Abstract
N-Alloc-protected furfuryl amino alcohols derived from furfural and ʟ- or ᴅ-valinol were subjected to Torii-type ester electrosynthesis to obtain the corresponding unsaturated esters. These served as key intermediates to prepare (S)- and (R)-enantioenriched unsaturated amides by N-Alloc deprotection which induced concomitant methoxymethyl group cleavage, O-to-N rearrangement, and isomerization of the double bond. An oxazoline ring formation in the resulting unsaturated amides provided the corresponding enantioenriched vinyloxazoline. The reactivity of the electron-deficient double bond in the vinyloxazoline was explored in several reactions. Out of these, the aza-Diels–Alder reaction with TsNCO was successful, leading to a highly diastereoselective formation of an oxazolo[3,2-c]pyrimidine derivative.

Graphical Abstract
Introduction
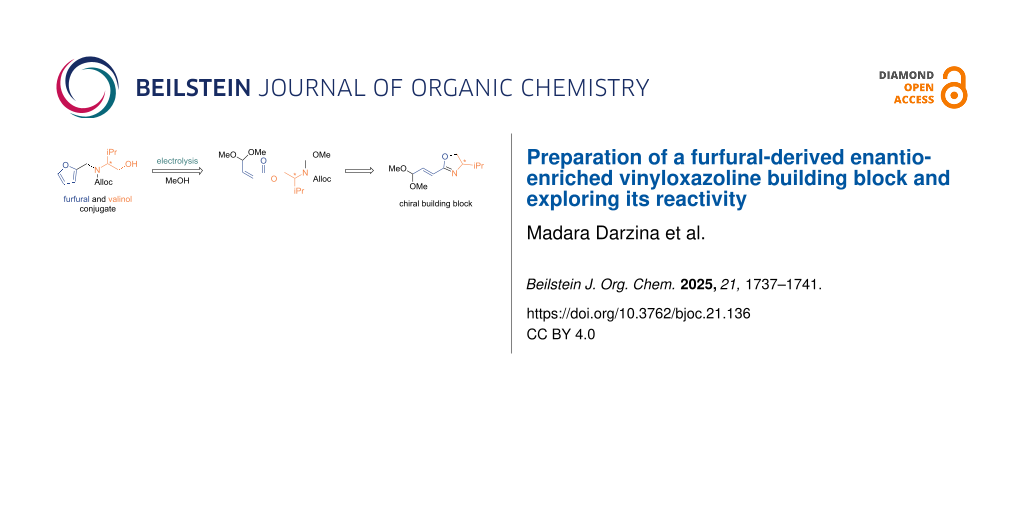
The utilization of biomass as an alternative to fossil feedstock is central to circular economy for the production of value-added products [1-7]. Furfural [furan-2-carbaldehyde (1)], a platform compound derived from lignocellulosic biomass, has been utilized to obtain a range of versatile chemicals with applications to functional materials, pharmaceutically relevant compounds, and agrochemicals [8-17]. In our recent work, we have developed a Torii-type electrosynthesis of unsaturated esters 3a–c starting from furfural (1) and amino alcohol conjugates 2a–c [18]. The process involves an electrooxidative dialkoxylation of the furan ring providing spirocycles 4a–c which undergo a further electrooxidative fragmentation to products 3a–c. However, the further transformation of products 3a–c was hampered by the problematic removal of the protecting groups (Ts, Boc, Ac) under the conditions compatible with the double bond and acetal functions. In this work, we present Torii-type electrosynthesis of ester 3d (PG = Alloc) and its transformation to the enantioenriched vinyloxazoline building block 6, which can be used for the asymmetric synthesis of complex molecules [19-24] (Scheme 1).
![[1860-5397-21-136-i1]](/bjoc/content/inline/1860-5397-21-136-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed approach for the preparation of vinyloxazoline 6.
Scheme 1: Proposed approach for the preparation of vinyloxazoline 6.
The proposed strategy relied on the N-deprotection of the intermediate ester 3d inducing O-to-N rearrangement to form amide 5 as a precursor of vinyloxazoline 6. For this purpose, Alloc (allyloxycarbonyl) turned out to be a suitable N-protecting group as it was compatible with the electrolysis conditions and its removal was compatible with the double bond and acetal functions in ester 3d (see Supporting Information File 1 for attempts of other protecting groups such as Boc, Troc, and tfa). However, Pd-catalyzed N-Alloc deprotection induced isomerization of the double bond leading to the trans-isomer of amide 5.
