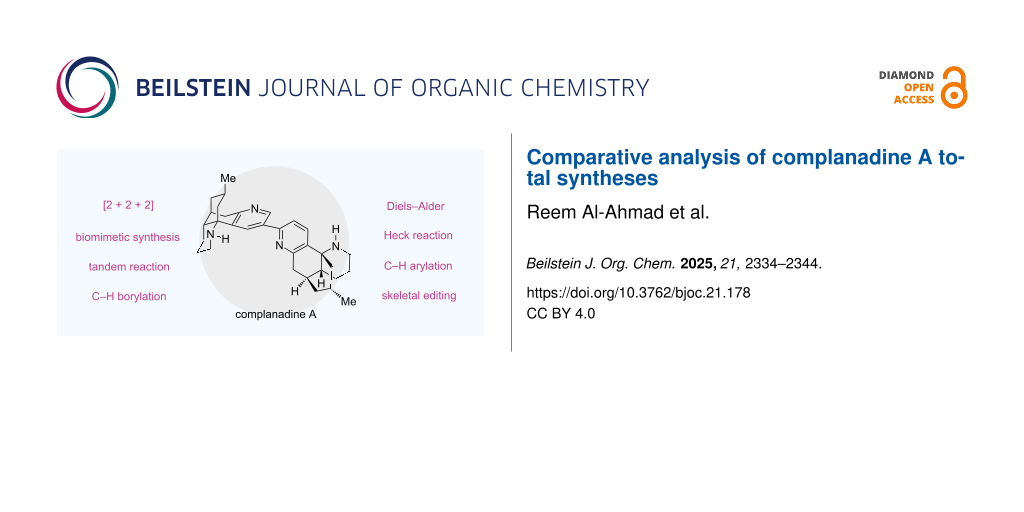
Abstract
In this review, we summarize and compare four total syntheses of complanadine A, a complex and pseudo-dimeric lycopodium alkaloid with promising neurotrophic activity and potential for pain management. These four total syntheses are from the groups of Siegel, Sarpong, Tsukano, and Dai. Each of the four total syntheses contains innovative strategies and creative tactics, reflecting how emerging synthetic capabilities and concepts can positively impact natural product total synthesis.

Graphical Abstract
Introduction
Natural products, owing to their structural complexity, diversity, and therapeutic potential, have continued to serve as inspirations for the development of novel synthetic methodologies and strategies. In turn, these methodological and strategic advancements have significantly improved the efficiency and step-economy of natural product total synthesis. This symbiotic relationship has also helped to accelerate natural product biological evaluation and the subsequent biomedical development [1]. Lycopodium alkaloids are one of the largest families of natural products [2], from which famous molecules such as the huperzines have been discovered and advanced into human clinical trials as acetylcholinesterase inhibitors for treating Alzheimer’s disease [3]. Among the lycopodium family, complanadine A (1, Scheme 1) and its natural congeners such as complanadines B (2), D (3), and E (4) were isolated by Kobayashi and co-workers from the club moss Lycopodium complanatum [4-7]. They discovered that complanadine A exhibited neurotrophic activity by enhancing the mRNA expression level for nerve growth factor (NGF) biosynthesis in 1321N1 human astrocytoma cells and NGF production in human glial cells, rendering complanadine A a promising lead compound for neurological disorder treatment. Later, complanadine A was also identified as a lead compound for pain management by Siegel and co-workers [8]. They discovered one of its potential cellular targets as the Mas-related G protein-coupled receptor (GPCR) X2 (MrgprX2), which is highly expressed in neurons and functions as a modulator of pain. Complanadine A serves as a selective agonist of MrgprX2.
![[1860-5397-21-178-i1]](/bjoc/content/inline/1860-5397-21-178-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Complanadine natural products and their plausible biosynthesis.
Scheme 1: Complanadine natural products and their plausible biosynthesis.
