Abstract
The influence of transition-state aromaticity on the barrier heights of concerted pericyclic reactions is summarized herein. To this end, selected representative examples ranging from fundamental processes such as Diels–Alder or Alder–ene reactions to double-group transfer reactions or 1,3-dipolar cycloadditions involving metal complexes are presented. It is found that while more synchronous processes tend to exhibit greater aromatic character in their transition states, this increased aromaticity does not necessarily correlate with lower activation barriers. State-of-the-art computational methods on reactivity, such as the combined activation strain model (ASM)–energy decomposition analysis (EDA) method, reveal that factors other than aromaticity govern the barrier heights of these pericyclic reactions.

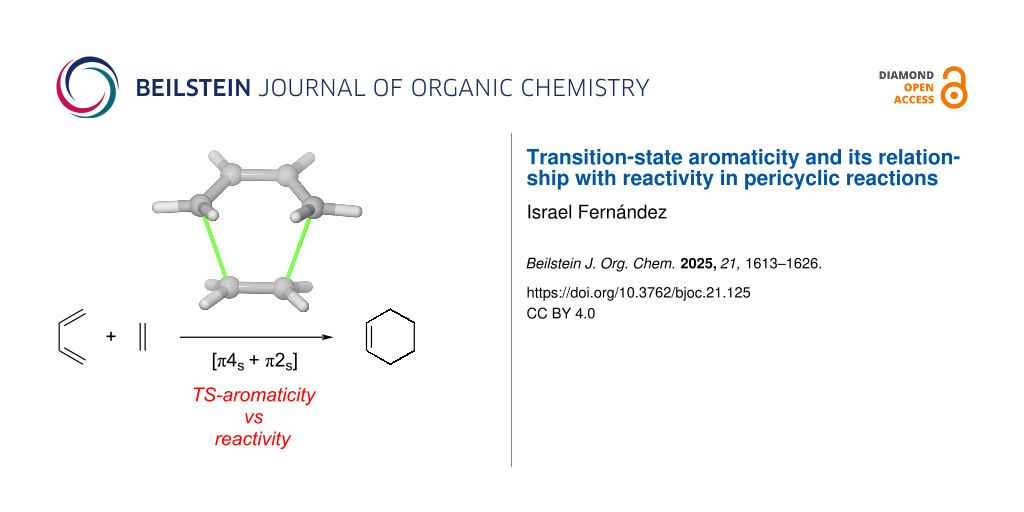
Graphical Abstract
Introduction
Aromaticity is arguably one of the most fundamental and extensively studied concepts in chemistry [1-3]. Initially introduced to account for the remarkable stability, low reactivity, and unique structural features of benzene and related aromatic hydrocarbons, the concept has undergone a significant evolution [4]. Since the introduction of the famous Hückel’s (4n + 2) rule [5], first clearly defined by Doering and Detert [6], this concept has been extended well beyond benzenoid molecules to organometallic, inorganic, and even saturated molecules. As a result, besides classical π-aromaticity, other aromaticity types such as Möebius, spherical, or excited-state aromaticity (to name a few) have been introduced [7-11].
The concept of aromaticity was also extended to the transition states (TSs) of concerted pericyclic reactions as early as 1938 when Evans and Warhurst [12] recognized the relationship between the six π-electrons of benzene and the six delocalized electrons in the transition structure of the Diels–Alder cycloaddition reaction between butadiene and ethylene “…Very qualitatively, we may say that whereas in the initial state the mobile electrons are those characteristics of an ethylene and a butadiene structure, in the TS they simulate the behavior of a benzene molecule”. This initial recognition was later generalized by Zimmerman and Dewar [13,14] who extended the Hückel–Möbius aromaticity concepts to different pericyclic reactions including not only cycloaddition reactions but also electrocyclizations or sigmatropic reactions, linking them to the Woodward–Hoffmann rules [15]. In this regard, it has been suggested that thermally allowed pericyclic reactions take place preferentially through concerted aromatic transition states, which are favored energetically and therefore display faster rates (i.e., lower barriers) [16,17]. This is in line with the seminal conclusion by Evans in 1939 [18] indicating that “...the lowering of the activation energy arises from the increased mobility which the π electrons of such reactions possess in the TS.” Indeed, it was found, both experimentally and computationally, that the concerted pathway (i.e., involving an aromatic TS) of the butadiene + ethylene reaction proceeds with a lower barrier (up to 7 kcal/mol) than the corresponding stepwise pathway leading to a diradical intermediate (Scheme 1a) [19,20]. Similar preferences for concerted pathways over stepwise mechanisms were also found for dipolar cycloadditions and Cope rearrangements [20]. More recently, Alabugin and co-workers reported that the gold(I)-catalyzed propargyl rearrangement depicted in Scheme 1b also follows a concerted oxonia Claisen pathway (via an aromatic TS) rather than through a higher barrier 6-endo-dig cyclization [21], which provides further support to the barrier-lowering effect induced by the aromaticity of the transition state.
![[1860-5397-21-125-i1]](/bjoc/content/inline/1860-5397-21-125-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: (a) Diels–Alder cycloaddition reaction between butadiene and ethylene. (b) Gold(I)-catalyzed propargyl rearrangement.
Scheme 1: (a) Diels–Alder cycloaddition reaction between butadiene and ethylene. (b) Gold(I)-catalyzed propar...
While the purported ‘‘aromatic stabilization’’ is mainly established based on comparisons to transition states of alternative stepwise reaction routes, its extension to highly related processes following concerted mechanisms has not been considered. For instance, very little is known about the influence of TS-aromaticity (if any) on the barrier heights of catalyzed pericyclic reactions and related transformations. Our group has been focused on this aspect in recent years by exploring the interplay between the aromaticity of the transition state and the activation barriers of different pericyclic reactions spanning from the parent Diels–Alder reactions to higher-order cycloadditions or even transition-metal-mediated transformations [22-29]. By selecting illustrative examples, in this perspective article, we shall show the relationship between transition-state aromaticity and reactivity in representative pericyclic reactions.
Perspective
Lewis acid-catalyzed Diels–Alder cycloadditions
It is well-established that catalytic amounts of a Lewis acid (LA) can significantly accelerate Diels–Alder cycloaddition reactions [30,31]. In addition, these LA-catalyzed cycloadditions not only exhibit higher reaction rates but also become, in many cases, more regio- and stereoselective than the corresponding uncatalyzed reactions. For these reasons, LAs have been (and still are) widely used in the synthesis of a good number of target molecules, including complex natural products [32-35].
Typically, the LA binds the dienophile through a donor–acceptor interaction (usually involving a carbonyl group) which results in a significant stabilization of the dienophile’s LUMO. As a consequence, the corresponding HOMO(diene)–LUMO(dienophile) energy gap becomes smaller, which, according to the frontier molecular orbital (FMO) theory, constitutes the origin of the observed acceleration (following the so-called LUMO-lowering concept in catalysis) [36-39]. This widely accepted rationalization of the effect of the LA in the Diels–Alder reaction therefore does not consider a possible influence of the aromaticity of the transition state on the barrier heights. For this reason, and to understand the ultimate origin of the faster rates observed in the LA-catalyzed Diels–Alder reactions, we focused on the textbook reaction involving isoprene and (s-trans) methyl acrylate catalyzed by different LAs [40].
Our results confirm that the LAs significantly accelerate the cycloaddition reaction, which in all cases proceeds in a concerted manner (Table 1). The computed barriers follow the same trend as the relative Lewis acidity of the catalyst (measured by the Child’s method [41-43]), but do not follow the same trend as the energy of the LUMO(dienophile). This finding therefore challenges the traditionally used LUMO-lowering concept as the ultimate factor controlling the catalysis in these Diels–Alder reactions.
Table 1: LA-catalyzed Diels–Alder reactions between isoprene and methyl acrylate.
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-125-i4.svg?max-width=637&scale=1.0)
|
|||
| LA | ΔE≠ [kcal mol−1]a | Relative Lewis acidityb | εLUMO (dienophile) [eV] |
| none | 16.2 | −2.6 | |
| SnCl4 | 12.3 | 0.52 ± 0.04 | −3.7 |
| TiCl4 | 11.8 | 0.66 ± 0.03 | −4.0 |
| ZnCl2 | 10.8 | −4.3 | |
| BF3 | 10.0 | 0.77 ± 0.02 | −3.8 |
| AlCl3 | 7.6 | 0.82 | −4.2 |
aComputed activation barriers at the (TightPNO)DLPNO-CCSD-(T) /CBS(3,4/def2)//ZORA-BP86/TZ2P level; brelative Lewis-acidity scale based on Δδ-values of H3 resonances of various bases related to methyl crotonate, data taken from reference [41].
We then explored whether the aromaticity of the involved transition states plays a role in the observed reduction of the barriers. To this end, we computed the nuclear independent chemical shift (NICS) [44] values at the (3, +1) ring critical point of the electron density of the TSs. This point was recommended due to its high sensitivity to diamagnetic effects and its unambiguous character [45,46], which is crucial for the considered transition structures due to their highly asynchronous nature. As shown in Figure 1, all the TSs can be considered aromatic in view of their negative NICS(3, +1) values. Interestingly, the TS-aromaticity of all species is not only comparable (ca. −15 ppm), but slightly decreases from the uncatalyzed reaction to the analogous AlCl3-catalyzed cycloaddition. If we take into account that aromaticity is usually associated with bond-length equalization [2,3], this finding is not surprising as the uncatalyzed reaction is clearly more synchronous than their catalyzed counterparts (see bond lengths in Figure 1). Therefore, it becomes evident that the trend in the TS-aromaticity is opposite to the reactivity trend, which suggests that aromaticity is not behind the observed acceleration in the catalyzed reactions.