Results and Discussion
The protected furfuryl amino alcohols S-2d and R-2d were prepared by reductive amination of furfural (1) with ʟ- and ᴅ-valinol followed by N-protection with Alloc-Cl (Scheme 2). The amino alcohols S-2d and R-2d were then subjected to electrochemical oxidation in methanol in batch electrolysis conditions, providing unsaturated esters S-3d and R-3d, respectively (Scheme 2). The previously used one-reactor two-step conditions were found to be productive for the electrosynthesis of S-3d, requiring the addition of acetic acid for the intermediate spiroketal 4d oxidation (Table 1, entry 1). We found that substrate S-2d can be transformed to ester S-3d in a single step with a reduced amount of HFIP as the only additive (Table 1, entries 2 and 3). Given the high cost of HFIP, we examined if it could be replaced with other proton donors for the cathode reaction, and we found that acetic acid served well for this purpose (Table 1, entry 4). Using the reaction conditions with acetic acid as additive, the reaction could be performed also in a 0.5 g scale for the synthesis of both ester enantiomers S-3d and R-3d (Table 1, entries 5 and 6). In the absence of acetic acid or HFIP the electrochemical oxidation also provided the desired product 3d, although in reduced yield (Table 1, entry 7). An increased amount of acetic acid (1 mL or ≈ 18 equiv) also reduced the yield (Table 1, entry 8). The attempt to use LiOAc as electrolyte instead of LiClO4 was not successful as the reaction was stopped at the spirocycle 4d formation stage (Table 1, entry 9).
![[1860-5397-21-136-i2]](/bjoc/content/inline/1860-5397-21-136-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of furfuryl amino alcohols S-2d and R-2d and their electrochemical oxidation to esters S-3d and R-3d.
Scheme 2: Synthesis of furfuryl amino alcohols S-2d and R-2d and their electrochemical oxidation to esters S-...
Table 1: Electrochemical oxidation of protected amino alcohol 2d to ester 3d.
| Entry | Conditionsa | Yield of 3d |
|---|---|---|
| 1b |
1st step: MeOH/HFIP (10:4 mL), LiClO4 (1 equiv), 2.0 F;
2nd step: add AcOH (4 equiv), 3.5 F |
72% (S-3d) |
| 2 | single step: MeOH/HFIP (13:1 mL), LiClO4 (1 equiv), 5.5 F | 71% (S-3d) |
| 3 | single step: MeOH/HFIP (13.5:0.5 mL), LiClO4 (1 equiv), 5.5 F | 70% (S-3d) |
| 4 | single step: MeOH (14 mL), AcOH (3 equiv), LiClO4 (1 equiv), 5.5 F | 72% (S-3d) |
| 5b | single step: MeOH (14 mL), AcOH (3 equiv), LiClO4 (0.5 equiv), 5.5 F | 68%c (S-3d) |
| 6b | single step: MeOH (14 mL), AcOH (3 equiv), LiClO4 (0.5 equiv), 5.5 F | 68% (R-3d) |
| 7 | single step: MeOH (14 mL), LiClO4 (1 equiv), 5.5 F | 60% (S-3d) |
| 8 | single step: MeOH/AcOH (13:1 mL), LiClO4 (1 equiv), 5.5 F | 55% (S-3d) |
| 9 | single step: MeOH (14 mL), AcOH (3 equiv), LiOAc (1 equiv), 5.5 F | 0%d (S-3d) |
aScale: 1 mmol. bScale: 2 mmol. cFaradaic efficiency 49.5%; cell productivity 0.09 mmol/h. dMajor product is spirocycle 4d (PG = Alloc).
The removal of the N-Alloc group in unsaturated ester S-3d was performed using a Pd catalyst and pyrrolidine as a nucleophile. The use of Pd(PPh3)4 as the catalyst led to a fast consumption of the starting material S-3d but provided a mixture of cis- and trans-amides cis-S-5 and trans-S-5 (Scheme 3). The use of PdCl2(S-BINAP) complex as a precatalyst resulted in a longer reaction time and an exclusive formation of amide trans-S-5 with isomerized double bond (Scheme 4). The amide trans-R-5 was prepared analogously from ester R-3d.
Thus, Alloc was validated as non-expensive and relatively small N-protecting group, removal of which is compatible with double bond and acetal function of amides S-5 and R-5. The removal of the Pd catalyst at laboratory scale was done by chromatography. For large scale synthesis, Pd scavengers have to be considered at the work-up.