Structurally, complanadine A is an unsymmetrical dimer of the tetracyclic lycodine (5) via a C2–C3’ linkage [9,10]. Complanadine B is a mono-oxidized analog and complanadines D and E are partially reduced analogs. In addition, natural analogs such as lycoplatyrine A (6), isolated as a mixture of diastereomers, were discovered as derivatives of lycodine and an amino acid [11]. Biosynthetically, lysine was proposed to be the starting point of these lycopodium alkaloids [12]. Lysine could be advanced to 4-(2-piperidyl)acetoacetate (7) and pelletierine (8), which would react with each other to deliver phlegmarine (9). Double oxidation of 9 would give 10 for a subsequent intramolecular Mannich-type cyclization to forge the C4–C13 bond and produce 11, which could be further converted to 12 and 13 for an intermolecular Mannich-type dimerization to form the C2–C3’ linkage [13]. Further oxidation state adjustment would give complanadines A, B, D, and E.
Since their isolation, the complanadines, especially complanadine A, have attracted a significant amount of synthetic attention due to their unique structural features and promising therapeutic potential. To date, four total syntheses of complanadine A have been reported from the groups of Siegel [14,15], Sarpong [16], Tsukano [17], and Dai [18], together with one synthetic study from Lewis and co-workers [19]. In this review article, we summarize these four total syntheses, comparatively analyze their strategic novelty and differences, and highlight the impact of enabling methodologies and concepts on the overall efficiency and economy of each total synthesis [20].
Review
The Siegel total synthesis – 2010
In 2010, Siegel and co-workers reported their total synthesis of complanadine A (Scheme 2). Their synthesis centres on two transition metal-catalyzed alkyne–alkyne–nitrile [2 + 2 + 2] cycloadditions to forge the two pyridine rings encoded by complanadine A [21]. Notably, the C2–C3’ linkage of complanadine A is embedded within the symmetrical bis(trimethylsilyl)butadiyne starting material, elegantly circumventing the challenges associated with the direct construction of this key connection. This strategic maneuver differentiates Siegel’s synthesis from the other three reported approaches. Overall, their synthesis highlights the impact of enabling transition metal catalysis on natural product total synthesis.
![[1860-5397-21-178-i2]](/bjoc/content/inline/1860-5397-21-178-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The Siegel total synthesis of complanadine A enabled by [2 + 2 + 2] cycloadditions.
Scheme 2: The Siegel total synthesis of complanadine A enabled by [2 + 2 + 2] cycloadditions.
The Siegel synthesis starts with chiral pool molecule (+)-pulegone (14), which encodes the first stereocenter of the entire sequence. (+)-Pulegone was converted to compound 15 in four steps. Subsequent conjugate addition of the lithium anion of TMS-acetonitrile to 15, followed by careful one-pot protonation of the resulting enolate with ethyl salicylate and TMS removal with CsF, gave 16 bearing three properly arranged substituents. Grignard 1,2-addition to ketone 16, followed by acetylation of the resulting propargylic alcohol afforded 17 which was further advanced to 18 via copper-catalyzed selective displacement of the propargyl acetate with benzylamine and hydrolysis of the primary acetate. The primary alcohol of 18 was activated with PPh3/CCl4, triggering an intramolecular cyclization to afford alkyne–nitrile 19 for the first [2 + 2 + 2] cycloaddition with bis(trimethylsilyl)butadiyne (20). This transformation proceeded smoothly under thermal conditions with CpCo(CO)2 to afford 21 as the major regioisomer (rr = 25:1) [22]. Removal of the two TMS groups and reinstallation of a single TMS on the alkyne provided pyridyl-alkyne 22 for the second [2 + 2 + 2] cycloaddition reaction which proved nontrivial, with the protecting group on the secondary amine of the alkyne-nitrile moiety and the choice of ligand playing crucial roles. Specifically, when using 19 as the alkyne-nitrile partner, the undesired regioisomer (2,2’-bipyridyl product) was obtained as the major product. Switching the alkyne-nitrile partner from 19, bearing a benzyl group, to the formyl-substituted partner 23, combined with addition of 8 equivalents of PPh3, successfully inverted the regioselectivity and gave the desired cycloadduct 24 as the major product in 56% yield. From here on, removal of the TMS group with TBAF, followed by hydrogenolytic removal of the benzyl group and acidic hydrolysis of the formyl group, completed Siegel’s total synthesis of complanadine A. In addition, this synthetic route enabled Siegel and co-workers to determine the first biological target of complanadine A. Screening against a panel of cell surface GPCRs revealed that complanadine A acts as a selective agonist of MrgprX2, while the monomer lycodine showed no significant activity towards this target. This finding highlights the importance of complanadine A’s pseudo-dimeric structural motif for its biological function.