![[1860-5397-21-125-1]](/bjoc/content/figures/1860-5397-21-125-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Transition states computed for the Diels–Alder cycloaddition reaction between isoprene and methyl acrylate catalyzed by Lewis acids. Bond distances are given in angstroms and NICS(3, +1) values in ppm.
Figure 1: Transition states computed for the Diels–Alder cycloaddition reaction between isoprene and methyl a...
Once we found that neither aromaticity nor favorable orbital (HOMO(diene)–LUMO(dienophile)) interactions are responsible for the faster rates exhibited by the catalyzed reactions, we applied the activation strain model (ASM) of reactivity [47,48] to quantitatively understand the origin of the acceleration in these pericyclic reactions. This model decomposes the electronic energy (ΔE) into two terms, namely the strain (ΔEstrain) that results from the distortion of the individual reactants and the interaction (ΔEint) between the deformed reactants along the reaction coordinate, defined in this case by the shortest C···C bond-forming distance. From the data in Figure 2a, which shows the computed activation strain diagrams (ASDs) for the uncatalyzed and AlCl3-catalyzed reactions from the beginning of the process up to the respective TSs, it becomes clear that the lower barrier computed for the catalyzed cycloaddition is due to a lower strain (which derives from the higher asynchronicity of the process) and, mainly, to a much stronger interaction between the deformed reactants along the entire reaction coordinate. According to the energy decomposition analysis (EDA) method [49,50], which further decomposes the crucial ΔEint term into three physically meaningful energy terms, namely the classical electrostatic interaction (ΔVelstat), the Pauli repulsion (ΔEPauli) arising from the repulsion between occupied closed-shell orbitals of both deformed reactants, and the orbital interaction (ΔEorb) that accounts for charge transfer and polarization, the stronger interaction computed for the AlCl3-catalyzed does not result from stronger orbital interactions (as traditionally viewed) but exclusively from a less destabilizing Pauli repulsion between the deformed reactants (Figure 2b).
![[1860-5397-21-125-2]](/bjoc/content/figures/1860-5397-21-125-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Comparative activation strain analyses (a) and energy decomposition analysis (b) of the Diels–Alder cycloaddition reaction between isoprene and methyl acrylate (black lines) and the analogous process catalyzed by AlCl3 (red lines) and projected onto the C···C bond-forming distance.
Figure 2: Comparative activation strain analyses (a) and energy decomposition analysis (b) of the Diels–Alder...
The less destabilizing Pauli repulsion computed for the catalyzed cycloaddition directly originates from the reduction of the electron density at the reactive C–C double bond of the dienophile, which translates into a lower <π(diene)|π(dienophile)> molecular orbital overlap (S = 0.13 vs S = 0.07 for the uncatalyzed and catalyzed reactions, respectively). In other words, there occurs a significant polarization of the conjugated π-system away from the C–C double bond when the LA binds the carbonylic oxygen of methyl acrylate, which results in a less destabilizing 4-electron interaction with the diene and ultimately, in a lower activation barrier. The term “Pauli-repulsion lowering in catalysis” [51] was coined to describe this effect, which has been proven to be general and applicable not only to related LA-catalyzed pericyclic reactions (as shown in the following subchapter) but also to other processes such as aza-Michael additions or processes catalyzed by species able to establish noncovalent interactions with the reactants [52-57].
Lewis acid-catalyzed carbonyl–ene reactions
Similar to the Diels–Alder cycloaddition reaction, the Alder–ene reaction [58,59] constitutes a fundamental reaction in organic chemistry that has been applied to the synthesis of a number of target molecules owing to its remarkable functional group tolerance [60,61]. Despite that, the main shortcoming of this transformation is its relatively high barrier, which is translated into high reaction temperatures that severely limit the scope of the process. Nevertheless, as Diels–Alder cycloadditions, Alder–ene reactions can be efficiently accelerated (i.e., having lower barriers) upon the addition of catalytic amounts of a Lewis acid [62], which again, typically binds a carbonyl group in the enophile partner. In this sense, the acceleration is even more significant in the so-called carbonyl–ene reaction [63], an analogous process where a carbonyl group directly acts as the enophile. This transformation has also been applied to the synthesis of complex natural products such as (+)-steenkrotin A [64] or (±)-andrastin C [65], among others.
In order to understand the factors leading to the observed reactivity enhancement, we first compared the parent carbonyl–ene reaction between 1-butene and acetaldehyde with the analogous processes catalyzed by different LAs [66] (Table 2). As expected, we found that the reduction of the activation barrier (up to ca. 25 kcal/mol) directly correlates with the relative Lewis acidity of the catalyst. In addition, the process becomes more and more asynchronous as the acidity of the catalyst increases, which strongly resembles the trends found for Diels–Alder reactions discussed above.
Table 2: Computed free activation energies (ΔG≠, in kcal/mol), synchronicity (Sy), and NICS(3, +1) values in the corresponding TSs (in ppm) of the considered carbonyl–ene reactions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-21-125-i5.svg?max-width=637&scale=1.0)
|
||||
| Catalyst | ΔG≠ [kcal mol−1] | Sya | NICS(3, +1) [ppm] | Lewis acidityb |
| none | 44.1 | 0.87 | −19.9 | |
| BMe3 | 34.8 | 0.62 | −13.5 | |
| BPh3 | 28.0 | 0.58 | −11.2 | |
| SnCl4 | 27.4 | 0.61 | −9.9 | 0.52 ± 0.04 |
| AlMe2Cl | 25.8 | 0.59 | −10.3 | 0.59 ± 0.03c |
| InCl3 | 24.7 | 0.61 | −10.5 | |
| TiCl4 | 24.0 | 0.55 | −6.5 | 0.66 ± 0.03 |
| BF3 | 22.4 | 0.58 | −8.8 | 0.77 ± 0.02 |
| AlCl3 | 19.3 | 0.56 | −7.8 | 0.82 |
aSy stands for the computed synchronicity (for a perfectly synchronous process, Sy = 1). All data were computed at the PCM(dichloromethane)-ωB97xD/def2-TZVPP//PCM-(dichloromethane)-ωB97xD/def2-SVP level; brelative Lewis-acidity scale based on Δδ-values of H3 resonances of various bases related to methyl crotonate, data taken from reference [41]; cvalue for AlEt2Cl.
For the parent Alder–ene reaction, which involves an alkene as the enophile, we previously reported that the corresponding six-membered transition state can be considered as in-plane aromatic in view of its highly negative NICS(3, +1) value (ca. −20 ppm) [67]. Not surprisingly, the analogous, uncatalyzed carbonyl–ene reaction features a similar TS-aromaticity as confirmed by the evolution of the NICS values along a z-axis perpendicular to the molecular plane which exhibits the bell-shape expected for an in-plane aromatic molecule [45,46] (Figure 3a). Similar to the LA-catalyzed Diels–Alder reactions, the asynchronicity induced by the catalysts results in a reduced TS-aromaticity, which results from the reduction of the six-electron delocalization in the uncatalyzed reaction, as confirmed by the electron density of delocalized bonds (EDDB) method [68] (Figure 3b). Indeed, the trend in the computed NICS(3, +1) values is opposite to the trend in the reactivity (see Table 2), which once again indicates that the TS-aromaticity has practically a negligible influence on the barriers of these pericyclic reactions.
![[1860-5397-21-125-3]](/bjoc/content/figures/1860-5397-21-125-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (a) Evolution of the NICS(3, +1) values along a z-axis perpendicular to the molecular plane of the TS involved in the 1-butene and acetaldehyde reaction. (b) EDDB plots and NICS(3, +1) values for the carbonyl–ene reaction between 1-butene and acetaldehyde (left) and the analogous process catalyzed by AlCl3 (right).
Figure 3: (a) Evolution of the NICS(3, +1) values along a z-axis perpendicular to the molecular plane of the ...
We also applied the combined ASM-EDA approach to gain quantitative insight into the ultimate factors controlling the catalysis in this particular reaction. Once again, the ASM method indicates that the catalyzed process benefits from both a less destabilizing strain (due to the higher asynchronicity) and, to a greater extent, from a much stronger interaction between the deformed reactants along the entire reaction coordinate (see Figure 4a for the extreme situations represented by the uncatalyzed and AlCl3-catalyzed reactions). According to the EDA method (Figure 4b), this is almost exclusively due to a significant reduction of the Pauli repulsion between the reactants in the catalyzed reaction. Therefore, once again we found that the LA catalyst induces a significant polarization in the reactive carbonyl group which (i) renders the process more asynchronous and therefore, less aromatic, but (ii) diminishes the destabilizing 4-electron interaction with the C–C double bond of the alkene partner, dramatically reducing the barrier of the process.
![[1860-5397-21-125-4]](/bjoc/content/figures/1860-5397-21-125-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Comparative activation strain analyses (a) and energy decomposition analysis (b) of the carbonyl–ene reaction between 1-butene and acetaldehyde (black lines) and the analogous process catalyzed by AlCl3 (red lines) and projected onto the C···C bond-forming distance.
Figure 4: Comparative activation strain analyses (a) and energy decomposition analysis (b) of the carbonyl–en...