![[1860-5397-21-136-i3]](/bjoc/content/inline/1860-5397-21-136-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cleavage of the N-Alloc group leading to a mixture of isomers cis-S-5 and trans-S-5.
Scheme 3: Cleavage of the N-Alloc group leading to a mixture of isomers cis-S-5 and trans-S-5.
![[1860-5397-21-136-i4]](/bjoc/content/inline/1860-5397-21-136-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cleavage of the N-Alloc group with PdCl2(S-BINAP) leading to trans-S-5 and trans-R-5.
Scheme 4: Cleavage of the N-Alloc group with PdCl2(S-BINAP) leading to trans-S-5 and trans-R-5.
Unsaturated amides trans-S-5 and trans-R-5 were transformed to oxazolines S-6 and R-6 in good yields by mesylation of the hydroxy group (Scheme 5). Having both enantiomers in hand, the enantiomeric excess of oxazolines S-6 and R-6 was determined by chiral HPLC. This confirmed that no erosion of enantiomeric purity had happened during the deprotection stage.
![[1860-5397-21-136-i5]](/bjoc/content/inline/1860-5397-21-136-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Cyclization of amides trans-S-5 and trans-R-5 to oxazolines S-6 and R-6.
Scheme 5: Cyclization of amides trans-S-5 and trans-R-5 to oxazolines S-6 and R-6.
With the enantioenriched vinyloxazoline S-6 in hand, we explored the reaction scope involving its electron-deficient double bond. Unfortunately, the olefin appeared unreactive or gave a mixture of products in copper-catalyzed 1,4-addition of phenylmagnesium bromide, Giese reaction with 2-iodopropane, Simmons‒Smith or Johnson–Corey–Chaykovsky cyclopropanation, hydroboration reaction with 9-BBN, and Diels–Alders reaction with Danishevsky diene. Gratifyingly, it was found that vinyloxazoline S-6 is a good substrate for an aza-Diels–Alder reaction with tosylisocyanate (TsNCO) providing the oxazolo[3,2-c]pyrimidine derivative 7 as the only detectable diastereomer (Scheme 6) [22-24]. Oxazolo[3,2-c]pyrimidines are substructures in several pharmaceutically relevant compounds such as potent gonadotropin-releasing hormone receptor antagonists with potential application as anticancer drugs [25] and as nucleoside analogs with antiviral potency [26].
![[1860-5397-21-136-i6]](/bjoc/content/inline/1860-5397-21-136-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: aza-Diels–Alder reaction of vinyloxazoline S-6 with TsNCO.
Scheme 6: aza-Diels–Alder reaction of vinyloxazoline S-6 with TsNCO.
According to the reaction mechanism proposed by Elliott et al., the aza-Diels–Alder reaction of vinyloxazoline S-6 with TsNCO is a step-wise process [22]. The first step involves addition of the oxazoline nitrogen to TsNCO leading to a zwitterionic intermediate A, which undergoes 1,4-conjugate addition forming a cyclic intermediate B. Subsequently, the electron-rich double bond in intermediate B reacts with a second equivalent of TsNCO to form oxazolo[3,2-c]pyrimidine derivative 7 (Scheme 7).
![[1860-5397-21-136-i7]](/bjoc/content/inline/1860-5397-21-136-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: The proposed mechanism of product 7 formation.
Scheme 7: The proposed mechanism of product 7 formation.
The highly diastereoselective formation of product 7 can be explained based on the theoretical calculations by Elliott et al. of aza-Diels–Alder reaction of vinyloxazoline with TsNCO [19]. According to their investigations, the aza-nucleophile attack to the double bond in intermediate A is kinetically preferred from the face forming the R-configured carbon of the C–N bond as a result of a minimized steric interaction of the oxygen in zwitterion with iPr substituent of oxazoline.
Conclusion
Unsaturated ester obtained by Torii-type ester electrosynthesis from the conjugate of two biobased starting materials furfural and valinol serves as an intermediate to prepare an enantioenriched vinyl oxazoline 6 with atom economy 24.5% over 5 steps (>75% average per step). The electrochemical oxidation of N-Alloc can be performed as a single step operation in MeOH with acetic acid as an additive. This is a step toward the large scale synthesis of enantioenriched vinyloxazolines 6 from biomass-derived furfural, however, several challenges remain such as replacing LiClO4 as electrolyte at the oxidation step and avoiding chromatography for purification. Further scale-up could be achieved by switching to flow conditions, or designing a continuous tank reactor [27].
