun, M.; Lowry, G. V.; Gregory, K. B. Water Res. 2013, 47, 3723–3731. doi:10.1016/j.watres.2013.04.041
Return to citation in text: [1] -
Bruckner, R. Ar-SE Reactions via Sigma Complexes: Individual Reactions. In Organic Mechanisms Reactions Stereochemistry and Synthesis; Harmata, M., Ed.; Springer: Berlin, Heidelberg, 2010; pp 215–233.
Return to citation in text: [1] -
Tang, N.; Song, X.; Yang, T.; Qiu, R.; Yin, S.-F. J. Organomet. Chem. 2021, 942, 121820. doi:10.1016/j.jorganchem.2021.121820
Return to citation in text: [1] -
Xie, Z.; Yang, B.; Liu, L.; Li, M.; Lin, D.; Ma, Y.; Cheng, G.; Liu, S. J. Phys. Org. Chem. 2005, 18, 962–973. doi:10.1002/poc.935
Return to citation in text: [1] -
Mikroyannidis, J. A. Macromolecules 2002, 35, 9289–9295. doi:10.1021/ma021242h
Return to citation in text: [1] -
Saikia, I.; Borah, A. J.; Phukan, P. Chem. Rev. 2016, 116, 6837–7042. doi:10.1021/acs.chemrev.5b00400
Return to citation in text: [1] [2] -
Djerassi, C. Chem. Rev. 1948, 43, 271–317. doi:10.1021/cr60135a004
Return to citation in text: [1] -
Cantillo, D.; de Frutos, O.; Rincon, J. A.; Mateos, C.; Kappe, C. O. J. Org. Chem. 2014, 79, 223–229. doi:10.1021/jo402409k
Return to citation in text: [1] [2] -
Srisailas, M.; Rajakumar, P. J. Chem. Res. 2006, 671–674. doi:10.3184/030823406779173677
Return to citation in text: [1] -
Pramanick, P. K.; Hou, Z.-L.; Yao, B. Tetrahedron 2017, 73, 7105–7114. doi:10.1016/j.tet.2017.10.073
Return to citation in text: [1] -
Ni, S.; El Remaily, M. A. E. A. A. A.; Franzén, J. Adv. Synth. Catal. 2018, 360, 4197–4204. doi:10.1002/adsc.201800788
Return to citation in text: [1] -
Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 1097–1099. doi:10.1016/j.tetlet.2005.12.040
Return to citation in text: [1] -
Bodzioch, A.; Owsianik, K.; Skalik, J.; Kowalska, E.; Stasiak, A.; Różycka-Sokołowska, E.; Marciniak, B.; Bałczewski, P. Synthesis 2016, 48, 3509–3514. doi:10.1055/s-0035-1561651
Return to citation in text: [1] -
Nabuurs, R. J. A.; Kapoerchan, V. V.; Metaxas, A.; Hafith, S.; de Backer, M.; Welling, M. M.; Jiskoot, W.; van den Nieuwendijk, A. M. C. H.; Windhorst, A. D.; Overkleeft, H. S.; van Buchem, M. A.; Overhand, M.; van der Weerd, L. Bioorg. Med. Chem. 2016, 24, 6139–6148. doi:10.1016/j.bmc.2016.05.022
Return to citation in text: [1] -
Levine, D. R.; Caruso, A.; Siegler, M. A.; Tovar, J. D. Chem. Commun. 2012, 48, 6256. doi:10.1039/c2cc32500d
Return to citation in text: [1] -
Suarez, D.; Laval, G.; Tu, S.-M.; Jiang, D.; Robinson, C. L.; Scott, R.; Golding, B. T. Synthesis 2009, 1807–1810. doi:10.1055/s-0029-1216793
Return to citation in text: [1] -
Beneto, A. J.; Sivamani, J.; Ashokkumar, V.; Balasaravanan, R.; Duraimurugan, K.; Siva, A. New J. Chem. 2015, 39, 3098–3104. doi:10.1039/c4nj02395a
Return to citation in text: [1] -