Other non-catalyzed pericyclic reactions
Double group transfer reactions
Double group transfer reactions (DGTRs) are a particular type of pericyclic reaction involving the simultaneous migration of two atoms or groups from one compound to another, very often in a concerted manner [69]. Textbook reactions such as the diimide reduction of unsaturated bonds [70-72], the Meerwein–Ponndorf–Verley reduction of carbonyl groups [73] and some type II dyotropic reactions [74,75] constitute representative DGTRs.
In general, concerted DGRTs are thermally allowed [σ2s + σ2s + π2s] transformations according to the Woodward–Hoffmann rules [15], and therefore proceed through a highly synchronous six-membered transition state. Not surprisingly and similar to the ene reactions discussed above, the delocalization of the involved six electrons within the molecular plane makes the corresponding DGTR transition state in-plane aromatic [76,77]. This is again confirmed not only by the highly negative NICS value computed at the (3, +1) ring critical point but also by the diatropic (i.e., aromatic) ring current observed upon applying the anisotropy of the induced current density (ACID) method [78] or the delocalization observed with the EDDB method (see Figure 5 for the parent process involving the reaction between ethane and ethene).
![[1860-5397-21-125-5]](/bjoc/content/figures/1860-5397-21-125-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: AICD (a) and EDDB (b) plots for the transition state involved in the DGRT between ethene and ethane.
Figure 5: AICD (a) and EDDB (b) plots for the transition state involved in the DGRT between ethene and ethane....
Typically, DGRTs involving C–H bonds are associated with high barriers (>40 kcal/mol). However, we found that the Meerwein–Ponndorf–Verley-type reaction involving the double hydrogen migration from CH3–OH to O=CH2 proceeds with a much lower activation barrier of ca. 25 kcal/mol [79]. Interestingly, although the analogous reaction involving CH3–CH3 and O=CH2 proceeds with a much higher barrier, both transformations feature transition states whose in-plane aromaticity is nearly identical according to their NICS(3, +1) values (−24.5 ppm vs −27.2 ppm, respectively). Therefore, it is found once again that there is no clear relationship between the TS-aromaticity and the barrier heights.
The origin of the computed decrease of the activation barrier in the CH3–OH to O=CH2 as compared to the analogous reaction involving ethane as hydrogen donor can be initially traced to different C–H vs O–H bond strengths. Indeed, we found that the total bond-dissociation energy required to liberate the two migrating hydrogen atoms is approximately 7 kcal/mol lower in CH3–OH than in CH3–CH3.
More quantitative insights into the factors leading to the barrier reduction were gained by applying the combined ASM-EDA method [79]. As graphically shown in Figure 6a, which compares the corresponding ASDs for both DGTRs from the beginning of the process up to the respective (aromatic) transition states, it becomes clear that the lower barrier computed for the process involving CH3–OH derives solely from the stronger interaction between the deformed reactants along the entire reaction coordinate, and particularly, at the transition state region. This results from the formation of a stabilizing, intramolecular CH3–OH···O=CH2 hydrogen bond at the initial stages of the process which brings together both reactants, significantly enhancing the orbital interactions (mainly HOMO–LUMO) between them (as confirmed by the EDA method, Figure 6b). Obviously, this stabilizing noncovalent interaction is lacking in the process involving ethane as hydrogen donor and as a consequence, the activation barrier computed for this DGTR is substantially higher (although the in-plane aromaticity of the corresponding transition states is comparable).
![[1860-5397-21-125-6]](/bjoc/content/figures/1860-5397-21-125-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Comparative activation strain analyses (a) and energy decomposition analysis (b) of the DGRT between ethane and formaldehyde (black lines) and the analogous process involving methanol (red lines) and projected onto the C···H bond-forming distance.
Figure 6: Comparative activation strain analyses (a) and energy decomposition analysis (b) of the DGRT betwee...
Thermal cycloisomerization of 1,3-hexadien-5-ynes (Hopf cyclization)
Since the original report by Hopf and Musso in 1969 [80], the thermal cycloisomerization reactions of 1,3-haxedien-5-ynes have been widely applied to the synthesis of aromatic six-membered rings [81]. From a mechanistic point of view, this process involves the initial formation of a bent-allene intermediate, which leads to the final reaction product via hydrogen shifts (Scheme 2) [82].
![[1860-5397-21-125-i2]](/bjoc/content/inline/1860-5397-21-125-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Representative cycloisomerization reaction of 1,3-hexadien-5-yne.
Scheme 2: Representative cycloisomerization reaction of 1,3-hexadien-5-yne.
The so-called Bergman cyclization of cis-3-hexene-1,5-diynes [83,84], which is suggested to proceed through an in-plane aromatic transition state, is analogous to the first step of this transformation, known as Hopf cyclization. Indeed, our calculations [85] indicate that the parent Hopf cyclization involving cis-hexa-1,3-diene-5-yne occurs in a concerted manner through a transition state that features in-plane aromaticity in view of the computed NICS(3, +1) value of −12.0 ppm and diatropic induced ring current (Figure 7a). Interestingly, the analogous reaction involving cis-pent-2-en-4-yn-1-imine, where the terminal CH=CH2 group in the parent system was replaced by an imine CH=NH group, proceeds with a much lower barrier (ΔE≠ = 23.8 kcal/mol vs 36.8 kcal/mol, respectively). However, the corresponding six-membered transition state is essentially non-aromatic according to the computed NICS(3, +1) value of −1.3 ppm, which results from the interruption of the six-electron delocalization (Figure 7b). A similar behavior was observed in the analogous aldehyde counterpart (cis-pent-2-en-4-ynal) whose cyclization proceeds with a lower barrier than the parent system (ΔE≠ = 30.3 kcal/mol) although the corresponding transition state is also non-aromatic (NICS(3, +1) = −1.5 ppm. Therefore, we found once again that the gain in (in-plane) aromaticity in the parent reaction is not translated into a gain in stability (i.e., a more aromatic transition structure does not translate into a lower-barrier process).
![[1860-5397-21-125-7]](/bjoc/content/figures/1860-5397-21-125-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: AICD plots of the transition states associated with the Hopf cyclization reactions involving cis-hexa-1,3-diene-5-yne (a) and the analogous process involving its imine counterpart (b).
Figure 7: AICD plots of the transition states associated with the Hopf cyclization reactions involving cis-he...
Following the same methodology, the ASM of reactivity was applied then to understand the reasons behind the lower barriers computed for the Hopf cyclizations of ene–ene–ynes E=CH–CH=CH–C≡CH where the terminal CH2 group was replaced by a heteroatom (E = NH or O). Figure 8 shows the corresponding ASDs for the parent cyclization (E = CH2) and the analogous process involving the imine system (E = NH). In this case, as the reactions are intramolecular, the ASM terms are referred to the initial reactants and are computed by considering the interaction between E=CH• and •CH=CH–C≡CH radicals. As shown in Figure 8, although the change in the interaction (ΔΔEint) between the fragments is stronger for the imine system at the initial stages of the cyclization, this term becomes rather similar for both systems at the proximity of the corresponding transition states and therefore, is not responsible for the different barriers. Instead, the cyclization involving the parent system is associated with a much more destabilizing strain energy. This derives from the rotation of the CH2 group required to maximize the π-HOMO(CH=CH2)/π*-LUMO(C≡CH) interaction. As the HOMO of the imine system is located on the lone pair (LP) of the nitrogen atom, such rotation is no longer required, which translates into the computed lower strain energy and ultimately into the lower barrier.
![[1860-5397-21-125-8]](/bjoc/content/figures/1860-5397-21-125-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Comparative activation strain analyses of the Hopf cyclization involving ene–ene–ynes E=CH–CH=CH–C≡CH (E = CH2, black lines; E = NH, red lines) ethane and formaldehyde (black lines) and projected onto the C···E bond-forming distance.
Figure 8: Comparative activation strain analyses of the Hopf cyclization involving ene–ene–ynes E=CH–CH=CH–C≡...
1,3-Dipolar cycloaddition reactions between azides and metal cyaphide complexes
Similar to the Diels–Alder cycloaddition reaction, the 1,3-dipolar cycloaddition between a 1,3-dipole (acting as 4π system) and a 2π dipolarophile is a widely used transformation in synthesis, typically leading to five-membered heterocycles [86,87]. Although introduced by Huisgen in 1960 [88-90], this process has experienced a new renaissance in the last decades due to its countless applications in different fields, and particularly in bioorthogonal chemistry [91-93].
Among the number of dipolarophiles compatible with this [3 + 2]-cycloaddition, phosphaalkynes [94-96] and arsaalkynes [97] have been recently used because they allow easy access to novel heterocycles. For instance, when using organic azides as dipoles, the transformation leads to the preparation of triazaphospholes and triazaarsoles, π-conjugated species with potential applications in materials science due to their luminescent properties [98]. However, these heavier alkyne analogues are typically kinetically unstable, which limits their use as dipolarophiles. Despite that, very recently it was found that the C≡P moiety, in particular, can be stabilized in the form of a cyaphide ligand bonded to a metal fragment [99,100]. These cyaphide complexes are proven to readily undergo 1,3-dipolar cycloaddition reactions with organic azides [99-101], affording novel metal triazaphospholes (Scheme 3) which can be further transformed into protio- and iodotriazaphospholes [101].
![[1860-5397-21-125-i3]](/bjoc/content/inline/1860-5397-21-125-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: 1,3-Dipolar cycloaddition reactions between t-BuN3 and cyaphide complexes.