Several attempts were made to explore enantioenriched vinyloxazoline 6 as a chiral building block. While additions to the double bond were not successful, vinyloxazoline 6 was found to be a competent substrate for the aza-Diels–Alder reaction in with TsNCO to give oxazolo[3,2-c]pyrimidine derivative 7 as a single diastereomer.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data and copies of NMR spectra. | ||
| Format: PDF | Size: 3.0 MB | Download |
Funding
This project has received funding from the European Union’s Horizon Europe research and innovation program under grant agreement No 101057816 – TRANSPHARM. M. Darzina acknowledges financial support from LIOS internal student grant IG-2021-03 and Recovery and Resilience Facility (RRF) (5.2.1.1.i.) grant Nr. 34/OSI/DG (2025).
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Corma, A.; Iborra, S.; Velty, A. Chem. Rev. 2007, 107, 2411–2502. doi:10.1021/cr050989d
Return to citation in text: [1] -
Li, H.; Guo, H.; Fang, Z.; Aida, T. M.; Smith, R. L. Green Chem. 2020, 22, 582–611. doi:10.1039/c9gc03655e
Return to citation in text: [1] -
He, J.; Chen, L.; Liu, S.; Song, K.; Yang, S.; Riisager, A. Green Chem. 2020, 22, 6714–6747. doi:10.1039/d0gc01869d
Return to citation in text: [1] -
Hommes, A.; Heeres, H. J.; Yue, J. ChemCatChem 2019, 11, 4671–4708. doi:10.1002/cctc.201900807
Return to citation in text: [1] -
Iglesias, J.; Martínez-Salazar, I.; Maireles-Torres, P.; Martin Alonso, D.; Mariscal, R.; López Granados, M. Chem. Soc. Rev. 2020, 49, 5704–5771. doi:10.1039/d0cs00177e
Return to citation in text: [1] -
Sun, Z.; Barta, K. Chem. Commun. 2018, 54, 7725–7745. doi:10.1039/c8cc02937g
Return to citation in text: [1] -
Li, X.-L.; Zhang, K.; Jiang, J.-L.; Zhu, R.; Wu, W.-P.; Deng, J.; Fu, Y. Green Chem. 2018, 20, 362–368. doi:10.1039/c7gc03125d
Return to citation in text: [1] -
Liu, X.; Li, Y.; Deng, J.; Fu, Y. Green Chem. 2019, 21, 4532–4540. doi:10.1039/c9gc01767d
Return to citation in text: [1] -
Song, S.; Fung Kin Yuen, V.; Di, L.; Sun, Q.; Zhou, K.; Yan, N. Angew. Chem., Int. Ed. 2020, 59, 19846–19850. doi:10.1002/anie.202006315
Return to citation in text: [1] -
Scodeller, I.; Mansouri, S.; Morvan, D.; Muller, E.; de Oliveira Vigier, K.; Wischert, R.; Jérôme, F. Angew. Chem., Int. Ed. 2018, 57, 10510–10514. doi:10.1002/anie.201803828
Return to citation in text: [1] -
Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; López Granados, M. Energy Environ. Sci. 2016, 9, 1144–1189. doi:10.1039/c5ee02666k
Return to citation in text: [1] -
Lange, J.-P.; Wadman, S. H. ChemSusChem 2020, 13, 5329–5337. doi:10.1002/cssc.202001376
Return to citation in text: [1] -
Kuznetsov, A.; Kumar, G.; Ardagh, M. A.; Tsapatsis, M.; Zhang, Q.; Dauenhauer, P. J. ACS Sustainable Chem. Eng. 2020, 8, 3273–3282. doi:10.1021/acssuschemeng.9b06881
Return to citation in text: [1] -
Li, X.; Jia, P.; Wang, T. ACS Catal. 2016, 6, 7621–7640. doi:10.1021/acscatal.6b01838
Return to citation in text: [1] -
Li, M.; Dong, X.; Zhang, N.; Jérôme, F.; Gu, Y. Green Chem. 2019, 21, 4650–4655. doi:10.1039/c9gc02206f