Shang, Q.; Zeng, T.; Gao, K.; Liu, N.; Cheng, Q.; Liao, G.; Pan, Z.; Zhou, H. New J. Chem. 2019, 43, 16595–16603. doi:10.1039/c9nj04371c
Return to citation in text: [1] [2] -
Soundararajan, R.; Krishnamurthy, S.; Srinivasan, V. S.; Balasubramanian, T. R. J. Organomet. Chem. 1983, 255, 295–297. doi:10.1016/s0022-328x(00)99321-5
Return to citation in text: [1] -
Eissen, M.; Lenoir, D. Chem. – Eur. J. 2008, 14, 9830–9841. doi:10.1002/chem.200800462
Return to citation in text: [1] -
Kikuchi, D.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 1998, 63, 6023–6026. doi:10.1021/jo972263q
Return to citation in text: [1] -
Adimurthy, S.; Ghosh, S.; Patoliya, P. U.; Ramachandraiah, G.; Agrawal, M.; Gandhi, M. R.; Upadhyay, S. C.; Ghosh, P. K.; Ranu, B. C. Green Chem. 2008, 10, 232–237. doi:10.1039/b713829f
Return to citation in text: [1] -
Joshi, G.; Adimurthy, S. Ind. Eng. Chem. Res. 2011, 50, 12271–12275. doi:10.1021/ie2004863
Return to citation in text: [1] -
Zhao, M.; Li, M.; Lu, W. Synthesis 2018, 50, 4933–4939. doi:10.1055/s-0037-1610651
Return to citation in text: [1] -
Bray, W. C.; Livingston, R. S. J. Am. Chem. Soc. 1923, 45, 1251–1271. doi:10.1021/ja01658a021
Return to citation in text: [1] [2] -
Leulier, A. Bull. Soc. Chim. Fr. 1924, 35, 1325–1330.
Return to citation in text: [1] -
Amati, A.; Dosualdo, G.; Zhao, L.; Bravo, A.; Fontana, F.; Minisci, F.; Bjørsvik, H.-R. Org. Process Res. Dev. 1998, 2, 261–269. doi:10.1021/op980028j
Return to citation in text: [1] [2] [3] -
Turner, P. J.; Jeff, M. Process for the bromination of alkyl arenes. Eur. Pat. Appl. EP0336567A1, Oct 11, 1989.
Return to citation in text: [1] -
Mestres, R.; Palenzuela, J. Green Chem. 2002, 4, 314–316. doi:10.1039/b203055a
Return to citation in text: [1] -
Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 7245–7247. doi:10.1016/j.tetlet.2006.07.109
Return to citation in text: [1] [2] -
Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron 2009, 65, 4429–4439. doi:10.1016/j.tet.2009.03.034
Return to citation in text: [1] [2] [3] [4] [5] -
Lima, H. H. L. B.; da Silva, G. R.; Pena, J. M.; Cella, R. ChemistrySelect 2017, 2, 9624–9627. doi:10.1002/slct.201702023
Return to citation in text: [1] [2] -
Podgoršek, A.; Zupan, M.; Iskra, J. Angew. Chem., Int. Ed. 2009, 48, 8424–8450. doi:10.1002/anie.200901223
Return to citation in text: [1] -
Hubinger, S.; Nee, J. B. J. Photochem. Photobiol., A 1995, 86, 1–7. doi:10.1016/1010-6030(94)03949-u
Return to citation in text: [1] -
Stephenson, E. F. M. Org. Synth. 1954, 34, 100. doi:10.15227/orgsyn.034.0100
Return to citation in text: [1] -
Pearson, G. S.; Magee, R. S. Pure Appl. Chem. 2002, 74, 187–316. doi:10.1351/pac200274020187
Return to citation in text: [1] -
Shaw, H.; Perlmutter, H. D.; Gu, C.; Arco, S. D.; Quibuyen, T. O. J. Org. Chem. 1997, 62, 236–237. doi:10.1021/jo950371b
