The cycloaddition of 1,3-dipoles at C=N bonds is a relatively rare process, in contrast to the widespread cycloaddition reactions at C=C, C≡C, and C=S bonds. In this study, we present the syntheses of novel hydantoin/1,2,4-oxadiazoline spiro-compounds using a 1,3-dipolar cycloaddition of nitrile oxides to C=N bonds of 5-iminohydantoins. The efficiency of the approach was demonstrated by varying the substituents at four positions of the resulting spirocyclic molecules. Cytotoxicity of the target hydantoin/1,2,4-oxadiazolines was shown to exceed previously known spiro-compounds bearing only hydantoins or 1,2,4-oxadiazolines (IC50 values were 30–50 μM, HCT116 cell lines).
The 1,2,4-oxadiazole fragment is a common pharmacophore, and molecules containing this group exhibit a wide range of biological activities, including antitumor, anti-HIV, anti-obesity, anti-inflammatory, antidiabetic, anticancer, and antitubercular properties [1,2]. Among these molecules, bicyclic compounds with the dihydrooxadiazole connected to another cycle via the single quaternary carbon atom C5 demonstrated significant biological activity, greater than analogs with non-spiro-bonded cyclic fragments [3,4].
Hydantoin derivatives also exhibit a wide range of biological activities. Compounds containing the hydantoin pharmacophore group, are known for their anticancer, anti-inflammatory, antidiabetic, antimicrobial, adrenoreceptor modulating, anticonvulsant, antiplatelet, anti-HIV, and other activities [5,6]. Modifying hydantoins at the N1, N3, and C5 positions make it possible to achieve better pharmacological properties.
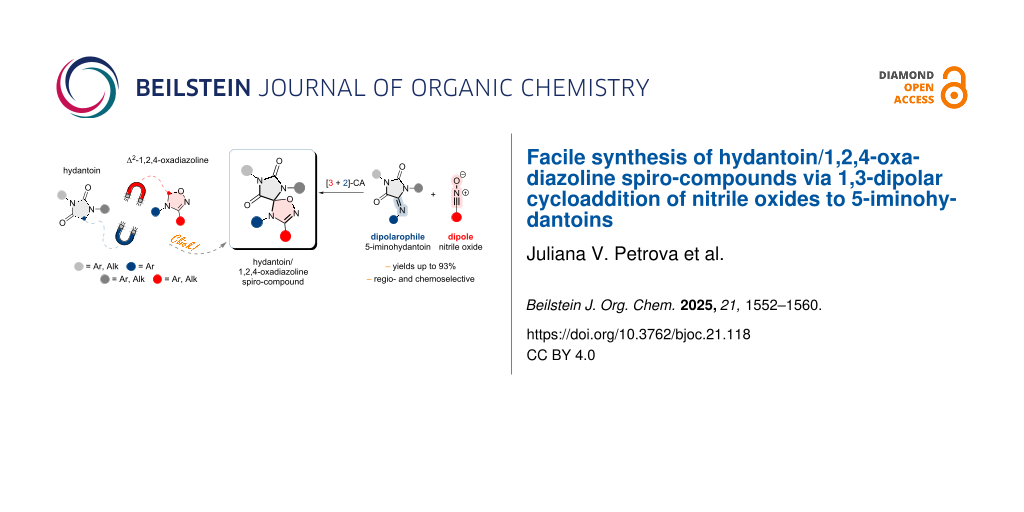
In this paper, we firstly report the synthesis of compounds comprising two pharmacophoric fragments, hydantoin and 1,2,4-oxadiazoline, joined through the C5 atom of each cycle forming spiro-fused structures. We propose an efficient method for the synthesis of such spiro-compounds based on a [3 + 2]-dipolar cycloaddition (32CA) of nitrile oxides with 5-iminohydantoins (Figure 1). The proposed method involves the use of readily available starting materials, avoids chromatographic purification of the final products which are obtained in good yields. We have also tested the cytotoxicity of some synthesized spiro-hydantoin-1,2,4-oxadiazolines against the HCT116 human prostate cancer cell line, and compared it with that of compounds containing either spiro-hydantoins or spiro-oxadiazolines alone.
Figure 1:
Design and synthetic strategies for the target hydantoin/1,2,4-oxadiazoline spiro-compounds.
Figure 1:
Design and synthetic strategies for the target hydantoin/1,2,4-oxadiazoline spiro-compounds.
Although there are many examples in the literature demonstrating the ability of nitrile oxides to react with various types of double and triple bonds, most of the described reactions are related to dipolarophiles with C=C, C≡C or C=S bonds [7]. However, the [3 + 2]-cycloaddition of nitrile oxides to exocyclic C=N bonds, is a much less explored area. There are few known reactions of these dipoles with imino derivatives of oxindole [3,8,9], chrysenequinone [10], cycloheptatriene [11,12], thiazole [13], and matrine-type alkaloids [14]. The hybrid pharmacophore design is a frequently employed approach in the development of potential antitumor and other drugs [15,16]. This method involves the merging of two distinct bioactive fragments into a single molecule through a spacer. An alternative strategy of combining heterocyclic pharmacophores is to incorporate them into a spiro-jointed structure that lacks any intermediate link between the active parts [17]. Despite the fact that these strategies share numerous similarities, the molecules produced by the first method are markedly different from those produced by the latter. This is primarily due to the presence of conformational flexibility, which has a substantial impact on the compounds’ biological properties. Hybrid compounds are believed to exhibit enhanced therapeutic efficacy (lower IC50 values) [18] but greater toxicity to healthy cells in comparison with spiro compounds [19]. Such hybrid-designed molecules may contain a third heterocycle as a linker, spiro-joined with one of the pharmacophore moieties. In this case, another pharmacophore fragment is included in the compound as one of the substituents in the central cycle [8,20]. We believe that this approach has the potential to be a successful combination of the presented design strategies. It enables the pharmacophore fragments to be positioned relative to one another in a specific manner, as determined by the configuration of the spiro-core, while still allowing for the possibility of relative rotation to facilitate better binding to the targets. Furthermore, it appears to be as exceedingly advantageous from a synthesis perspective, as it enables the application of methods that have been developed for existing spiro systems to the new hybrid drugs.