Scheme 3: 1,3-Dipolar cycloaddition reactions between t-BuN3 and cyaphide complexes.
We first compared these metal cyaphide/azide cycloadditions with the analogous non-metallic process involving t-BuC≡P as dipolarophile [102,103]. Interestingly, we found that, although in all cases the process occurs in a concerted manner through the corresponding five-membered transition state, the germanium and magnesium cyaphide complexes are significantly more reactive (i.e., leading to a lower-barrier process, ΔG≠ ≈ 20 kcal/mol) than their platinum or gold counterparts, which exhibit barriers comparable to that computed for the non-metallic process (ΔG≠ ≈ 26 kcal/mol). Once again, we explored the impact of the aromaticity of the involved transition structures on the computed barriers. As related [3 + 2] cycloaddition reactions [22,45,104,105], the transition states associated with these cycloadditions involving metal cyaphides should be in-plane aromatic species due to the cyclic delocalization of 6-electrons in the molecular plane. Indeed, the evolution of the NICS(3, +1) values along a z-axis perpendicular to the molecular plane shows the expected bell shape with a maximum value at z = 0 Å (Figure 9). Despite that, the computed NICS(3, +1) values are rather similar for all the considered transition structures (ca. −25 ppm), which suggests that, once again, there is no clear relationship between in-plane aromaticity and reactivity.
![[1860-5397-21-125-9]](/bjoc/content/figures/1860-5397-21-125-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Evolution of the NICS(3, +1) values along a z-axis perpendicular to the molecular plane of the TSs involved in the 1,3-dipolar cycloaddition reactions between t-BuN3 and cyaphide complexes.
Figure 9: Evolution of the NICS(3, +1) values along a z-axis perpendicular to the molecular plane of the TSs ...
To understand the factors leading to the lower barriers computed for the 1,3-dipolar cycloadditions involving Mg and Ge cyaphide complexes, the ASM of reactivity was applied. Figure 10a shows the ASDs for the [3 + 2]-cycloaddition reactions between t-BuN3 and [Au]C≡P and [Mg]C≡P cyaphide complexes, as representative systems, from the initial stages of the process up to the corresponding transition states. It is found that the lower barrier computed for the cycloaddition involving the Mg cyaphide complex does not originate from the strain energy, which is actually much less destabilizing for the analogous reaction involving the gold(I)–cyaphide complex. Instead, the former reaction benefits from a much stronger interaction between the deformed reactants along the entire reaction coordinate, which compensates for the destabilization provoked by the significant deformation required in this transformation. According to the EDA method, this stronger interaction derives from both electrostatic and orbital interactions, which are markedly more stabilizing for the process involving the magnesium cyaphide complex. The stronger electrostatic interactions mainly result from the significant polarization of the cyaphide ligand induced by the highly electropositive magnesium atom whereas the stronger orbital interactions derive from a more stabilizing direct π(C≡P)→π*(azide) molecular orbital interaction coupled with a stronger reverse π(azide)→π*(C≡P) interaction.
![[1860-5397-21-125-10]](/bjoc/content/figures/1860-5397-21-125-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Comparative activation strain analyses (a) and energy decomposition analysis (b) of the 1,3-dipolar cycloaddition reactions between t-BuN3 and [Au]CP complex (black lines) and the analogous process involving [Mg]CP complex and projected onto the N···P bond-forming distance.
Figure 10: Comparative activation strain analyses (a) and energy decomposition analysis (b) of the 1,3-dipolar...
Conclusion
Through selected representative examples, in this perspective article the interplay between transition-state aromaticity and reactivity in concerted pericyclic reactions has been discussed. Not surprisingly, the strength of the aromaticity of the transition states strongly depends on their geometry in the sense that a higher degree of synchronicity is typically associated with a more aromatic species. Although aromaticity and energetic stabilization are traditionally connected in stable species (i.e., minima on the potential energy surface), it is found that processes featuring more asynchronous (less aromatic) transition states exhibit lower barriers than their more synchronous counterparts. This finding, which is systematically found regardless of the nature of the pericyclic reaction (from catalyzed Diels–Alder cycloaddition reactions to uncatalyzed double group transfer reactions) therefore indicates that the influence of the TS-aromaticity on the barrier heights of these transformations is not significant (or even, negligible). Instead, other factors such as the reduction of the Pauli repulsion or more favorable electrostatic/orbital interactions, as revealed by the ASM-EDA method, become the ultimate factors controlling the barriers of these reactions.
Data Availability Statement
Data sharing is not applicable as no new data was generated or analyzed in this study.
References
-
Minkin, V. I.; Glukhovtsev, M. N.; Simkin, B. Y. Aromaticity and Antiaromaticity: Electronic and Structural Aspects; John Wiley & Sons: New York, NY, USA, 1994.
Return to citation in text: [1] -
Fernández, I., Ed. Aromaticity: Modern Computational Methods and Applications; Elsevier: Amsterdam, Netherlands, 2021. doi:10.1016/c2019-0-04193-3
Return to citation in text: [1] [2] -
Solà, M.; Boldyrev, A. I.; Cyranski, M. K.; Krygowski, T. M.; Merino, G. Aromaticity and Antiaromaticity: Basics and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2023. doi:10.1002/9781119085928
Return to citation in text: [1] [2] -
Merino, G.; Solà, M.; Fernández, I.; Foroutan-Nejad, C.; Lazzeretti, P.; Frenking, G.; Anderson, H. L.; Sundholm, D.; Cossío, F. P.; Petrukhina, M. A.; Wu, J.; Wu, J. I.; Restrepo, A. Chem. Sci. 2023, 14, 5569–5576. doi:10.1039/d2sc04998h
Return to citation in text: [1] -
Hückel, E. Z. Elektrochem. Angew. Phys. Chem. 1937, 43, 752–788. doi:10.1002/bbpc.19370430907
Return to citation in text: [1] -
von E. Doering, W.; Detert, F. L. J. Am. Chem. Soc. 1951, 73, 876–877. doi:10.1021/ja01146a537
Return to citation in text: [1] -
Solà, M. Nat. Chem. 2022, 14, 585–590. doi:10.1038/s41557-022-00961-w
Return to citation in text: [1] -
Heilbronner, E. Tetrahedron Lett. 1964, 5, 1923–1928. doi:10.1016/s0040-4039(01)89474-0
Return to citation in text: [1] -
Baird, N. C. J. Am. Chem. Soc. 1972, 94, 4941–4948. doi:10.1021/ja00769a025
Return to citation in text: [1] -
Garcia-Borràs, M.; Osuna, S.; Luis, J. M.; Swart, M.; Solà, M. Chem. Soc. Rev. 2014, 43, 5089–5105. doi:10.1039/c4cs00040d
Return to citation in text: [1] -
Fernández, I.; Frenking, G.; Merino, G. Chem. Soc. Rev. 2015, 44, 6452–6463. doi:10.1039/c5cs00004a
Return to citation in text: [1] -
Evans, M. G.; Warhurst, E. Trans. Faraday Soc. 1938, 34, 614–624. doi:10.1039/tf9383400614
Return to citation in text: [1] -
Zimmerman, H. E. Acc. Chem. Res. 1971, 4, 272–280. doi:10.1021/ar50044a002
Return to citation in text: [1] -
Dewar, M. J. S. Angew. Chem., Int. Ed. Engl. 1971, 10, 761–776. doi:10.1002/anie.197107611
Return to citation in text: [1] -
Woodward, R. B.; Hoffmann, R. The Conservation of Orbital Symmetry; Verlag Chemie: Weinheim, Germany, 1970. doi:10.1016/b978-1-4832-3290-4.50006-4
Return to citation in text: [1] [2] -
Jiao, H.; Schleyer, P. v. R. J. Phys. Org. Chem. 1998, 11, 655–662. doi:10.1002/(sici)1099-1395(199808/09)11:8/9<655::aid-poc66>3.0.co;2-u
and references cited therein.