Return to citation in text: [1] -
Newkome, G. R.; Yao, Z.; Baker, G. R.; Gupta, V. K.; Russo, P. S.; Saunders, M. J. J. Am. Chem. Soc. 1986, 108, 849–850. doi:10.1021/ja00264a054
Return to citation in text: [1] [2] -
Tal, D. M.; Karlish, S. J. D. Tetrahedron 1995, 51, 3823–3830. doi:10.1016/0040-4020(95)00105-h
Return to citation in text: [1] -
Li, J.; Liu, D.; Li, Y.; Lee, C.-S.; Kwong, H.-L.; Lee, S. Chem. Mater. 2005, 17, 1208–1212. doi:10.1021/cm034731k
Return to citation in text: [1] -
Yang, X.; Liu, D.; Miao, Q. Angew. Chem., Int. Ed. 2014, 53, 6786–6790. doi:10.1002/anie.201403509
Return to citation in text: [1] -
Bonifacio, M. C.; Robertson, C. R.; Jung, J.-Y.; King, B. T. J. Org. Chem. 2005, 70, 8522–8526. doi:10.1021/jo051418o
Return to citation in text: [1] [2] -
Naresh, M.; Arun Kumar, M.; Mahender Reddy, M.; Swamy, P.; Nanubolu, J.; Narender, N. Synthesis 2013, 45, 1497–1504. doi:10.1055/s-0033-1338431
Return to citation in text: [1] -
Kajorinne, J. K.; Steers, J. C. M.; Merchant, M. E.; MacKinnon, C. D. Can. J. Chem. 2018, 96, 1087–1091. doi:10.1139/cjc-2018-0259
Return to citation in text: [1] [2] -
Vyas, P. V.; Bhatt, A. K.; Ramachandraiah, G.; Bedekar, A. V. Tetrahedron Lett. 2003, 44, 4085–4088. doi:10.1016/s0040-4039(03)00834-7
Return to citation in text: [1] -
Sabuzi, F.; Pomarico, G.; Floris, B.; Valentini, F.; Galloni, P.; Conte, V. Coord. Chem. Rev. 2019, 385, 100–136. doi:10.1016/j.ccr.2019.01.013
Return to citation in text: [1] -
Dai, H.; Cai, Z.-B.; Lou, Q.-X.; Li, S.-L.; Tian, Y.-P. Tetrahedron 2021, 96, 132359. doi:10.1016/j.tet.2021.132359
Return to citation in text: [1] -
Auffray, M.; Charra, F.; Sosa Vargas, L.; Mathevet, F.; Attias, A.-J.; Kreher, D. New J. Chem. 2020, 44, 7665–7674. doi:10.1039/d0nj00110d
Return to citation in text: [1] -
Bose, A.; Mal, P. Tetrahedron Lett. 2014, 55, 2154–2156. doi:10.1016/j.tetlet.2014.02.064
Return to citation in text: [1] -
Rivera, J. M.; Martín, T.; Rebek, J. J. Am. Chem. Soc. 2001, 123, 5213–5220. doi:10.1021/ja004080i
Return to citation in text: [1] -
Hennrich, G.; Lynch, V. M.; Anslyn, E. V. Chem. – Eur. J. 2002, 8, 2274–2278. doi:10.1002/1521-3765(20020517)8:10<2274::aid-chem2274>3.0.co;2-t
Return to citation in text: [1]
| 24. | Saikia, I.; Borah, A. J.; Phukan, P. Chem. Rev. 2016, 116, 6837–7042. doi:10.1021/acs.chemrev.5b00400 |
| 39. | Kikuchi, D.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 1998, 63, 6023–6026. doi:10.1021/jo972263q |
| 40. | Adimurthy, S.; Ghosh, S.; Patoliya, P. U.; Ramachandraiah, G.; Agrawal, M.; Gandhi, M. R.; Upadhyay, S. C.; Ghosh, P. K.; Ranu, B. C. Green Chem. 2008, 10, 232–237. doi:10.1039/b713829f |
| 41. | Joshi, G.; Adimurthy, S. Ind. Eng. Chem. Res. 2011, 50, 12271–12275. doi:10.1021/ie2004863 |