In this work a similar modification of imidazolidine derivatives was performed for the first time for the synthesis of spiro-hydantoins. The title reactions were carried out using two alternative techniques for the generation of the reactive 1,3-dipoles. These techniques differed in the method of introducing the base necessary for generating the 1,3-dipole into the reaction medium, and included the base addition “drop by drop” or using a diffusion mixing procedure [21]. We found that the formation of spiro-compounds occurs chemo- and regioselectively for both methods. In most cases, the reactions proceeded with high yield and without the formation of significant byproducts.
Results and Discussion
Synthesis
Hydantoins 2, containing an exocyclic C=N group, were synthesized according to the methodology described in [22] (Scheme 1). In brief, N,N'-disubstituted ureas were initially reacted with oxalyl chloride to form imidazolidinetriones 1a,b, which were then added to an iminophosphorane formed in situ from an aryl azide and triphenylphosphine. As a result of the aza-Wittig reaction, 5-iminohydantoins 2a–i were then used as dipolarophiles in the 32CA reactions with nitrile oxides.
Scheme 1:
Synthesis of dipolarophiles (5-iminohydantoins 2a–i).
Scheme 1:
Synthesis of dipolarophiles (5-iminohydantoins 2a–i).
The nitrile oxide precursors, hydroxyimidoyl chlorides 4a–d were prepared according to known methods [23,24] (Scheme 2). Conversion of benzaldehydes to the corresponding benzaldoximes 3a–c was achieved through a reaction with hydroxylamine hydrochloride and a base. The dipole precursors 4a–c were then prepared by the reaction of compounds 3a–c with N-chlorosuccinimide (NCS) in dimethylformamide (DMF). The ester group-containing chloro oxime 4d was obtained by the treatment of glycine ethyl ester hydrochloride with sodium nitrite and hydrochloric acid [25] and used thereafter as carbethoxyformonitrile oxide (CEFNO) precursor.
Scheme 2:
Preparation of the dipole precursors 4a–d.
Scheme 2:
Preparation of the dipole precursors 4a–d.
The nitrile oxides were generated in situ by the action of a base (Et3N) to compounds 4a–d in the dipolarophile-containing solution to prevent high concentrations of dipole molecules and to minimize the inevitable competition between the 32CA reaction and the isomerization of nitrile oxides to isocyanates or their dimerization [26,27]. Since isomerization is believed to be the primary direction of molecule degradation only for bulky o,o'-disubstituted aromatic dipoles [28-30], the main efforts in developing a methodology were focused on suppressing dimerization processes. It is worth noting that, in some cases, due to the high reactivity of nitrile oxides towards certain types of dipolarophiles, such as monosubstituted or activated by electron-acceptor groups olefins [31], no additional attention to this is required. However, when introducing obstructed dipolarophiles into the 32CA, it is reasonable to utilize conditions that minimize the side reactions of the dipole. This could include lowering the temperature [32] and slowly adding a base [33]. In this work we have tested two alternative techniques for the dipolar cycloaddition. In the first method, a triethylamine solution was slowly added dropwise to the reaction mixture at 0 °C under an inert gas atmosphere to avoid moisture from the air (“classical” method). In another method, the base was inserted via diffusion of NEt3 into a solution containing the dipolarophile and dipole precursor (see Figure S1 in Supporting Information File 1 for a schematic reaction setup) [21]. This method, previously tested in other reactions of nitrile oxides [21] and nitrile imines [22], was chosen as a reliable technique for the slow and consistent generation of the reactive dipole. In all cases the dipole precursor 4 was used in small excess (1.1 equiv) over the dipolarophile 2 (1 equiv). Using both methods, a series of products 5a–l containing spiro-conjugated fragments of hydantoin and 1,2,4-oxadiazole was obtained (Scheme 3).
Scheme 3:
32CA reactions of nitrile oxides with 5-iminohydantoins (synthesis of spiro-compounds 5a–l). Isolated yields are shown. Unless otherwise noted dipolarophile 2 (0.15 mmol) and chloro oxime 4 (0.165 mmol) were used, and the product 5 was isolated via Et2O trituration. aTriethylamine dropwise addition (1.2 equiv, 0.06 M CH2Cl2 solution, Ar, 0–5 °C); bdiffusion mixing technique (for details see Supporting Information File 1), cusing diffusion mixing N-(2-bromophenyl)formamide (6) was obtained with 80% yield; dproduct was isolated using column chromatography.