The Sarpong total synthesis – 2010
In the same year (2010), Sarpong and co-workers reported their total synthesis of complanadine A (Scheme 3). Their synthesis features a biomimetic cascade reaction to rapidly establish the tetracyclic skeleton of complanadine A and an iridium-catalyzed site-selective pyridine C–H borylation followed by a Suzuki–Miyaura cross coupling to forge the C2–C3’ linkage. Their synthesis achieves a high degree of synergy between classic transformations and modern synthetic capabilities, highlighting the importance of biomimetic strategies in total synthesis [23-25].
![[1860-5397-21-178-i3]](/bjoc/content/inline/1860-5397-21-178-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The Sarpong total synthesis of complanadine A enabled by a biomimetic strategy and C–H activation.
Scheme 3: The Sarpong total synthesis of complanadine A enabled by a biomimetic strategy and C–H activation.
The Sarpong synthesis utilized (+)-pulegone (14) as the starting point as well. They first advanced (+)-pulegone to primary amine 25 with a masked enone moiety, which upon treatment with HClO4 underwent ketone release and amine-ketone condensation to form iminium ion 27. Under the same acidic conditions, enamide 26 underwent hydrolysis and tautomerization to form enol 28. Conjugate addition of enol 28 to iminium ion 27 gave 29 for the subsequent intramolecular Mannich cyclization to deliver 30, which continued with an amide-ketone condensation to finally produce 31 in this highly efficient one-step biomimetic cascade sequence. The secondary amine of 31 was selectively protected as a Boc carbamate and the dihydropyridone moiety was oxidized to a pyridone with Pb(OAc)4. Pyridone 32 was prepared in 55% from 25 and 26 in three steps. Triflation of the pyridone gave 33 with a triflate at the C2 position for cross coupling to form the C2–C3’ linkage. At this stage, the Sarpong group needed to install a functional group at the C3 position of the pyridine. They creatively solved this challenge with an Ir-catalyzed regioselective C–H borylation developed simultaneously by Ishiyama, Miyaura, Hartwig, and co-workers and Smith and co-workers [26,27]. First, the triflate group of 33 was removed by a Pd-catalyzed reduction with ammonium formate as the reducing reagent. The resulting Boc-protected lycodine 34 underwent Ir-catalyzed C3–H borylation mainly guided by steric factors to provide boronic ester 35 in 75% yield. With the boronic ester handle at the C3 position, the subsequent Suzuki–Miyaura cross coupling between 35 and 33 occurred smoothly to deliver pseudo-dimer 36, which upon acidic removal of the two Boc protecting groups completed the Sarpong total synthesis of complanadine A.
Efficient access of key coupling intermediates 33 and 34 further enabled Sarpong and co-workers to successfully synthesize other lycopodium alkaloids. For example, to synthesize complanadine B with mono-oxidation at one of the two benzylic positions, they started with benzylic oxidation of 34 using SeO2 to provide 37, which was further oxidized to pyridine N-oxide 38. Treatment of 38 with POCl3 in DMF delivered 2-chloropyridine 39 for the subsequent Suzuki–Miyaura cross coupling with 35 to form the C2–C3’ linkage. Boc removal then completed their total synthesis of complanadine B [28]. Notably, while complanadine B could be derived from complanadine A via a selective enzymatic oxidation, attempts to achieve this transformation using chemical methods were unsuccessful. For instance, treatment of 36 with SeO2 gave a mono-oxidation product at the undesired benzylic position. In addition, from 33 or 35, Sarpong and co-workers prepared several other lycopodium alkaloids including 8,15-dihydrohuperzine A (40), casuarinine H (41), lycoplatyrines B (42), A (6), and F (43, existing as a mixture of diastereomers) via either a creative “degradation” of the piperidine ring or cross-coupling reactions at the pyridine C3 position [29].