| 50. | Lima, H. H. L. B.; da Silva, G. R.; Pena, J. M.; Cella, R. ChemistrySelect 2017, 2, 9624–9627. doi:10.1002/slct.201702023 |
| 51. | Podgoršek, A.; Zupan, M.; Iskra, J. Angew. Chem., Int. Ed. 2009, 48, 8424–8450. doi:10.1002/anie.200901223 |
| 48. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 7245–7247. doi:10.1016/j.tetlet.2006.07.109 |
| 49. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron 2009, 65, 4429–4439. doi:10.1016/j.tet.2009.03.034 |
| 45. | Amati, A.; Dosualdo, G.; Zhao, L.; Bravo, A.; Fontana, F.; Minisci, F.; Bjørsvik, H.-R. Org. Process Res. Dev. 1998, 2, 261–269. doi:10.1021/op980028j |
| 46. | Turner, P. J.; Jeff, M. Process for the bromination of alkyl arenes. Eur. Pat. Appl. EP0336567A1, Oct 11, 1989. |
| 47. | Mestres, R.; Palenzuela, J. Green Chem. 2002, 4, 314–316. doi:10.1039/b203055a |
| 48. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 7245–7247. doi:10.1016/j.tetlet.2006.07.109 |
| 49. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron 2009, 65, 4429–4439. doi:10.1016/j.tet.2009.03.034 |
| 45. | Amati, A.; Dosualdo, G.; Zhao, L.; Bravo, A.; Fontana, F.; Minisci, F.; Bjørsvik, H.-R. Org. Process Res. Dev. 1998, 2, 261–269. doi:10.1021/op980028j |
| 42. | Zhao, M.; Li, M.; Lu, W. Synthesis 2018, 50, 4933–4939. doi:10.1055/s-0037-1610651 |
| 43. | Bray, W. C.; Livingston, R. S. J. Am. Chem. Soc. 1923, 45, 1251–1271. doi:10.1021/ja01658a021 |
| 45. | Amati, A.; Dosualdo, G.; Zhao, L.; Bravo, A.; Fontana, F.; Minisci, F.; Bjørsvik, H.-R. Org. Process Res. Dev. 1998, 2, 261–269. doi:10.1021/op980028j |
| 52. | Hubinger, S.; Nee, J. B. J. Photochem. Photobiol., A 1995, 86, 1–7. doi:10.1016/1010-6030(94)03949-u |
| 49. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron 2009, 65, 4429–4439. doi:10.1016/j.tet.2009.03.034 |
| 59. | Yang, X.; Liu, D.; Miao, Q. Angew. Chem., Int. Ed. 2014, 53, 6786–6790. doi:10.1002/anie.201403509 |
| 60. | Bonifacio, M. C.; Robertson, C. R.; Jung, J.-Y.; King, B. T. J. Org. Chem. 2005, 70, 8522–8526. doi:10.1021/jo051418o |
| 49. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron 2009, 65, 4429–4439. doi:10.1016/j.tet.2009.03.034 |
| 50. | Lima, H. H. L. B.; da Silva, G. R.; Pena, J. M.; Cella, R. ChemistrySelect 2017, 2, 9624–9627. doi:10.1002/slct.201702023 |
| 61. | Naresh, M.; Arun Kumar, M.; Mahender Reddy, M.; Swamy, P.; Nanubolu, J.; Narender, N. Synthesis 2013, 45, 1497–1504. doi:10.1055/s-0033-1338431 |
| 57. | Tal, D. M.; Karlish, S. J. D. Tetrahedron 1995, 51, 3823–3830. doi:10.1016/0040-4020(95)00105-h |
| 36. | Shang, Q.; Zeng, T.; Gao, K.; Liu, N.; Cheng, Q.; Liao, G.; Pan, Z.; Zhou, H. New J. Chem. 2019, 43, 16595–16603. doi:10.1039/c9nj04371c |
| 58. | Li, J.; Liu, D.; Li, Y.; Lee, C.-S.; Kwong, H.-L.; Lee, S. Chem. Mater. 2005, 17, 1208–1212. doi:10.1021/cm034731k |