Scheme 3:
32CA reactions of nitrile oxides with 5-iminohydantoins (synthesis of spiro-compounds 5a–l). Isolat...
In most cases, the use of the “classical” drop-by-drop method gave compounds 5a–l in yields near or superior to those obtained by diffusion mixing. However, there were three exceptions (5d, 5g and 5h) where we observed a significant difference in the results. The most striking example was compound 5h, which exhibited a yield decrease of more than a third when the diffusion mixing technique was employed. The substantial impact of the reaction conditions can be attributed to the stability of the dipole formed. The presence of mesomeric donor substituent (such as 4-OMe group) has been shown to render nitrile oxides more susceptible to dimerization in comparison to other dipoles [27,34]. The most dramatic discrepancy in spiro-compound yields was observed during the reaction with this nitrile oxide, suggesting that classical conditions (low temperature and inert atmosphere) may prove more fruitful in preventing the degradation of the dipole. The influence of the concentration of the generated nitrile oxide seems to be less significant.
It was found that the low solubility of products containing aromatic substituents in non-polar solvents allows for their isolation by washing the crude reaction mixture with diethyl ether to remove organic impurities and then with water to eliminate triethylamine hydrochloride. This method has proven to be simple and effective in most cases, with the exception of compound 5g. The moderate solubility of this compound in diethyl ether led to partial precipitation from the reaction mixture. It is worth noting that chromatographic purification is also possible for compounds 5a–l. However, in most cases, this method is more laborious due to the similarity in the retention factor values of the initial imines 2 and the reaction products. In this way, products 5i and 5j with good solubility in diethyl ester were isolated.
A comparative analysis of compounds 5a–l yields revealed that in the absence of aromatic substituents with strong electron-donating or electron-accepting properties, the reaction occurred with near-complete conversion of the initial dipolarophile and high yields of the products (as, for example, 5a–d and 5f). The best result, however, was observed for the spiro-compound 5l, which contains a p-nitro group in the aromatic core of the nitrile oxide fragment. This compound was obtained in excellent yield of 93% and 88% using the dropwise and diffusion mixing technique, respectively. This result may be explained by the optimal congruence of the electronic properties of the substituents in the dipole and dipolarophile, as well as very low solubility of the product 5l in diethyl ether.
An unexpected result was obtained during the 1,3-dipolar cycloaddition reaction between 2f and chloro oxime 4d. In contrast to reactions involving benzonitrile oxides, in this case the dropwise addition method of triethylamine resulted in complete inactivity of the dipole towards the dipolarophile 2f, which was quantitatively recovered from the reaction mixture. The low reactivity of CEFNO can be explained by considering nitrile oxides in terms of frontier molecular orbital (FMO) theory. Nitrile oxides are considered to be "type II" dipoles capable of reacting both through the interaction of HOMOdipole–LUMOdipolarophile and LUMOdipole–HOMOdipolarophile[35]. According to the results presented in [36], reactions with benzonitrile oxide can satisfy both normal and inverse-electron demands, depending on the electronic properties of substituents in the aromatic fragments of the dipole and dipolarophile. Our result may indicate a predominant overlap of HOMOdipole and LUMOdipolarophile (inverse-electron demands) in the reactions of the imine 2f. In this case, the strong electron-withdrawing properties of the carboxyethyl group in CEFNO can significantly inhibit the reaction with 2f, lowering the energy of the HOMOdipole involved in the interaction. Instead, the reaction can proceed towards dipole dimerization [37], which is generally much easier for nitrile oxides that are not stabilized by an aromatic group [28]. By performing the diffusion mixing technique, we were also unable to detect the cycloaddition product. Instead, after 3 weeks of reaction, exceptionally N-(2-bromophenyl)formamide (6) was isolated with 80% yield. Apparently, it is the product of decomposition of the initial imine 2f. No other substances could be identified in this case.
In all cases, the 32CA reactions occurred regiospecifically, forming a 1,2,4-oxadiazoline ring, which is consistent with the results of the reactions for similar substrates [36,38]. The regioselectivity of the cycloaddition reaction between nitrile oxides and C=N bonds can be rationalized by considering the relative electronegativities of the terminal elements and the distribution of electron density in the frontier orbitals of the reagents [35]. The electrons are preferentially located on the more electronegative oxygen or nitrogen atoms, both in the HOMO of dipole and dipolarophile [36,39]. In contrast, the carbon atoms have a significantly higher orbital coefficient than the heteroatoms, both in the LUMOdipolarophile and the LUMOdipole. Thus, nitrile oxide tends to react with an imine to form two new carbon–heteroatom bonds in a 1,2,4-oxadiazoline ring through both variants of orbital overlap. It should be noted that, in this case, NMR spectroscopy is not applicable to assign the product structure due to the presence of only quaternary carbon atoms in the tetrasubstituted 1,2,4-oxadiazolines [40]. Therefore, single crystal X-ray diffraction analysis is typically used to confirm the structure, as we have applied for the hydantoin/1,2,4-oxadiazoline spiro-compounds 5k (CCDC 2432465) and 5l (CCDC 2432466) (for details see Supporting Information File 1).