Return to citation in text: [1] -
Schleyer, P. v. R.; Wu, J. I.; Cossío, F. P.; Fernández, I. Chem. Soc. Rev. 2014, 43, 4909–4921. doi:10.1039/c4cs00012a
Return to citation in text: [1] -
Evans, M. G. Trans. Faraday Soc. 1939, 35, 824–834. doi:10.1039/tf9393500824
Return to citation in text: [1] -
Li, Y.; Houk, K. N. J. Am. Chem. Soc. 1993, 115, 7478–7485. doi:10.1021/ja00069a055
Return to citation in text: [1] -
Houk, K. N.; González, J.; Li, Y. Acc. Chem. Res. 1995, 28, 81–90. doi:10.1021/ar00050a004
Return to citation in text: [1] [2] -
Vidhani, D. V.; Cran, J. W.; Krafft, M. E.; Alabugin, I. V. Org. Biomol. Chem. 2013, 11, 1624–1630. doi:10.1039/c2ob27231h
Return to citation in text: [1] -
Fernández, I.; Sierra, M. A.; Cossío, F. P. J. Org. Chem. 2006, 71, 6178–6184. doi:10.1021/jo060918u
Return to citation in text: [1] [2] -
Fernández, I.; Sierra, M. A.; Cossío, F. P. J. Org. Chem. 2008, 73, 2083–2089. doi:10.1021/jo702257s
Return to citation in text: [1] -
Fernández, I.; Cossío, F. P.; de Cózar, A.; Lledós, A.; Mascareñas, J. L. Chem. – Eur. J. 2010, 16, 12147–12157. doi:10.1002/chem.201001714
Return to citation in text: [1] -
Fernández, I.; Cossío, F. P.; Bickelhaupt, F. M. J. Org. Chem. 2011, 76, 2310–2314. doi:10.1021/jo102572x
Return to citation in text: [1] -
Faza, O. N.; López, C. S.; Fernández, I. J. Org. Chem. 2013, 78, 5669–5676. doi:10.1021/jo400837n
Return to citation in text: [1] -
Sosa Carrizo, E. D.; Fernández, I. Phys. Chem. Chem. Phys. 2016, 18, 11677–11682. doi:10.1039/c5cp06523b
Return to citation in text: [1] -
Cabrera-Trujillo, J. J.; Fernández, I. Chem. Commun. 2019, 55, 675–678. doi:10.1039/c8cc09777a
Return to citation in text: [1] -
Cabrera-Trujillo, J. J.; Fernández, I. Chem. Commun. 2022, 58, 6801–6804. doi:10.1039/d2cc02013k
Return to citation in text: [1] -
Yates, P.; Eaton, P. J. Am. Chem. Soc. 1960, 82, 4436–4437. doi:10.1021/ja01501a085
Return to citation in text: [1] -
Pindur, U.; Lutz, G.; Otto, C. Chem. Rev. 1993, 93, 741–761. doi:10.1021/cr00018a006
Return to citation in text: [1] -
Kagan, H. B.; Riant, O. Chem. Rev. 1992, 92, 1007–1019. doi:10.1021/cr00013a013
Return to citation in text: [1] -
Nicolaou, K. C.; Snyder, S. A.; Montagnon, T.; Vassilikogiannakis, G. Angew. Chem., Int. Ed. 2002, 41, 1668–1698. doi:10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z
Return to citation in text: [1] -
Takao, K.-i.; Munakata, R.; Tadano, K.-i. Chem. Rev. 2005, 105, 4779–4807. doi:10.1021/cr040632u
Return to citation in text: [1] -
Juhl, M.; Tanner, D. Chem. Soc. Rev. 2009, 38, 2983–2992. doi:10.1039/b816703f
Return to citation in text: [1] -
Fleming, I. Molecular Orbitals and Organic Chemical Reactions; John Wiley & Sons: Hoboken, NJ, USA, 2009. doi:10.1002/9780470684306
Return to citation in text: [1] -
Houk, K. N. Acc. Chem. Res. 1975, 8, 361–369. doi:10.1021/ar50095a001
Return to citation in text: [1] -
Dewar, M. J. R. Molecular Orbital Theory for Organic Chemists; Prentice Hall: Englewood Cliffs, NJ, USA, 1975.
Return to citation in text: [1] -
Borden, W. T. Modern Molecular Orbital Theory for Organic Chemists; Prentice Hall: Englewood Cliffs, NJ, USA, 1975.
Return to citation in text: [1] -
Vermeeren, P.; Hamlin, T. A.; Fernández, I.; Bickelhaupt, F. M. Angew. Chem., Int. Ed. 2020, 59, 6201–6206. doi:10.1002/anie.201914582
Return to citation in text: [1] -
Childs, R. F.; Mulholland, D. L.; Nixon, A. Can. J. Chem. 1982, 60, 801–808. doi:10.1139/v82-117
Return to citation in text: [1] [2] [3] -
Laszlo, P.; Teston, M. J. Am. Chem. Soc. 1990, 112, 8750–8754. doi:10.1021/ja00180a017
Return to citation in text: [1] -
González-Pinardo, D.; Fernández, I. Chem. – Asian J. 2025, 20, e202401212. doi:10.1002/asia.202401212
Return to citation in text: [1] -
Chen, Z.; Wannere, C. S.; Corminboeuf, C.; Puchta, R.; Schleyer, P. v. R. Chem. Rev. 2005, 105, 3842–3888. doi:10.1021/cr030088+
Return to citation in text: [1] -
Cossío, F. P.; Morao, I.; Jiao, H.; Schleyer, P. v. R. J. Am. Chem. Soc. 1999, 121, 6737–6746. doi:10.1021/ja9831397
Return to citation in text: [1] [2] [3] -
Morao, I.; Cossío, F. P. J. Org. Chem. 1999, 64, 1868–1874. doi:10.1021/jo981862+
Return to citation in text: [1] [2] -
Fernández, I.; Bickelhaupt, F. M. Chem. Soc. Rev. 2014, 43, 4953–4967. doi:10.1039/c4cs00055b
Return to citation in text: [1] -
Bickelhaupt, F. M.; Houk, K. N. Angew. Chem., Int. Ed. 2017, 56, 10070–10086. doi:10.1002/anie.201701486
Return to citation in text: [1] -
Bickelhaupt, F. M.; Baerends, E. J. Rev. Comput. Chem. 2000, 15, 1–86. doi:10.1002/9780470125922.ch1
and references cited therein.
Return to citation in text: [1] -
Zhao, L.; von Hopffgarten, M.; Andrada, D. M.; Frenking, G. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1345. doi:10.1002/wcms.1345
Return to citation in text: [1] -
Hamlin, T. A.; Bickelhaupt, F. M.; Fernández, I. Acc. Chem. Res. 2021, 54, 1972–1981. doi:10.1021/acs.accounts.1c00016
Return to citation in text: [1] -
Hamlin, T. A.; Fernández, I.; Bickelhaupt, F. M. Angew. Chem., Int. Ed. 2019, 58, 8922–8926. doi:10.1002/anie.201903196
Return to citation in text: [1] -
Vermeeren, P.; Hamlin, T. A.; Fernández, I.; Bickelhaupt, F. M. Chem. Sci. 2020, 11, 8105–8112. doi:10.1039/d0sc02901g
Return to citation in text: [1] -
Vermeeren, P.; Hamlin, T. A.; Bickelhaupt, F. M.; Fernández, I. Chem. – Eur. J. 2021, 27, 5180–5190. doi:10.1002/chem.202004496
Return to citation in text: [1] -
Portela, S.; Cabrera-Trujillo, J. J.; Fernández, I. J. Org. Chem. 2021, 86, 5317–5326. doi:10.1021/acs.joc.1c00534
Return to citation in text: [1] -
Portela, S.; Fernández, I. Eur. J. Org. Chem. 2021, 6102–6110. doi:10.1002/ejoc.202101244
Return to citation in text: [1] -
Portela, S.; Fernández, I. Tetrahedron Chem 2022, 1, 100008. doi:10.1016/j.tchem.2022.100008
Return to citation in text: [1] -
Alder, K.; Pascher, F.; Schmitz, A. Ber. Dtsch. Chem. Ges. B 1943, 76B, 27–53. doi:10.1002/cber.19430760105
Return to citation in text: [1] -
Alder, K.; Noble, T. Ber. Dtsch. Chem. Ges. B 1943, 76B, 54–57. doi:10.1002/cber.19430760106
Return to citation in text: [1] -
Hoffmann, H. M. R. Angew. Chem., Int. Ed. Engl. 1969, 8, 556–577. doi:10.1002/anie.196905561
Return to citation in text: [1] -
Mikami, K.; Shimizu, M. Chem. Rev. 1992, 92, 1021–1050. doi:10.1021/cr00013a014
Return to citation in text: [1] -
Snider, B. B. Acc. Chem. Res. 1980, 13, 426–432. doi:10.1021/ar50155a007
Return to citation in text: [1] -
Clarke, M. L.; France, M. B. Tetrahedron 2008, 64, 9003–9031. doi:10.1016/j.tet.2008.06.075
Return to citation in text: [1] -
Pan, S.; Gao, B.; Hu, J.; Xuan, J.; Xie, H.; Ding, H. Chem. – Eur. J. 2016, 22, 959–970. doi:10.1002/chem.201503831
Return to citation in text: [1] -
Okamoto, R.; Takeda, K.; Tokuyama, H.; Ihara, M.; Toyota, M. J. Org. Chem. 2013, 78, 93–103. doi:10.1021/jo301948h
Return to citation in text: [1] -
Rodríguez, H. A.; Cruz, D. A.; Padrón, J. I.; Fernández, I. J. Org. Chem. 2023, 88, 11102–11110. doi:10.1021/acs.joc.3c01059
Return to citation in text: [1] -
Fernández, I.; Bickelhaupt, F. M. J. Comput. Chem. 2012, 33, 509–516. doi:10.1002/jcc.22877
Return to citation in text: [1] -
Szczepanik, D. W.; Andrzejak, M.; Dominikowska, J.; Pawełek, B.; Krygowski, T. M.; Szatylowicz, H.; Solà, M. Phys. Chem. Chem. Phys. 2017, 19, 28970–28981. doi:10.1039/c7cp06114e
Return to citation in text: [1] -
Sankararaman, S. Pericyclic Reactions – A Textbook: Reactions, Applications and Theory; Wiley-VCH: Weinheim, Germany, 2005; pp 326–329.
and references cited therein.
Return to citation in text: [1] -
Hünig, S.; Müller, H. R.; Thier, W. Angew. Chem., Int. Ed. Engl. 1965, 4, 271–280. doi:10.1002/anie.196502711
Return to citation in text: [1] -
Franck-Neumann, M.; Dietrich-Buchecker, C. Tetrahedron Lett. 1980, 21, 671–674. doi:10.1016/s0040-4039(01)85589-1
Return to citation in text: [1] -
Garbisch, E. W., Jr.; Schildcrout, S. M.; Patterson, D. B.; Sprecher, C. M. J. Am. Chem. Soc. 1965, 87, 2932–2944. doi:10.1021/ja01091a025
Return to citation in text: [1] -
Sominsky, L.; Rozental, E.; Gottlieb, H.; Gedanken, A.; Hoz, S. J. Org. Chem. 2004, 69, 1492–1496. doi:10.1021/jo035251f
and references cited therein.