The Tsukano total synthesis – 2013
In 2013, Tsukano and co-workers reported their total synthesis of complanadines A and B (Scheme 4). Their synthesis utilizes a Diels–Alder reaction and an intramolecular Heck reaction to build the two six-membered carbocycles embedded in the bicyclo[3.3.1]nonane ring system of complanadine A and a pyridine N-oxide directed ortho C–H arylation to forge the C2–C3’ linkage.
![[1860-5397-21-178-i4]](/bjoc/content/inline/1860-5397-21-178-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The Tsukano total synthesis of complanadine A enabled by Diels–Alder cycloaddition, Heck cyclization, and directed C–H arylation.
Scheme 4: The Tsukano total synthesis of complanadine A enabled by Diels–Alder cycloaddition, Heck cyclizatio...
As shown in Scheme 4, they first prepared diene 44 and dienophile 45 for a thermal Diels–Alder cycloaddition, which afforded a mixture of stereo- (endo/exo) and regioisomers, among which the desired product 46 was obtained in 45% yield as a racemic mixture. After triflation of the free hydroxy group of 46 to provide 47, an intramolecular Heck reaction was employed to close the second six-membered carbocycle to deliver 48 in 73% yield. The Diels–Alder reaction and Heck reaction quickly set up the tetracyclic skeleton for subsequent peripheral modifications. First, the ketone functionality of 48 was reduced to a methylene group via a sequence of Luche reduction and Barton–McCombie deoxygenation. The extra ethyl carboxylate was removed via a sequence of LiOH hydrolysis and a Curtius rearrangement using DPPA to form the corresponding acyl azide. Ketone 50 was produced in 98% yield over two steps. The newly formed ketone functionality enabled the introduction of the desired methyl group at its α-position to afford 51 in racemic form. This seemingly straightforward α-methylation turned out to be quite challenging. Tsukano and co-workers eventually utilized a two-step sequence to solve this problem, namely, TMS enol ether formation followed by trapping the enol ether with MeI in presence of benzyltrimethylammonium fluoride (BTAF). At this stage, to prepare optically active natural product, the racemic mixture of 51 was separated using chiral HPLC to afford (+)-51 and (−)-51, which were used to prepare both enantiomers of complanadine A for biological evaluations. With optically active 51 in hand, its extra ketone functionality was reduced via thioacetalization (51 → 52) and radical reduction (52 → 53) to provide 53, a diverging point to access C–H arylation partners 54 and 55. mCPBA oxidation of 53 afforded pyridine N-oxide 54. The Ir-catalyzed C–H borylation used in the Sarpong synthesis was again utilized here to introduce a boronic ester at the C3 position, which was further converted to 3-bromopyridine 55 with CuBr2. With both 54 and 55, Tsukano and co-workers employed a remarkable pyridine N-oxide directed C–H arylation method developed by Fagnou et al. to forge the C2–C3’ bipyridyl linkage and produce 56 in good yield [30]. From 56, a one-pot Cbz removal and pyridine N-oxide reduction completed their total synthesis of complanadine A. In addition, 56 also served as a key intermediate for their synthesis of complanadine B, which was achieved via a sequence of Boekelheide rearrangement (56 → 57), acetate hydrolysis, DMP oxidation and Cbz removal. Based on these results, Tsukano and co-workers suggested that a mono-N-oxide intermediate could be involved in the biosynthesis of these dimeric complanadine alkaloids. Importantly, access to both enantiomers of 51 allowed Tsukano and co-workers to prepare both enantiomers of complanadine A. Their further biological evaluation of the complanadines and several synthetic intermediates revealed that the pseudo-dimeric structure, absolute configuration, and oxidation level are important for the observed neurotrophic activity, providing a strong foundation for future analog design and synthesis [31].