The effect of the substituents on the N1 and N3 nitrogen atoms in the hydantoin core was studied in the reactions of hydroxyimidoyl chloride 4a with dipolarophiles 2g, 2h, and 2i, resulting in the products 5c (83% yield), 5i (91%), and 5j (25%), respectively (Scheme 3). The replacement of a phenyl (2g) with an alkyl substituent at the N1 nitrogen atom (2i) led to a sharp decrease in the yield of the cycloaddition product 5j, whereas the presence of the N3-CH2COOEt (2h) fragment slightly enhanced the yield of 5i compared to the bis-phenyl substituted dipolarophile 2g. The low yield observed for product 5j may be accounted for by a greater involvement of the N1 electron pair in conjugation with the imide fragment of the dipolarophile and its deactivation in the reaction with nitrile oxide. This pattern is consistent with the one previously observed when studying the reactions of these dipolarophiles with nitrile imines [22].
It is known that nitrile oxides are capable of reacting with various multiple carbon–heteroatom bonds [28]. In contrast to the products of nitrile oxide cycloaddition to imino groups, 1,4,2-oxathiazoles, formed by addition to C=S bonds, are unstable and undergo decomposition into isothiocyanate and a carbonyl-containing compound [38]; the stability of these compounds is contingent on the nature of the substituents bonded to the thiocarbonyl group, which reacts with nitrile oxide [41]. It has been demonstrated [7] that 1,4,2-oxathiazoles derived from thiourea are among the least stable compounds, gradually decomposing even at room temperature. Taking these into account, we have synthesized compound 5c using an alternative route starting from the chloro oxime 4a and 5-iminothiohydantoin 2j (Scheme 4), obtained similarly to other dipolarophiles [22]. The total yield of the product 5c in this case turned out to be comparable to that obtained for all stages of the initial scheme (synthesis of 5c from 2g, Scheme 3). The yield of the desulfurized product was moderate both with triethylamine dropwise addition (51%) and with diffusion mixing technique (46%).
Scheme 4:
Cycloaddition of nitrile oxide to 5-iminothiohydantoin 2j. aTriethylamine dropwise addition (2.4 equiv, 0.06 M CH2Cl2 solution, Ar, 0–5 °C); bdiffusion mixing technique.
Scheme 4:
Cycloaddition of nitrile oxide to 5-iminothiohydantoin 2j. aTriethylamine dropwise addition (2.4 eq...
Interestingly, for spiro-compounds 5b and 5d containing an ortho-substituted aromatic group, we have observed atropoisomerism (Figure 2) what is manifested by the appearance of a second set of signals in their NMR spectra. As the previously studied similar structures (Y = N–R) [22], products 5b and 5d (Y = O) were formed through the addition of a dipole molecule, resulting in the formation of two new carbon–heteroatom bonds and the formation of a five-membered ring. Thus, the aryl attached to the nitrogen atom from the dipolarophile fragment is brought closer to the substituent from the C-end of the dipole. This proximity in the presence of ortho-substituents in the aromatic core hinders rotation around the C–N bond.
Figure 2:
Atropoisomerism of ortho-substituted spiro-compounds 5b and 5d.
Figure 2:
Atropoisomerism of ortho-substituted spiro-compounds 5b and 5d.
The cytotoxic properties of hydantoin/1,2,4-oxadiazoline spiro-compounds were investigated using the MTT [42] assay on human colorectal carcinoma cell line HCT116.
To evaluate the cytotoxicity of the compounds in vitro, cells were placed in 96-well culture plates at a concentration of 4–7 × 103 cells/mL and incubated at 37 °C for 24 hours. The cells were counted after treatment with trypan blue solution (0.4%) in a Goryaev chamber. Then, after incubation at 37 °C for 72 h, the cells were exposed to various concentrations of the studied compounds in two-fold serial (50–100 μM) dilutions. Only DMSO + PBS (phosphate-buffered saline) was used as a control, since the studied compounds were soluble only in DMSO. Cell viability was measured using the standard MTT test [42]. Absorbance was measured at 540 nm using a Multiskan™ FC microplate reader and Skanlt 6.1 RE software for a microplate reader, both from Thermo Scientific (Waltham, MA, USA). In vitro experiments were performed in triplicate. Graphpad prism version 9.0 was used to determine the IC50. IC50 data are presented as mean ± standard deviation (SD) (Figure 3b) [20,22].
Figure 3:
Cytotoxicity investigation of hydantoin/1,2,4-oxadiazolines 5 (MTT test, HCT116 cell line) and selected examples of oxindole/1,2,4-oxadiazole [20] and hydantoin/1,2,4-triazoline [22] spiro-compounds.
Figure 3:
Cytotoxicity investigation of hydantoin/1,2,4-oxadiazolines 5 (MTT test, HCT116 cell line) and sele...
Notwithstanding the moderate cytotoxicity exhibited by the obtained compounds, they demonstrated a slight improvement over those previously obtained for the hydantoin/1,2,4-triazoline spiro-compounds [22] (Figure 3c, IC50 ≈ 35–45 µM) and were more prominent than oxindolo/1,2,4-oxadiazoles (Figure 3a, IC50 ≈ 50–5 µM) tested on this cell line in the reference [20].