Return to citation in text: [1] -
Reetz, M. T. Angew. Chem., Int. Ed. Engl. 1972, 11, 130–131. doi:10.1002/anie.197201301
Return to citation in text: [1] -
Fernández, I.; Cossío, F. P.; Sierra, M. A. Chem. Rev. 2009, 109, 6687–6711. doi:10.1021/cr900209c
Return to citation in text: [1] -
Fernández, I.; Sierra, M. A.; Cossío, F. P. J. Org. Chem. 2007, 72, 1488–1491. doi:10.1021/jo062310r
Return to citation in text: [1] -
Frenking, G.; Cossío, F. P.; Sierra, M. A.; Fernández, I. Eur. J. Org. Chem. 2007, 5410–5415. doi:10.1002/ejoc.200700547
Return to citation in text: [1] -
Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Chem. Rev. 2005, 105, 3758–3772. doi:10.1021/cr0300901
Return to citation in text: [1] -
Fernández, I.; Bickelhaupt, F. M.; Cossío, F. P. Chem. – Eur. J. 2009, 15, 13022–13032. doi:10.1002/chem.200902024
Return to citation in text: [1] [2] -
Hopf, H.; Musso, H. Angew. Chem., Int. Ed. Engl. 1969, 8, 680. doi:10.1002/anie.196906801
Return to citation in text: [1] -
Zimmermann, G. Eur. J. Org. Chem. 2001, 457–471. doi:10.1002/1099-0690(200102)2001:3<457::aid-ejoc457>3.0.co;2-b
Return to citation in text: [1] -
Litovitz, A. E.; Carpenter, B. K.; Hopf, H. Org. Lett. 2005, 7, 507–510. doi:10.1021/ol047447n
Return to citation in text: [1] -
Jones, R. R.; Bergman, R. G. J. Am. Chem. Soc. 1972, 94, 660–661. doi:10.1021/ja00757a071
Return to citation in text: [1] -
Bergman, R. G. Acc. Chem. Res. 1973, 6, 25–31. doi:10.1021/ar50061a004
Return to citation in text: [1] -
Fernández, I.; Bickelhaupt, F. M.; Cossío, F. P. Chem. – Eur. J. 2014, 20, 10791–10801. doi:10.1002/chem.201303874
Return to citation in text: [1] -
Padwa, A., Ed. 1,3-Dipolar Cycloaddition Chemistry; John Wiley & Sons: New York, NY, USA, 1984; Vol. 1.
Return to citation in text: [1] -
Padwa, A.; Pearson, W. H., Eds. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; John Wiley & Sons: New York, NY, USA, 2002.
Return to citation in text: [1] -
Huisgen, R. Naturwiss. Rundsch. 1960, 73–102.
Return to citation in text: [1] -
Huisgen, R. Proc. Chem. Soc., London 1961, 357–369. doi:10.1039/ps9610000357
Return to citation in text: [1] -
Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565–598. doi:10.1002/anie.196305651
Return to citation in text: [1] -
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5
Return to citation in text: [1] -
Sletten, E. M.; Bertozzi, C. R. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. doi:10.1002/anie.200900942
Return to citation in text: [1] -
Sletten, E. M.; Bertozzi, C. R. Acc. Chem. Res. 2011, 44, 666–676. doi:10.1021/ar200148z
Return to citation in text: [1] -
Rösch, W.; Regitz, M. Angew. Chem., Int. Ed. Engl. 1984, 23, 900–901. doi:10.1002/anie.198409001
Return to citation in text: [1] -
Rösch, W.; Facklam, T.; Regitz, M. Tetrahedron 1987, 43, 3247–3256. doi:10.1016/s0040-4020(01)90292-3
Return to citation in text: [1] -
Choong, S. L.; Nafady, A.; Stasch, A.; Bond, A. M.; Jones, C. Dalton Trans. 2013, 42, 7775–7780. doi:10.1039/c3dt50505g
Return to citation in text: [1] -
Pfeifer, G.; Papke, M.; Frost, D.; Sklorz, J. A. W.; Habicht, M.; Müller, C. Angew. Chem., Int. Ed. 2016, 55, 11760–11764. doi:10.1002/anie.201605143
Return to citation in text: [1] -
Sklorz, J. A. W.; Hoof, S.; Rades, N.; De Rycke, N.; Könczöl, L.; Szieberth, D.; Weber, M.; Wiecko, J.; Nyulászi, L.; Hissler, M.; Müller, C. Chem. – Eur. J. 2015, 21, 11096–11109. doi:10.1002/chem.201500988
Return to citation in text: [1] -
Görlich, T.; Frost, D. S.; Boback, N.; Coles, N. T.; Dittrich, B.; Müller, P.; Jones, W. D.; Müller, C. J. Am. Chem. Soc. 2021, 143, 19365–19373. doi:10.1021/jacs.1c07370
Return to citation in text: [1] [2] -
Wilson, D. W. N.; Urwin, S. J.; Yang, E. S.; Goicoechea, J. M. J. Am. Chem. Soc. 2021, 143, 10367–10373. doi:10.1021/jacs.1c04417
Return to citation in text: [1] [2] -
Yang, E. S.; Mapp, A.; Taylor, A.; Beer, P. D.; Goicoechea, J. M. Chem. – Eur. J. 2023, 29, e202301648. doi:10.1002/chem.202301648
Return to citation in text: [1] [2] -
González‐Pinardo, D.; Goicoechea, J. M.; Fernández, I. Chem. – Eur. J. 2024, 30, e202303977. doi:10.1002/chem.202303977
Return to citation in text: [1] -
González-Pinardo, D.; Fernández, I. Inorg. Chem. 2025, 64, 5628–5636. doi:10.1021/acs.inorgchem.5c00110
Return to citation in text: [1] -
Liu, L. L.; Zhu, J.; Zhao, Y. Chem. Commun. 2014, 50, 11347–11349. doi:10.1039/c4cc04610b
Return to citation in text: [1] -
Zhu, Q.; Chen, S.; Xu, F.; Zhu, J. Inorg. Chem. 2020, 59, 7318–7324. doi:10.1021/acs.inorgchem.0c00754
Return to citation in text: [1]
| 58. | Alder, K.; Pascher, F.; Schmitz, A. Ber. Dtsch. Chem. Ges. B 1943, 76B, 27–53. doi:10.1002/cber.19430760105 |
| 59. | Alder, K.; Noble, T. Ber. Dtsch. Chem. Ges. B 1943, 76B, 54–57. doi:10.1002/cber.19430760106 |
| 60. | Hoffmann, H. M. R. Angew. Chem., Int. Ed. Engl. 1969, 8, 556–577. doi:10.1002/anie.196905561 |
| 61. | Mikami, K.; Shimizu, M. Chem. Rev. 1992, 92, 1021–1050. doi:10.1021/cr00013a014 |
| 45. | Cossío, F. P.; Morao, I.; Jiao, H.; Schleyer, P. v. R. J. Am. Chem. Soc. 1999, 121, 6737–6746. doi:10.1021/ja9831397 |
| 46. | Morao, I.; Cossío, F. P. J. Org. Chem. 1999, 64, 1868–1874. doi:10.1021/jo981862+ |
| 68. | Szczepanik, D. W.; Andrzejak, M.; Dominikowska, J.; Pawełek, B.; Krygowski, T. M.; Szatylowicz, H.; Solà, M. Phys. Chem. Chem. Phys. 2017, 19, 28970–28981. doi:10.1039/c7cp06114e |
| 41. | Childs, R. F.; Mulholland, D. L.; Nixon, A. Can. J. Chem. 1982, 60, 801–808. doi:10.1139/v82-117 |
| 67. | Fernández, I.; Bickelhaupt, F. M. J. Comput. Chem. 2012, 33, 509–516. doi:10.1002/jcc.22877 |
| 65. | Okamoto, R.; Takeda, K.; Tokuyama, H.; Ihara, M.; Toyota, M. J. Org. Chem. 2013, 78, 93–103. doi:10.1021/jo301948h |
| 66. | Rodríguez, H. A.; Cruz, D. A.; Padrón, J. I.; Fernández, I. J. Org. Chem. 2023, 88, 11102–11110. doi:10.1021/acs.joc.3c01059 |
| 63. | Clarke, M. L.; France, M. B. Tetrahedron 2008, 64, 9003–9031. doi:10.1016/j.tet.2008.06.075 |
| 64. | Pan, S.; Gao, B.; Hu, J.; Xuan, J.; Xie, H.; Ding, H. Chem. – Eur. J. 2016, 22, 959–970. doi:10.1002/chem.201503831 |
| 69. |
Sankararaman, S. Pericyclic Reactions – A Textbook: Reactions, Applications and Theory; Wiley-VCH: Weinheim, Germany, 2005; pp 326–329.