Return to citation in text: [1] -
Simeonov, S. P.; Ravutsov, M. A.; Mihovilovic, M. D. ChemSusChem 2019, 12, 2748–2754. doi:10.1002/cssc.201900601
Return to citation in text: [1] -
Babij, N. R.; Choy, N.; Cismesia, M. A.; Couling, D. J.; Hough, N. M.; Johnson, P. L.; Klosin, J.; Li, X.; Lu, Y.; McCusker, E. O.; Meyer, K. G.; Renga, J. M.; Rogers, R. B.; Stockman, K. E.; Webb, N. J.; Whiteker, G. T.; Zhu, Y. Green Chem. 2020, 22, 6047–6054. doi:10.1039/d0gc02063j
Return to citation in text: [1] -
Darzina, M.; Lielpetere, A.; Jirgensons, A. Eur. J. Org. Chem. 2021, 4224–4228. doi:10.1002/ejoc.202100605
Return to citation in text: [1] -
Barluenga, J.; Suárez-Sobrino, A. L.; Tomás, M.; García-Granda, S.; Santiago-García, R. J. Am. Chem. Soc. 2001, 123, 10494–10501. doi:10.1021/ja010719m
Return to citation in text: [1] [2] -
Mitsui, K.; Sato, T.; Urabe, H.; Sato, F. Angew. Chem., Int. Ed. 2004, 43, 490–492. doi:10.1002/anie.200352769
Return to citation in text: [1] -
Meyers, A. I.; Stoianova, D. J. Org. Chem. 1997, 62, 5219–5221. doi:10.1021/jo970429h
Return to citation in text: [1] -
Elliott, M. C.; Kruiswijk, E.; Willock, D. J. Tetrahedron Lett. 1998, 39, 8911–8914. doi:10.1016/s0040-4039(98)01949-2
Return to citation in text: [1] [2] [3] -
Elliott, M. C.; Kruiswijk, E. J. Chem. Soc., Perkin Trans. 1 1999, 3157–3166. doi:10.1039/a905700e
Return to citation in text: [1] [2] -
Elliott, M. C.; Kruiswijk, E. Chem. Commun. 1997, 2311–2312. doi:10.1039/a705571d
Return to citation in text: [1] [2] -
Pontillo, J.; Chen, C. Bioorg. Med. Chem. Lett. 2005, 15, 1407–1411. doi:10.1016/j.bmcl.2005.01.009
Return to citation in text: [1] -
Folkers, G.; Junginger, G.; Müller, C. E.; Schloz, U.; Eger, K. Arch. Pharm. (Weinheim, Ger.) 1989, 322, 119–123. doi:10.1002/ardp.19893220213
Return to citation in text: [1] -
Lehnherr, D.; Chen, L. Org. Process Res. Dev. 2024, 28, 338–366. doi:10.1021/acs.oprd.3c00340
Return to citation in text: [1]
| 1. | Corma, A.; Iborra, S.; Velty, A. Chem. Rev. 2007, 107, 2411–2502. doi:10.1021/cr050989d |
| 2. | Li, H.; Guo, H.; Fang, Z.; Aida, T. M.; Smith, R. L. Green Chem. 2020, 22, 582–611. doi:10.1039/c9gc03655e |
| 3. | He, J.; Chen, L.; Liu, S.; Song, K.; Yang, S.; Riisager, A. Green Chem. 2020, 22, 6714–6747. doi:10.1039/d0gc01869d |
| 4. | Hommes, A.; Heeres, H. J.; Yue, J. ChemCatChem 2019, 11, 4671–4708. doi:10.1002/cctc.201900807 |
| 5. | Iglesias, J.; Martínez-Salazar, I.; Maireles-Torres, P.; Martin Alonso, D.; Mariscal, R.; López Granados, M. Chem. Soc. Rev. 2020, 49, 5704–5771. doi:10.1039/d0cs00177e |
| 6. | Sun, Z.; Barta, K. Chem. Commun. 2018, 54, 7725–7745. doi:10.1039/c8cc02937g |
| 7. | Li, X.-L.; Zhang, K.; Jiang, J.-L.; Zhu, R.; Wu, W.-P.; Deng, J.; Fu, Y. Green Chem. 2018, 20, 362–368. doi:10.1039/c7gc03125d |
| 22. | Elliott, M. C.; Kruiswijk, E.; Willock, D. J. Tetrahedron Lett. 1998, 39, 8911–8914. doi:10.1016/s0040-4039(98)01949-2 |
| 23. | Elliott, M. C.; Kruiswijk, E. J. Chem. Soc., Perkin Trans. 1 1999, 3157–3166. doi:10.1039/a905700e |