Two of the five compounds studied in this work (5d and 5g) demonstrated IC50 values of about 30 µM. The data obtained indicates that products 5 exhibit more pronounced cytotoxic properties when they contain a p-methoxy group in the dipole fragment (Figure 3b, red ring) than 4-NO2 or 4-Cl substituents (IC50 of 5h was lower than 5l and 5f). The presence of a methoxy group in the imine aromatic core (blue ring) also had a significant impact on cytotoxicity. This can be concluded from the results obtained for compounds 5f and 5g: the substitution of -CH3 to -OCH3 led to a decrease in IC50 by at least 20 µM and turned out to be comparable to that obtained for 5d.
Conclusion
In this study, we firstly investigated the 1,3-dipolar cycloaddition reactions of nitrile oxides to 5-iminohydantoins at their exocyclic C=N bond. A convenient preparative synthesis of hydantoin/1,2,4-oxadiazoline spiro-compounds using two different methods for introducing a tertiary amine into the reaction mixture containing the dipolarophile and precursor of the 1,3-dipole (chloro oxime), either by dropwise addition or by diffusion mixing. By varying the substituents on N1, N3, and the imino group of the dipolarophile as well as the dipole structure (aryl and alkyl), we determined the synthetic potential of the method and identified the most favorable electronic and steric combinations of substituents that affect the course of the studied 32CA reactions. The cytotoxicity of the obtained compounds was evaluated using the HCT116 cell line. It has been demonstrated that the combination of the hydantoin and 1,2,4-oxadiazoline moieties in a single spiro-fused system leads to increased cytotoxic activity compared to other spiro-derivatives of hydantoin and 1,2,4-oxadiazole.
Experimental
General procedure for the synthesis of the spirocyclic products
Triethylamine dropwise addition: A solution of dipolarophile 2 (1 equiv, 0.150 mmol) and N-hydroxybenzimidoyl chloride 4 (1.1 equiv, 0.165 mmol) in 4.5 mL of DCM was added to a 25 mL round-bottomed flask equipped with a magnetic stirring bar and a dripping funnel. After a solution of Et3N (1.2 equiv, 0.180 mmol, 25 µL) in DCM (3 mL) had been added to the funnel, the system was filled with argon and placed in an ice bath. A few minutes later, the Et3N solution was added dropwise to the mixture in the flask, while it was stirred. After that, the reaction mixture was left to stir in the ice bath for 24 hours, allowing it to slowly reach room temperature.
Diffusion mixing technique: A small vial (15 mL volume, diameter 1.3 cm) equipped with a magnetic stirring bar was charged with a mixture of dipolarophile 2 (1 equiv, 0.150 mmol) and hydroxyimidoyl chloride 4 (1.1 equiv, 0.165 mol) in 4.5 mL of DCM and then placed in larger vial (50 mL volume, diameter 3.5 cm) containing TEA (35.85 mmol, 5 mL). The outer vial was tightly closed with a lid and the reaction mixture was stirred at room temperature for 24 h.
In both cases, after the reaction time had expired, the solvent was removed in vacuo. The residue was washed with 2–3 small portions (0.5–1 mL) of Et2O and water giving a solid precipitate of the product (compounds 5a–d, 5f–h and 5k–l). Compounds 5i and 5j were isolated from the reaction mixtures by column chromatography on silica gel using DCM as eluent.
Supporting Information
Supporting Information File 1:
Detailed experimental procedures, characterization data and X-ray crystallographic details.
The synthetic research was funded by the Russian Science Foundation, grant number 24-23-00173. Сell viability tests were supported by the Ministry of Science and Higher Education of Russia no. 075-01551-23-00 (FSSF-2023-0006). Single crystal X-ray diffraction analysis study was supported by the RUDN University Scientific Projects Grant System, project №021408-2-000.
Author Contributions
Juliana V. Petrova: investigation; methodology; visualization; writing – original draft. Varvara T. Tkachenko: investigation; validation. Victor A. Tafeenko: investigation; methodology. Anna S. Pestretsova: investigation; visualization; writing – review & editing. Vadim S. Pokrovsky: methodology; resources; validation; writing – review & editing. Maxim E. Kukushkin: conceptualization; funding acquisition; methodology; project administration; validation. Elena K. Beloglazkina: conceptualization; project administration; writing – review & editing.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