and references cited therein. |
| 70. | Hünig, S.; Müller, H. R.; Thier, W. Angew. Chem., Int. Ed. Engl. 1965, 4, 271–280. doi:10.1002/anie.196502711 |
| 71. | Franck-Neumann, M.; Dietrich-Buchecker, C. Tetrahedron Lett. 1980, 21, 671–674. doi:10.1016/s0040-4039(01)85589-1 |
| 72. | Garbisch, E. W., Jr.; Schildcrout, S. M.; Patterson, D. B.; Sprecher, C. M. J. Am. Chem. Soc. 1965, 87, 2932–2944. doi:10.1021/ja01091a025 |
| 73. |
Sominsky, L.; Rozental, E.; Gottlieb, H.; Gedanken, A.; Hoz, S. J. Org. Chem. 2004, 69, 1492–1496. doi:10.1021/jo035251f
and references cited therein. |
| 80. | Hopf, H.; Musso, H. Angew. Chem., Int. Ed. Engl. 1969, 8, 680. doi:10.1002/anie.196906801 |
| 81. | Zimmermann, G. Eur. J. Org. Chem. 2001, 457–471. doi:10.1002/1099-0690(200102)2001:3<457::aid-ejoc457>3.0.co;2-b |
| 79. | Fernández, I.; Bickelhaupt, F. M.; Cossío, F. P. Chem. – Eur. J. 2009, 15, 13022–13032. doi:10.1002/chem.200902024 |
| 79. | Fernández, I.; Bickelhaupt, F. M.; Cossío, F. P. Chem. – Eur. J. 2009, 15, 13022–13032. doi:10.1002/chem.200902024 |
| 76. | Fernández, I.; Sierra, M. A.; Cossío, F. P. J. Org. Chem. 2007, 72, 1488–1491. doi:10.1021/jo062310r |
| 77. | Frenking, G.; Cossío, F. P.; Sierra, M. A.; Fernández, I. Eur. J. Org. Chem. 2007, 5410–5415. doi:10.1002/ejoc.200700547 |
| 78. | Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Chem. Rev. 2005, 105, 3758–3772. doi:10.1021/cr0300901 |
| 74. | Reetz, M. T. Angew. Chem., Int. Ed. Engl. 1972, 11, 130–131. doi:10.1002/anie.197201301 |
| 75. | Fernández, I.; Cossío, F. P.; Sierra, M. A. Chem. Rev. 2009, 109, 6687–6711. doi:10.1021/cr900209c |
| 15. | Woodward, R. B.; Hoffmann, R. The Conservation of Orbital Symmetry; Verlag Chemie: Weinheim, Germany, 1970. doi:10.1016/b978-1-4832-3290-4.50006-4 |
| 83. | Jones, R. R.; Bergman, R. G. J. Am. Chem. Soc. 1972, 94, 660–661. doi:10.1021/ja00757a071 |
| 84. | Bergman, R. G. Acc. Chem. Res. 1973, 6, 25–31. doi:10.1021/ar50061a004 |
| 85. | Fernández, I.; Bickelhaupt, F. M.; Cossío, F. P. Chem. – Eur. J. 2014, 20, 10791–10801. doi:10.1002/chem.201303874 |
| 82. | Litovitz, A. E.; Carpenter, B. K.; Hopf, H. Org. Lett. 2005, 7, 507–510. doi:10.1021/ol047447n |
| 1. | Minkin, V. I.; Glukhovtsev, M. N.; Simkin, B. Y. Aromaticity and Antiaromaticity: Electronic and Structural Aspects; John Wiley & Sons: New York, NY, USA, 1994. |
| 2. | Fernández, I., Ed. Aromaticity: Modern Computational Methods and Applications; Elsevier: Amsterdam, Netherlands, 2021. doi:10.1016/c2019-0-04193-3 |
| 3. | Solà, M.; Boldyrev, A. I.; Cyranski, M. K.; Krygowski, T. M.; Merino, G. Aromaticity and Antiaromaticity: Basics and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2023. doi:10.1002/9781119085928 |
| 7. | Solà, M. Nat. Chem. 2022, 14, 585–590. doi:10.1038/s41557-022-00961-w |
| 8. | Heilbronner, E. Tetrahedron Lett. 1964, 5, 1923–1928. doi:10.1016/s0040-4039(01)89474-0 |
| 9. | Baird, N. C. J. Am. Chem. Soc. 1972, 94, 4941–4948. doi:10.1021/ja00769a025 |
| 10. | Garcia-Borràs, M.; Osuna, S.; Luis, J. M.; Swart, M.; Solà, M. Chem. Soc. Rev. 2014, 43, 5089–5105. doi:10.1039/c4cs00040d |
| 11. | Fernández, I.; Frenking, G.; Merino, G. Chem. Soc. Rev. 2015, 44, 6452–6463. doi:10.1039/c5cs00004a |
| 30. | Yates, P.; Eaton, P. J. Am. Chem. Soc. 1960, 82, 4436–4437. doi:10.1021/ja01501a085 |
| 31. | Pindur, U.; Lutz, G.; Otto, C. Chem. Rev. 1993, 93, 741–761. doi:10.1021/cr00018a006 |
| 99. | Görlich, T.; Frost, D. S.; Boback, N.; Coles, N. T.; Dittrich, B.; Müller, P.; Jones, W. D.; Müller, C. J. Am. Chem. Soc. 2021, 143, 19365–19373. doi:10.1021/jacs.1c07370 |
| 100. | Wilson, D. W. N.; Urwin, S. J.; Yang, E. S.; Goicoechea, J. M. J. Am. Chem. Soc. 2021, 143, 10367–10373. doi:10.1021/jacs.1c04417 |
| 6. | von E. Doering, W.; Detert, F. L. J. Am. Chem. Soc. 1951, 73, 876–877. doi:10.1021/ja01146a537 |
| 32. | Kagan, H. B.; Riant, O. Chem. Rev. 1992, 92, 1007–1019. doi:10.1021/cr00013a013 |
| 33. | Nicolaou, K. C.; Snyder, S. A.; Montagnon, T.; Vassilikogiannakis, G. Angew. Chem., Int. Ed. 2002, 41, 1668–1698. doi:10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z |
| 34. | Takao, K.-i.; Munakata, R.; Tadano, K.-i. Chem. Rev. 2005, 105, 4779–4807. doi:10.1021/cr040632u |
| 35. | Juhl, M.; Tanner, D. Chem. Soc. Rev. 2009, 38, 2983–2992. doi:10.1039/b816703f |
| 5. | Hückel, E. Z. Elektrochem. Angew. Phys. Chem. 1937, 43, 752–788. doi:10.1002/bbpc.19370430907 |
| 21. | Vidhani, D. V.; Cran, J. W.; Krafft, M. E.; Alabugin, I. V. Org. Biomol. Chem. 2013, 11, 1624–1630. doi:10.1039/c2ob27231h |
| 97. | Pfeifer, G.; Papke, M.; Frost, D.; Sklorz, J. A. W.; Habicht, M.; Müller, C. Angew. Chem., Int. Ed. 2016, 55, 11760–11764. doi:10.1002/anie.201605143 |
| 4. | Merino, G.; Solà, M.; Fernández, I.; Foroutan-Nejad, C.; Lazzeretti, P.; Frenking, G.; Anderson, H. L.; Sundholm, D.; Cossío, F. P.; Petrukhina, M. A.; Wu, J.; Wu, J. I.; Restrepo, A. Chem. Sci. 2023, 14, 5569–5576. doi:10.1039/d2sc04998h |
| 22. | Fernández, I.; Sierra, M. A.; Cossío, F. P. J. Org. Chem. 2006, 71, 6178–6184. doi:10.1021/jo060918u |
| 23. | Fernández, I.; Sierra, M. A.; Cossío, F. P. J. Org. Chem. 2008, 73, 2083–2089. doi:10.1021/jo702257s |
| 24. | Fernández, I.; Cossío, F. P.; de Cózar, A.; Lledós, A.; Mascareñas, J. L. Chem. – Eur. J. 2010, 16, 12147–12157. doi:10.1002/chem.201001714 |
| 25. | Fernández, I.; Cossío, F. P.; Bickelhaupt, F. M. J. Org. Chem. 2011, 76, 2310–2314. doi:10.1021/jo102572x |
| 26. | Faza, O. N.; López, C. S.; Fernández, I. J. Org. Chem. 2013, 78, 5669–5676. doi:10.1021/jo400837n |
| 27. | Sosa Carrizo, E. D.; Fernández, I. Phys. Chem. Chem. Phys. 2016, 18, 11677–11682. doi:10.1039/c5cp06523b |
| 28. | Cabrera-Trujillo, J. J.; Fernández, I. Chem. Commun. 2019, 55, 675–678. doi:10.1039/c8cc09777a |
| 29. | Cabrera-Trujillo, J. J.; Fernández, I. Chem. Commun. 2022, 58, 6801–6804. doi:10.1039/d2cc02013k |
| 98. | Sklorz, J. A. W.; Hoof, S.; Rades, N.; De Rycke, N.; Könczöl, L.; Szieberth, D.; Weber, M.; Wiecko, J.; Nyulászi, L.; Hissler, M.; Müller, C. Chem. – Eur. J. 2015, 21, 11096–11109. doi:10.1002/chem.201500988 |
| 16. |
Jiao, H.; Schleyer, P. v. R. J. Phys. Org. Chem. 1998, 11, 655–662. doi:10.1002/(sici)1099-1395(199808/09)11:8/9<655::aid-poc66>3.0.co;2-u
and references cited therein. |
| 17. | Schleyer, P. v. R.; Wu, J. I.; Cossío, F. P.; Fernández, I. Chem. Soc. Rev. 2014, 43, 4909–4921. doi:10.1039/c4cs00012a |