| 24. | Elliott, M. C.; Kruiswijk, E. Chem. Commun. 1997, 2311–2312. doi:10.1039/a705571d |
| 19. | Barluenga, J.; Suárez-Sobrino, A. L.; Tomás, M.; García-Granda, S.; Santiago-García, R. J. Am. Chem. Soc. 2001, 123, 10494–10501. doi:10.1021/ja010719m |
| 20. | Mitsui, K.; Sato, T.; Urabe, H.; Sato, F. Angew. Chem., Int. Ed. 2004, 43, 490–492. doi:10.1002/anie.200352769 |
| 21. | Meyers, A. I.; Stoianova, D. J. Org. Chem. 1997, 62, 5219–5221. doi:10.1021/jo970429h |
| 22. | Elliott, M. C.; Kruiswijk, E.; Willock, D. J. Tetrahedron Lett. 1998, 39, 8911–8914. doi:10.1016/s0040-4039(98)01949-2 |
| 23. | Elliott, M. C.; Kruiswijk, E. J. Chem. Soc., Perkin Trans. 1 1999, 3157–3166. doi:10.1039/a905700e |
| 24. | Elliott, M. C.; Kruiswijk, E. Chem. Commun. 1997, 2311–2312. doi:10.1039/a705571d |
| 18. | Darzina, M.; Lielpetere, A.; Jirgensons, A. Eur. J. Org. Chem. 2021, 4224–4228. doi:10.1002/ejoc.202100605 |
| 8. | Liu, X.; Li, Y.; Deng, J.; Fu, Y. Green Chem. 2019, 21, 4532–4540. doi:10.1039/c9gc01767d |
| 9. | Song, S.; Fung Kin Yuen, V.; Di, L.; Sun, Q.; Zhou, K.; Yan, N. Angew. Chem., Int. Ed. 2020, 59, 19846–19850. doi:10.1002/anie.202006315 |
| 10. | Scodeller, I.; Mansouri, S.; Morvan, D.; Muller, E.; de Oliveira Vigier, K.; Wischert, R.; Jérôme, F. Angew. Chem., Int. Ed. 2018, 57, 10510–10514. doi:10.1002/anie.201803828 |
| 11. | Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; López Granados, M. Energy Environ. Sci. 2016, 9, 1144–1189. doi:10.1039/c5ee02666k |
| 12. | Lange, J.-P.; Wadman, S. H. ChemSusChem 2020, 13, 5329–5337. doi:10.1002/cssc.202001376 |
| 13. | Kuznetsov, A.; Kumar, G.; Ardagh, M. A.; Tsapatsis, M.; Zhang, Q.; Dauenhauer, P. J. ACS Sustainable Chem. Eng. 2020, 8, 3273–3282. doi:10.1021/acssuschemeng.9b06881 |
| 14. | Li, X.; Jia, P.; Wang, T. ACS Catal. 2016, 6, 7621–7640. doi:10.1021/acscatal.6b01838 |
| 15. | Li, M.; Dong, X.; Zhang, N.; Jérôme, F.; Gu, Y. Green Chem. 2019, 21, 4650–4655. doi:10.1039/c9gc02206f |
| 16. | Simeonov, S. P.; Ravutsov, M. A.; Mihovilovic, M. D. ChemSusChem 2019, 12, 2748–2754. doi:10.1002/cssc.201900601 |
| 17. | Babij, N. R.; Choy, N.; Cismesia, M. A.; Couling, D. J.; Hough, N. M.; Johnson, P. L.; Klosin, J.; Li, X.; Lu, Y.; McCusker, E. O.; Meyer, K. G.; Renga, J. M.; Rogers, R. B.; Stockman, K. E.; Webb, N. J.; Whiteker, G. T.; Zhu, Y. Green Chem. 2020, 22, 6047–6054. doi:10.1039/d0gc02063j |
| 19. | Barluenga, J.; Suárez-Sobrino, A. L.; Tomás, M.; García-Granda, S.; Santiago-García, R. J. Am. Chem. Soc. 2001, 123, 10494–10501. doi:10.1021/ja010719m |
| 22. | Elliott, M. C.; Kruiswijk, E.; Willock, D. J. Tetrahedron Lett. 1998, 39, 8911–8914. doi:10.1016/s0040-4039(98)01949-2 |
| 26. | Folkers, G.; Junginger, G.; Müller, C. E.; Schloz, U.; Eger, K. Arch. Pharm. (Weinheim, Ger.) 1989, 322, 119–123. doi:10.1002/ardp.19893220213 |
| 25. | Pontillo, J.; Chen, C. Bioorg. Med. Chem. Lett. 2005, 15, 1407–1411. doi:10.1016/j.bmcl.2005.01.009 |
| 27. | Lehnherr, D.; Chen, L. Org. Process Res. Dev. 2024, 28, 338–366. doi:10.1021/acs.oprd.3c00340 |
© 2025 Darzina et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.