Biernacki, K.; Daśko, M.; Ciupak, O.; Kubiński, K.; Rachon, J.; Demkowicz, S. Pharmaceuticals2020,13, 111. doi:10.3390/ph13060111
Return to citation in text:
[1]
Pace, A.; Buscemi, S.; Piccionello, A. P.; Pibiri, I. Adv. Heterocycl. Chem.2015,116, 85–136. doi:10.1016/bs.aihch.2015.05.001
Return to citation in text:
[1]
Ribeiro, C. J. A.; Amaral, J. D.; Rodrigues, C. M. P.; Moreira, R.; Santos, M. M. M. MedChemComm2016,7, 420–425. doi:10.1039/c5md00450k
Return to citation in text:
[1]
[2]
Ivanenkov, Y. A.; Vasilevski, S. V.; Beloglazkina, E. K.; Kukushkin, M. E.; Machulkin, A. E.; Veselov, M. S.; Chufarova, N. V.; Chernyaginab, E. S.; Vanzcool, A. S.; Zyk, N. V.; Skvortsov, D. A.; Khutornenko, A. A.; Rusanov, A. L.; Tonevitsky, A. G.; Dontsova, O. A.; Majouga, A. G. Bioorg. Med. Chem. Lett.2015,25, 404–409. doi:10.1016/j.bmcl.2014.09.070
Return to citation in text:
[1]
Wang, Y.; Deng, Y.; Pang, X.; Yu, J.; Fan, L.; Chen, Y.; Zhao, L. RSC Adv.2017,7, 31866–31874. doi:10.1039/c7ra02142a
Return to citation in text:
[1]
Hewitt, R. J.; Ong, M. J. H.; Lim, Y. W.; Burkett, B. A. Eur. J. Org. Chem.2015, 6687–6700. doi:10.1002/ejoc.201500909
Return to citation in text:
[1]
[2]
de Azevedo, L. D.; Leite, D. I.; de Oliveira, A. P.; Junior, F. P. S.; Dantas, R. F.; Bastos, M. M.; Boechat, N.; Pimentel, L. C. F. Arch. Pharm. (Weinheim, Ger.)2024,357, 2400029. doi:10.1002/ardp.202400029
Return to citation in text:
[1]
[2]
Kanchrana, M.; Gamidi, R. K.; Kumari, J.; Sriram, D.; Basavoju, S. Mol. Diversity2024,28, 3979–3991. doi:10.1007/s11030-023-10790-9
Return to citation in text:
[1]
Awad, W. I.; Sobhy, M. Can. J. Chem.1969,47, 1473–1477. doi:10.1139/v69-243
Return to citation in text:
[1]
Ito, K.; Saito, K. Bull. Chem. Soc. Jpn.1995,68, 3539–3547. doi:10.1246/bcsj.68.3539
Return to citation in text:
[1]
Rajanarendar, E.; Raju, S.; Reddy, A. S. R. Phosphorus, Sulfur Silicon Relat. Elem.2009,184, 3139–3148. doi:10.1080/10426500802705164
Return to citation in text:
[1]
Lv, M.; Ma, Q.; Zhang, S.; Xu, H. Bioorg. Med. Chem. Lett.2021,51, 128356. doi:10.1016/j.bmcl.2021.128356
Return to citation in text:
[1]
Upadhyay, N.; Tilekar, K.; Loiodice, F.; Anisimova, N. Y.; Spirina, T. S.; Sokolova, D. V.; Smirnova, G. B.; Choe, J.-y.; Meyer-Almes, F.-J.; Pokrovsky, V. S.; Lavecchia, A.; Ramaa, C. Bioorg. Chem.2021,107, 104527. doi:10.1016/j.bioorg.2020.104527
Return to citation in text:
[1]
El-Shafey, H. W.; Gomaa, R. M.; El-Messery, S. M.; Goda, F. E. Bioorg. Chem.2020,101, 103987. doi:10.1016/j.bioorg.2020.103987
Return to citation in text:
[1]
Torres, R. R., Ed. Spiro Compounds; John Wiley & Sons: Hoboken, NJ, USA, 2022. doi:10.1002/9781119567646
Return to citation in text:
[1]
Al-Sanea, M. M.; Hamdi, A.; Mohamed, A. A. B.; El-Shafey, H. W.; Moustafa, M.; Elgazar, A. A.; Eldehna, W. M.; Ur Rahman, H.; Parambi, D. G. T.; Elbargisy, R. M.; Selim, S.; Bukhari, S. N. A.; Magdy Hendawy, O.; Tawfik, S. S. J. Enzyme Inhib. Med. Chem.2023,38, 2166036. doi:10.1080/14756366.2023.2166036
Return to citation in text:
[1]
Romero-Hernández, L. L.; Ahuja-Casarín, A. I.; Merino-Montiel, P.; Montiel-Smith, S.; Vega-Báez, J. L.; Sandoval-Ramírez, J. Beilstein J. Org. Chem.2024,20, 1713–1745. doi:10.3762/bjoc.20.152
Return to citation in text:
[1]
Kanchrana, M.; Allaka, B. S.; Krishna, G. R.; Basavoju, S. ARKIVOC2023, No. vi, 202211940. doi:10.24820/ark.5550190.p011.940
Return to citation in text:
[1]
[2]
[3]
[4]
Shybanov, D. E.; Filkina, M. E.; Kukushkin, M. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Zyk, N. V.; Beloglazkina, E. K. New J. Chem.2022,46, 18575–18586. doi:10.1039/d2nj03756d
Return to citation in text:
[1]
[2]
[3]
Kuznetsova, J. V.; Tkachenko, V. T.; Petrovskaya, L. M.; Filkina, M. E.; Shybanov, D. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Tafeenko, V. A.; Pestretsova, A. S.; Yakovleva, V. A.; Pokrovsky, V. S.; Kukushkin, M. E.; Beloglazkina, E. K. Int. J. Mol. Sci.2023,25, 18. doi:10.3390/ijms25010018
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
Liu, K.-C.; Shelton, B. R.; Howe, R. K. J. Org. Chem.1980,45, 3916–3918. doi:10.1021/jo01307a039
Return to citation in text:
[1]
Castellano, S.; Kuck, D.; Viviano, M.; Yoo, J.; López-Vallejo, F.; Conti, P.; Tamborini, L.; Pinto, A.; Medina-Franco, J. L.; Sbardella, G. J. Med. Chem.2011,54, 7663–7677. doi:10.1021/jm2010404
Return to citation in text:
[1]