| 55. | Shaw, H.; Perlmutter, H. D.; Gu, C.; Arco, S. D.; Quibuyen, T. O. J. Org. Chem. 1997, 62, 236–237. doi:10.1021/jo950371b |
| 56. | Newkome, G. R.; Yao, Z.; Baker, G. R.; Gupta, V. K.; Russo, P. S.; Saunders, M. J. J. Am. Chem. Soc. 1986, 108, 849–850. doi:10.1021/ja00264a054 |
| 43. | Bray, W. C.; Livingston, R. S. J. Am. Chem. Soc. 1923, 45, 1251–1271. doi:10.1021/ja01658a021 |
| 53. | Stephenson, E. F. M. Org. Synth. 1954, 34, 100. doi:10.15227/orgsyn.034.0100 |
| 54. | Pearson, G. S.; Magee, R. S. Pure Appl. Chem. 2002, 74, 187–316. doi:10.1351/pac200274020187 |
| 63. | Vyas, P. V.; Bhatt, A. K.; Ramachandraiah, G.; Bedekar, A. V. Tetrahedron Lett. 2003, 44, 4085–4088. doi:10.1016/s0040-4039(03)00834-7 |
| 64. | Sabuzi, F.; Pomarico, G.; Floris, B.; Valentini, F.; Galloni, P.; Conte, V. Coord. Chem. Rev. 2019, 385, 100–136. doi:10.1016/j.ccr.2019.01.013 |
| 62. | Kajorinne, J. K.; Steers, J. C. M.; Merchant, M. E.; MacKinnon, C. D. Can. J. Chem. 2018, 96, 1087–1091. doi:10.1139/cjc-2018-0259 |
| 49. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron 2009, 65, 4429–4439. doi:10.1016/j.tet.2009.03.034 |
| 62. | Kajorinne, J. K.; Steers, J. C. M.; Merchant, M. E.; MacKinnon, C. D. Can. J. Chem. 2018, 96, 1087–1091. doi:10.1139/cjc-2018-0259 |
| 1. | La Manna, P.; Soriente, A.; De Rosa, M.; Buonerba, A.; Talotta, C.; Gaeta, C.; Neri, P. ChemSusChem 2019, 12, 1673–1683. doi:10.1002/cssc.201900137 |
| 8. | Dale, E. J.; Ferris, D. P.; Vermeulen, N. A.; Henkelis, J. J.; Popovs, I.; Juríček, M.; Barnes, J. C.; Schneebeli, S. T.; Stoddart, J. F. J. Am. Chem. Soc. 2016, 138, 3667–3670. doi:10.1021/jacs.6b01368 |
| 17. | Bruckner, R. Radical Halogenation of Hydrocarbons. In Organic Mechanisms Reactions Stereochemistry and Synthesis; Harmata, M., Ed.; Springer: Berlin, Heidelberg, 2010; pp 21–35. |
| 69. | Hennrich, G.; Lynch, V. M.; Anslyn, E. V. Chem. – Eur. J. 2002, 8, 2274–2278. doi:10.1002/1521-3765(20020517)8:10<2274::aid-chem2274>3.0.co;2-t |
| 4. | Al-Masri, M.; Kricheldorf, H. R.; Fritsch, D. Macromolecules 1999, 32, 7853–7858. doi:10.1021/ma9910742 |
| 5. | Yang, X.; Conrad, C. A.; Wan, W.; Bedford, M. S.; Hu, L.; Chumanov, G.; Smith, R. C. J. Mater. Chem. C 2015, 3, 4537–4544. doi:10.1039/c4tc02576h |
| 6. | Liu, Z.; Wang, Q. Polymer 2016, 94, 14–18. doi:10.1016/j.polymer.2016.04.057 |
| 7. | Liu, Z.; Wang, Q. Polymer 2016, 100, 56–59. doi:10.1016/j.polymer.2016.08.013 |
| 20. | Bruckner, R. Ar-SE Reactions via Sigma Complexes: Individual Reactions. In Organic Mechanisms Reactions Stereochemistry and Synthesis; Harmata, M., Ed.; Springer: Berlin, Heidelberg, 2010; pp 215–233. |
| 3. | Nakamura, H.; Lee, J.-D.; Ueno, M.; Miyajima, Y.; Ban, H. S. NanoBiotechnology 2007, 3, 135–145. doi:10.1007/s12030-008-9000-6 |