| 19. | Li, Y.; Houk, K. N. J. Am. Chem. Soc. 1993, 115, 7478–7485. doi:10.1021/ja00069a055 |
| 20. | Houk, K. N.; González, J.; Li, Y. Acc. Chem. Res. 1995, 28, 81–90. doi:10.1021/ar00050a004 |
| 91. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5 |
| 92. | Sletten, E. M.; Bertozzi, C. R. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. doi:10.1002/anie.200900942 |
| 93. | Sletten, E. M.; Bertozzi, C. R. Acc. Chem. Res. 2011, 44, 666–676. doi:10.1021/ar200148z |
| 15. | Woodward, R. B.; Hoffmann, R. The Conservation of Orbital Symmetry; Verlag Chemie: Weinheim, Germany, 1970. doi:10.1016/b978-1-4832-3290-4.50006-4 |
| 20. | Houk, K. N.; González, J.; Li, Y. Acc. Chem. Res. 1995, 28, 81–90. doi:10.1021/ar00050a004 |
| 94. | Rösch, W.; Regitz, M. Angew. Chem., Int. Ed. Engl. 1984, 23, 900–901. doi:10.1002/anie.198409001 |
| 95. | Rösch, W.; Facklam, T.; Regitz, M. Tetrahedron 1987, 43, 3247–3256. doi:10.1016/s0040-4020(01)90292-3 |
| 96. | Choong, S. L.; Nafady, A.; Stasch, A.; Bond, A. M.; Jones, C. Dalton Trans. 2013, 42, 7775–7780. doi:10.1039/c3dt50505g |
| 13. | Zimmerman, H. E. Acc. Chem. Res. 1971, 4, 272–280. doi:10.1021/ar50044a002 |
| 14. | Dewar, M. J. S. Angew. Chem., Int. Ed. Engl. 1971, 10, 761–776. doi:10.1002/anie.197107611 |
| 86. | Padwa, A., Ed. 1,3-Dipolar Cycloaddition Chemistry; John Wiley & Sons: New York, NY, USA, 1984; Vol. 1. |
| 87. | Padwa, A.; Pearson, W. H., Eds. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; John Wiley & Sons: New York, NY, USA, 2002. |
| 12. | Evans, M. G.; Warhurst, E. Trans. Faraday Soc. 1938, 34, 614–624. doi:10.1039/tf9383400614 |
| 88. | Huisgen, R. Naturwiss. Rundsch. 1960, 73–102. |
| 89. | Huisgen, R. Proc. Chem. Soc., London 1961, 357–369. doi:10.1039/ps9610000357 |
| 90. | Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565–598. doi:10.1002/anie.196305651 |
| 41. | Childs, R. F.; Mulholland, D. L.; Nixon, A. Can. J. Chem. 1982, 60, 801–808. doi:10.1139/v82-117 |
| 42. | Laszlo, P.; Teston, M. J. Am. Chem. Soc. 1990, 112, 8750–8754. doi:10.1021/ja00180a017 |
| 43. | González-Pinardo, D.; Fernández, I. Chem. – Asian J. 2025, 20, e202401212. doi:10.1002/asia.202401212 |
| 36. | Fleming, I. Molecular Orbitals and Organic Chemical Reactions; John Wiley & Sons: Hoboken, NJ, USA, 2009. doi:10.1002/9780470684306 |
| 37. | Houk, K. N. Acc. Chem. Res. 1975, 8, 361–369. doi:10.1021/ar50095a001 |
| 38. | Dewar, M. J. R. Molecular Orbital Theory for Organic Chemists; Prentice Hall: Englewood Cliffs, NJ, USA, 1975. |
| 39. | Borden, W. T. Modern Molecular Orbital Theory for Organic Chemists; Prentice Hall: Englewood Cliffs, NJ, USA, 1975. |
| 40. | Vermeeren, P.; Hamlin, T. A.; Fernández, I.; Bickelhaupt, F. M. Angew. Chem., Int. Ed. 2020, 59, 6201–6206. doi:10.1002/anie.201914582 |
| 102. | González‐Pinardo, D.; Goicoechea, J. M.; Fernández, I. Chem. – Eur. J. 2024, 30, e202303977. doi:10.1002/chem.202303977 |
| 103. | González-Pinardo, D.; Fernández, I. Inorg. Chem. 2025, 64, 5628–5636. doi:10.1021/acs.inorgchem.5c00110 |
| 22. | Fernández, I.; Sierra, M. A.; Cossío, F. P. J. Org. Chem. 2006, 71, 6178–6184. doi:10.1021/jo060918u |
| 45. | Cossío, F. P.; Morao, I.; Jiao, H.; Schleyer, P. v. R. J. Am. Chem. Soc. 1999, 121, 6737–6746. doi:10.1021/ja9831397 |
| 104. | Liu, L. L.; Zhu, J.; Zhao, Y. Chem. Commun. 2014, 50, 11347–11349. doi:10.1039/c4cc04610b |
| 105. | Zhu, Q.; Chen, S.; Xu, F.; Zhu, J. Inorg. Chem. 2020, 59, 7318–7324. doi:10.1021/acs.inorgchem.0c00754 |
| 99. | Görlich, T.; Frost, D. S.; Boback, N.; Coles, N. T.; Dittrich, B.; Müller, P.; Jones, W. D.; Müller, C. J. Am. Chem. Soc. 2021, 143, 19365–19373. doi:10.1021/jacs.1c07370 |
| 100. | Wilson, D. W. N.; Urwin, S. J.; Yang, E. S.; Goicoechea, J. M. J. Am. Chem. Soc. 2021, 143, 10367–10373. doi:10.1021/jacs.1c04417 |
| 101. | Yang, E. S.; Mapp, A.; Taylor, A.; Beer, P. D.; Goicoechea, J. M. Chem. – Eur. J. 2023, 29, e202301648. doi:10.1002/chem.202301648 |
| 101. | Yang, E. S.; Mapp, A.; Taylor, A.; Beer, P. D.; Goicoechea, J. M. Chem. – Eur. J. 2023, 29, e202301648. doi:10.1002/chem.202301648 |
| 51. | Hamlin, T. A.; Bickelhaupt, F. M.; Fernández, I. Acc. Chem. Res. 2021, 54, 1972–1981. doi:10.1021/acs.accounts.1c00016 |
| 52. | Hamlin, T. A.; Fernández, I.; Bickelhaupt, F. M. Angew. Chem., Int. Ed. 2019, 58, 8922–8926. doi:10.1002/anie.201903196 |
| 53. | Vermeeren, P.; Hamlin, T. A.; Fernández, I.; Bickelhaupt, F. M. Chem. Sci. 2020, 11, 8105–8112. doi:10.1039/d0sc02901g |
| 54. | Vermeeren, P.; Hamlin, T. A.; Bickelhaupt, F. M.; Fernández, I. Chem. – Eur. J. 2021, 27, 5180–5190. doi:10.1002/chem.202004496 |
| 55. | Portela, S.; Cabrera-Trujillo, J. J.; Fernández, I. J. Org. Chem. 2021, 86, 5317–5326. doi:10.1021/acs.joc.1c00534 |
| 56. | Portela, S.; Fernández, I. Eur. J. Org. Chem. 2021, 6102–6110. doi:10.1002/ejoc.202101244 |
| 57. | Portela, S.; Fernández, I. Tetrahedron Chem 2022, 1, 100008. doi:10.1016/j.tchem.2022.100008 |
| 47. | Fernández, I.; Bickelhaupt, F. M. Chem. Soc. Rev. 2014, 43, 4953–4967. doi:10.1039/c4cs00055b |
| 48. | Bickelhaupt, F. M.; Houk, K. N. Angew. Chem., Int. Ed. 2017, 56, 10070–10086. doi:10.1002/anie.201701486 |
| 49. |
Bickelhaupt, F. M.; Baerends, E. J. Rev. Comput. Chem. 2000, 15, 1–86. doi:10.1002/9780470125922.ch1
and references cited therein. |
| 50. | Zhao, L.; von Hopffgarten, M.; Andrada, D. M.; Frenking, G. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1345. doi:10.1002/wcms.1345 |
| 45. | Cossío, F. P.; Morao, I.; Jiao, H.; Schleyer, P. v. R. J. Am. Chem. Soc. 1999, 121, 6737–6746. doi:10.1021/ja9831397 |
| 46. | Morao, I.; Cossío, F. P. J. Org. Chem. 1999, 64, 1868–1874. doi:10.1021/jo981862+ |
| 2. | Fernández, I., Ed. Aromaticity: Modern Computational Methods and Applications; Elsevier: Amsterdam, Netherlands, 2021. doi:10.1016/c2019-0-04193-3 |
| 3. | Solà, M.; Boldyrev, A. I.; Cyranski, M. K.; Krygowski, T. M.; Merino, G. Aromaticity and Antiaromaticity: Basics and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2023. doi:10.1002/9781119085928 |
| 41. | Childs, R. F.; Mulholland, D. L.; Nixon, A. Can. J. Chem. 1982, 60, 801–808. doi:10.1139/v82-117 |
| 44. | Chen, Z.; Wannere, C. S.; Corminboeuf, C.; Puchta, R.; Schleyer, P. v. R. Chem. Rev. 2005, 105, 3842–3888. doi:10.1021/cr030088+ |
© 2025 Fernández; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.