Kozikowski, A. P.; Adamcz, M. J. Org. Chem.1983,48, 366–372. doi:10.1021/jo00151a017
Return to citation in text:
[1]
Bode, J. W.; Carreira, E. M. J. Org. Chem.2001,66, 6410–6424. doi:10.1021/jo015791h
Return to citation in text:
[1]
Barbaro, G.; Battaglia, A.; Dondoni, A. J. Chem. Soc. B1970, 588–592. doi:10.1039/j29700000588
Return to citation in text:
[1]
[2]
Belen'kii, L. I. Nitrile Oxides. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp 1–127. doi:10.1002/9780470191552.ch1
Return to citation in text:
[1]
[2]
[3]
Pasinszki, T.; Hajgató, B.; Havasi, B.; Westwood, N. P. C. Phys. Chem. Chem. Phys.2009,11, 5263–5272. doi:10.1039/b823406j
Return to citation in text:
[1]
Grundmann, C.; Dean, J. M. J. Org. Chem.1965,30, 2809–2812. doi:10.1021/jo01019a074
Return to citation in text:
[1]
Zhao, G.; Liang, L.; Wen, C. H. E.; Tong, R. Org. Lett.2019,21, 315–319. doi:10.1021/acs.orglett.8b03829
Return to citation in text:
[1]
Balsamini, C.; Bedini, A.; Galarini, R.; Spadoni, G.; Tarzia, G.; Hamdan, M. Tetrahedron1994,50, 12375–12394. doi:10.1016/s0040-4020(01)89546-6
Return to citation in text:
[1]
Jiang, H.; Zhao, J.; Han, X.; Zhu, S. Tetrahedron2006,62, 11008–11011. doi:10.1016/j.tet.2006.06.119
Return to citation in text:
[1]
Quilico, A. Experientia1970,26, 1169–1183. doi:10.1007/bf01897949
Return to citation in text:
[1]
Houk, K. N.; Sims, J.; Duke, R. E.; Strozier, R. W.; George, J. K. J. Am. Chem. Soc.1973,95, 7287–7301. doi:10.1021/ja00803a017
Return to citation in text:
[1]
[2]
Alcaide, B.; Mardomingo, C. L.; Plumet, J.; Cativiela, C.; Mayoral, J. A. Can. J. Chem.1987,65, 2050–2056. doi:10.1139/v87-340
Return to citation in text:
[1]
[2]
[3]
Caramella, P.; Gandour, R. W.; Hall, J. A.; Deville, C. G.; Houk, K. N. J. Am. Chem. Soc.1977,99, 385–392. doi:10.1021/ja00444a013
Return to citation in text:
[1]
Kesornpun, C.; Aree, T.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Angew. Chem.2016,128, 4065–4069. doi:10.1002/ange.201511730
Return to citation in text:
[1]
Belen'kii, L. I. Nitrile Oxides. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp 1–127. doi:10.1002/9780470191552.ch1
Kuznetsova, J. V.; Tkachenko, V. T.; Petrovskaya, L. M.; Filkina, M. E.; Shybanov, D. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Tafeenko, V. A.; Pestretsova, A. S.; Yakovleva, V. A.; Pokrovsky, V. S.; Kukushkin, M. E.; Beloglazkina, E. K. Int. J. Mol. Sci.2023,25, 18. doi:10.3390/ijms25010018
Belen'kii, L. I. Nitrile Oxides. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp 1–127. doi:10.1002/9780470191552.ch1
Kuznetsova, J. V.; Tkachenko, V. T.; Petrovskaya, L. M.; Filkina, M. E.; Shybanov, D. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Tafeenko, V. A.; Pestretsova, A. S.; Yakovleva, V. A.; Pokrovsky, V. S.; Kukushkin, M. E.; Beloglazkina, E. K. Int. J. Mol. Sci.2023,25, 18. doi:10.3390/ijms25010018
Kuznetsova, J. V.; Tkachenko, V. T.; Petrovskaya, L. M.; Filkina, M. E.; Shybanov, D. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Tafeenko, V. A.; Pestretsova, A. S.; Yakovleva, V. A.; Pokrovsky, V. S.; Kukushkin, M. E.; Beloglazkina, E. K. Int. J. Mol. Sci.2023,25, 18. doi:10.3390/ijms25010018
Kuznetsova, J. V.; Tkachenko, V. T.; Petrovskaya, L. M.; Filkina, M. E.; Shybanov, D. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Tafeenko, V. A.; Pestretsova, A. S.; Yakovleva, V. A.; Pokrovsky, V. S.; Kukushkin, M. E.; Beloglazkina, E. K. Int. J. Mol. Sci.2023,25, 18. doi:10.3390/ijms25010018
Kuznetsova, J. V.; Tkachenko, V. T.; Petrovskaya, L. M.; Filkina, M. E.; Shybanov, D. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Tafeenko, V. A.; Pestretsova, A. S.; Yakovleva, V. A.; Pokrovsky, V. S.; Kukushkin, M. E.; Beloglazkina, E. K. Int. J. Mol. Sci.2023,25, 18. doi:10.3390/ijms25010018
Kuznetsova, J. V.; Tkachenko, V. T.; Petrovskaya, L. M.; Filkina, M. E.; Shybanov, D. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Tafeenko, V. A.; Pestretsova, A. S.; Yakovleva, V. A.; Pokrovsky, V. S.; Kukushkin, M. E.; Beloglazkina, E. K. Int. J. Mol. Sci.2023,25, 18. doi:10.3390/ijms25010018
Ribeiro, C. J. A.; Amaral, J. D.; Rodrigues, C. M. P.; Moreira, R.; Santos, M. M. M. MedChemComm2016,7, 420–425. doi:10.1039/c5md00450k
8.
de Azevedo, L. D.; Leite, D. I.; de Oliveira, A. P.; Junior, F. P. S.; Dantas, R. F.; Bastos, M. M.; Boechat, N.; Pimentel, L. C. F. Arch. Pharm. (Weinheim, Ger.)2024,357, 2400029. doi:10.1002/ardp.202400029
9.