| 18. | Compare, for example, the SDS of benzyl chloride with that of benzyl bromide. Source: Sigma-Aldrich® site. SDS are up to date at 05/12/2023. https://www.sigmaaldrich.com/IT/ru/sds/aldrich/185558?userType=undefined, https://www.sigmaaldrich.com/IT/en/sds/aldrich/b17905?userType=undefined. |
| 68. | Rivera, J. M.; Martín, T.; Rebek, J. J. Am. Chem. Soc. 2001, 123, 5213–5220. doi:10.1021/ja004080i |
| 2. | Winter, A.; Friebe, C.; Hager, M. D.; Schubert, U. S. Eur. J. Org. Chem. 2009, 801–809. doi:10.1002/ejoc.200800857 |
| 19. | Sun, M.; Lowry, G. V.; Gregory, K. B. Water Res. 2013, 47, 3723–3731. doi:10.1016/j.watres.2013.04.041 |
| 56. | Newkome, G. R.; Yao, Z.; Baker, G. R.; Gupta, V. K.; Russo, P. S.; Saunders, M. J. J. Am. Chem. Soc. 1986, 108, 849–850. doi:10.1021/ja00264a054 |
| 14. | Kamali, E.; Mohammadkhani, A.; Pazoki, F.; Heydari, A. ChemistrySelect 2023, 8, e202204642. doi:10.1002/slct.202204642 |
| 16. | European Commission; Directorate-General for Communication; European green deal – delivering on our targets. Publications Office of the European Union: 2021. https://data.europa.eu/doi/10.2775/373022. |
| 67. | Bose, A.; Mal, P. Tetrahedron Lett. 2014, 55, 2154–2156. doi:10.1016/j.tetlet.2014.02.064 |
| 13. | Bratulescu, G. Synth. Commun. 2008, 38, 2748–2752. doi:10.1080/00397910802222795 |
| 17. | Bruckner, R. Radical Halogenation of Hydrocarbons. In Organic Mechanisms Reactions Stereochemistry and Synthesis; Harmata, M., Ed.; Springer: Berlin, Heidelberg, 2010; pp 21–35. |
| 60. | Bonifacio, M. C.; Robertson, C. R.; Jung, J.-Y.; King, B. T. J. Org. Chem. 2005, 70, 8522–8526. doi:10.1021/jo051418o |
| 12. | Zhu, T.; Shi, B.; Wu, H.; You, X.; Wang, X.; Fan, C.; Peng, Q.; Jiang, Z. Ind. Eng. Chem. Res. 2021, 60, 6337–6343. doi:10.1021/acs.iecr.1c00418 |
| 65. | Dai, H.; Cai, Z.-B.; Lou, Q.-X.; Li, S.-L.; Tian, Y.-P. Tetrahedron 2021, 96, 132359. doi:10.1016/j.tet.2021.132359 |
| 66. | Auffray, M.; Charra, F.; Sosa Vargas, L.; Mathevet, F.; Attias, A.-J.; Kreher, D. New J. Chem. 2020, 44, 7665–7674. doi:10.1039/d0nj00110d |
| 9. | Park, Y.-I.; Lee, J. S.; Kim, B. J.; Kim, B.; Lee, J.; Kim, D. H.; Oh, S.-Y.; Cho, J. H.; Park, J.-W. Chem. Mater. 2011, 23, 4038–4044. doi:10.1021/cm2016824 |
| 10. | Yun, J. H.; Han, S. H.; Lee, J. Y. Dyes Pigm. 2020, 173, 107947. doi:10.1016/j.dyepig.2019.107947 |
| 11. | Broløs, L.; Kilde, M. D.; Hammerich, O.; Nielsen, M. B. J. Org. Chem. 2020, 85, 3277–3286. doi:10.1021/acs.joc.9b03118 |
| 15. | Sasaki, Y. F.; Saga, A.; Akasaka, M.; Ishibashi, S.; Yoshida, K.; Su, Y. Q.; Matsusaka, N.; Tsuda, S. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 1998, 419, 13–20. doi:10.1016/s1383-5718(98)00114-4 |
| 24. | Saikia, I.; Borah, A. J.; Phukan, P. Chem. Rev. 2016, 116, 6837–7042. doi:10.1021/acs.chemrev.5b00400 |