Kanchrana, M.; Gamidi, R. K.; Kumari, J.; Sriram, D.; Basavoju, S. Mol. Diversity2024,28, 3979–3991. doi:10.1007/s11030-023-10790-9
Shybanov, D. E.; Filkina, M. E.; Kukushkin, M. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Zyk, N. V.; Beloglazkina, E. K. New J. Chem.2022,46, 18575–18586. doi:10.1039/d2nj03756d
Kuznetsova, J. V.; Tkachenko, V. T.; Petrovskaya, L. M.; Filkina, M. E.; Shybanov, D. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Tafeenko, V. A.; Pestretsova, A. S.; Yakovleva, V. A.; Pokrovsky, V. S.; Kukushkin, M. E.; Beloglazkina, E. K. Int. J. Mol. Sci.2023,25, 18. doi:10.3390/ijms25010018
Romero-Hernández, L. L.; Ahuja-Casarín, A. I.; Merino-Montiel, P.; Montiel-Smith, S.; Vega-Báez, J. L.; Sandoval-Ramírez, J. Beilstein J. Org. Chem.2024,20, 1713–1745. doi:10.3762/bjoc.20.152
Ribeiro, C. J. A.; Amaral, J. D.; Rodrigues, C. M. P.; Moreira, R.; Santos, M. M. M. MedChemComm2016,7, 420–425. doi:10.1039/c5md00450k
4.
Ivanenkov, Y. A.; Vasilevski, S. V.; Beloglazkina, E. K.; Kukushkin, M. E.; Machulkin, A. E.; Veselov, M. S.; Chufarova, N. V.; Chernyaginab, E. S.; Vanzcool, A. S.; Zyk, N. V.; Skvortsov, D. A.; Khutornenko, A. A.; Rusanov, A. L.; Tonevitsky, A. G.; Dontsova, O. A.; Majouga, A. G. Bioorg. Med. Chem. Lett.2015,25, 404–409. doi:10.1016/j.bmcl.2014.09.070
de Azevedo, L. D.; Leite, D. I.; de Oliveira, A. P.; Junior, F. P. S.; Dantas, R. F.; Bastos, M. M.; Boechat, N.; Pimentel, L. C. F. Arch. Pharm. (Weinheim, Ger.)2024,357, 2400029. doi:10.1002/ardp.202400029
Al-Sanea, M. M.; Hamdi, A.; Mohamed, A. A. B.; El-Shafey, H. W.; Moustafa, M.; Elgazar, A. A.; Eldehna, W. M.; Ur Rahman, H.; Parambi, D. G. T.; Elbargisy, R. M.; Selim, S.; Bukhari, S. N. A.; Magdy Hendawy, O.; Tawfik, S. S. J. Enzyme Inhib. Med. Chem.2023,38, 2166036. doi:10.1080/14756366.2023.2166036
Kuznetsova, J. V.; Tkachenko, V. T.; Petrovskaya, L. M.; Filkina, M. E.; Shybanov, D. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Tafeenko, V. A.; Pestretsova, A. S.; Yakovleva, V. A.; Pokrovsky, V. S.; Kukushkin, M. E.; Beloglazkina, E. K. Int. J. Mol. Sci.2023,25, 18. doi:10.3390/ijms25010018
Shybanov, D. E.; Filkina, M. E.; Kukushkin, M. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Zyk, N. V.; Beloglazkina, E. K. New J. Chem.2022,46, 18575–18586. doi:10.1039/d2nj03756d
Shybanov, D. E.; Filkina, M. E.; Kukushkin, M. E.; Grishin, Y. K.; Roznyatovsky, V. A.; Zyk, N. V.; Beloglazkina, E. K. New J. Chem.2022,46, 18575–18586. doi:10.1039/d2nj03756d
Belen'kii, L. I. Nitrile Oxides. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp 1–127. doi:10.1002/9780470191552.ch1
29.
Pasinszki, T.; Hajgató, B.; Havasi, B.; Westwood, N. P. C. Phys. Chem. Chem. Phys.2009,11, 5263–5272. doi:10.1039/b823406j



![[1860-5397-21-118-1]](/bjoc/content/figures/1860-5397-21-118-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-21-118-i1]](/bjoc/content/inline/1860-5397-21-118-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-21-118-i2]](/bjoc/content/inline/1860-5397-21-118-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-21-118-i3]](/bjoc/content/inline/1860-5397-21-118-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-21-118-i4]](/bjoc/content/inline/1860-5397-21-118-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-21-118-2]](/bjoc/content/figures/1860-5397-21-118-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-21-118-3]](/bjoc/content/figures/1860-5397-21-118-3.svg?scale=2.0&max-width=1024&background=FFFFFF)