| 21. | Tang, N.; Song, X.; Yang, T.; Qiu, R.; Yin, S.-F. J. Organomet. Chem. 2021, 942, 121820. doi:10.1016/j.jorganchem.2021.121820 |
| 22. | Xie, Z.; Yang, B.; Liu, L.; Li, M.; Lin, D.; Ma, Y.; Cheng, G.; Liu, S. J. Phys. Org. Chem. 2005, 18, 962–973. doi:10.1002/poc.935 |
| 23. | Mikroyannidis, J. A. Macromolecules 2002, 35, 9289–9295. doi:10.1021/ma021242h |
| 35. | Beneto, A. J.; Sivamani, J.; Ashokkumar, V.; Balasaravanan, R.; Duraimurugan, K.; Siva, A. New J. Chem. 2015, 39, 3098–3104. doi:10.1039/c4nj02395a |
| 36. | Shang, Q.; Zeng, T.; Gao, K.; Liu, N.; Cheng, Q.; Liao, G.; Pan, Z.; Zhou, H. New J. Chem. 2019, 43, 16595–16603. doi:10.1039/c9nj04371c |
| 37. | Soundararajan, R.; Krishnamurthy, S.; Srinivasan, V. S.; Balasubramanian, T. R. J. Organomet. Chem. 1983, 255, 295–297. doi:10.1016/s0022-328x(00)99321-5 |
| 38. | Eissen, M.; Lenoir, D. Chem. – Eur. J. 2008, 14, 9830–9841. doi:10.1002/chem.200800462 |
| 26. | Cantillo, D.; de Frutos, O.; Rincon, J. A.; Mateos, C.; Kappe, C. O. J. Org. Chem. 2014, 79, 223–229. doi:10.1021/jo402409k |
| 30. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 1097–1099. doi:10.1016/j.tetlet.2005.12.040 |
| 31. | Bodzioch, A.; Owsianik, K.; Skalik, J.; Kowalska, E.; Stasiak, A.; Różycka-Sokołowska, E.; Marciniak, B.; Bałczewski, P. Synthesis 2016, 48, 3509–3514. doi:10.1055/s-0035-1561651 |
| 32. | Nabuurs, R. J. A.; Kapoerchan, V. V.; Metaxas, A.; Hafith, S.; de Backer, M.; Welling, M. M.; Jiskoot, W.; van den Nieuwendijk, A. M. C. H.; Windhorst, A. D.; Overkleeft, H. S.; van Buchem, M. A.; Overhand, M.; van der Weerd, L. Bioorg. Med. Chem. 2016, 24, 6139–6148. doi:10.1016/j.bmc.2016.05.022 |
| 33. | Levine, D. R.; Caruso, A.; Siegler, M. A.; Tovar, J. D. Chem. Commun. 2012, 48, 6256. doi:10.1039/c2cc32500d |
| 34. | Suarez, D.; Laval, G.; Tu, S.-M.; Jiang, D.; Robinson, C. L.; Scott, R.; Golding, B. T. Synthesis 2009, 1807–1810. doi:10.1055/s-0029-1216793 |
| 28. | Pramanick, P. K.; Hou, Z.-L.; Yao, B. Tetrahedron 2017, 73, 7105–7114. doi:10.1016/j.tet.2017.10.073 |
| 29. | Ni, S.; El Remaily, M. A. E. A. A. A.; Franzén, J. Adv. Synth. Catal. 2018, 360, 4197–4204. doi:10.1002/adsc.201800788 |
| 17. | Bruckner, R. Radical Halogenation of Hydrocarbons. In Organic Mechanisms Reactions Stereochemistry and Synthesis; Harmata, M., Ed.; Springer: Berlin, Heidelberg, 2010; pp 21–35. |
| 25. | Djerassi, C. Chem. Rev. 1948, 43, 271–317. doi:10.1021/cr60135a004 |
| 26. | Cantillo, D.; de Frutos, O.; Rincon, J. A.; Mateos, C.; Kappe, C. O. J. Org. Chem. 2014, 79, 223–229. doi:10.1021/jo402409k |
| 27. | Srisailas, M.; Rajakumar, P. J. Chem. Res. 2006, 671–674. doi:10.3184/030823406779173677 |
© 2024 Roncaglia et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.