The Dai total synthesis – 2021
In 2021, eleven years after the first two total syntheses of complanadine A, Dai and co-workers reported their total synthesis of complanadine A (Scheme 5). Their synthesis features a novel single-atom skeletal editing strategy [32,33] to form the pyridine from a pyrrole and a similar pyridine N-oxide directed ortho C–H arylation to forge the C2–C3’ linkage as the Tsukano synthesis, but with a much less reactive 3-chloropyridine as the cross-coupling partner.
![[1860-5397-21-178-i5]](/bjoc/content/inline/1860-5397-21-178-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: The Dai total synthesis of complanadine A using single-atom skeletal editing.
Scheme 5: The Dai total synthesis of complanadine A using single-atom skeletal editing.
As shown in Scheme 5, the Dai synthesis starts with compound 58 which can be prepared from (+)-pulegone in three steps or via an organocatalyzed tandem sequence in one step. The terminal olefin of 58 was then converted to a primary azide via an anti-Markovnikov hydroazidation reaction with a combination of 59 and TMSN3 recently developed by Xu and co-workers [34]. Mukaiyama conjugate addition between 60 and 61 promoted by Tf2NH followed by a one-pot enol ether hydrolysis gave 62 as a mixture of inconsequential stereoisomers. Subsequent oxidative cleavage of the terminal olefin of 62 using ozonolysis followed by Paal–Knorr pyrrole synthesis delivered 63, which was unstable and spontaneously cyclized to provide 64. Compound 64 was then advanced to tetracyclic intermediate 67 in a one-pot tandem process, which initiated with Staudinger azide reduction with PPh3 to form a primary amine. After reversible hemiaminal opening and amine–ketone condensation, iminium ion 65 was produced for the next pyrrole nucleophilic addition to form a strategically important C–C bond and afford 66, which was protected as Boc carbamate in the same pot to give 67 in 96% yield from 64. In this tandem sequence, the nucleophilicity of the electron-rich pyrrole group is essential for the key C–C bond formation. In the next step, the pyrrole group was converted to the pyridine group encoded by the natural product. This single-atom skeletal editing step (67 → 68) was achieved using the Ciamician–Dennstedt rearrangement, a reaction discovered back in 1881 [35]. Furthermore, this rearrangement positioned a chloride handle at the desired site for the next C–H arylation. Part of 68 was then converted to pyridine N-oxide via Pd/C-catalyzed dechlorination and mCPBA oxidation. While a similar C–H arylation strategy was used in the Tsukano synthesis, the 3-chloropyridine 68 used in the Dai synthesis exhibited much lower reactivity compared to the 3-bromopyridine 55 used in the Tsukano synthesis. Thus, a set of new reaction conditions was needed. To solve this reactivity issue, a protocol developed by Stoltz et al. in their jorunnamycin synthesis [Pd(OAc)2, t-Bu2MePHBF4, Cs2CO3, and CsOPiv in toluene at 130 °C] was utilized to afford the C–H arylation in 78% yield with a 1:4 ratio of 68/69 [36]. Subsequent reduction of the pyridine N-oxide with Pd(OH)2/C and H2 followed by acidic Boc removal completed their total synthesis of complanadine A (1). In addition, 3-chloropyridine 68 enabled Dai and co-workers to prepare simplified analogs of complanadine A for biological evaluation [13].
Conclusion
As summarized in Scheme 6, four complanadine A total syntheses were reviewed here. Toward the same target molecule, four strategically unique and different approaches were developed. The Siegel synthesis harnesses the power and efficiency of two Co-mediated [2 + 2 + 2] cycloadditions to build the C2–C3’-bipyridyl moiety encoded by complanadine A. The Sarpong synthesis leverages a biomimetic approach to rapidly assemble the tetracyclic core skeleton and the newly developed C–H borylation to install a boronic ester handle at the desired position for a Suzuki–Miyaura cross coupling to build the C2–C3’ linkage which was hidden in bis(trimethylsilyl)butadiyne (20) of the Siegel synthesis. Both the groups of Tsukano and Dai utilized a pyridine N-oxide directed C–H arylation to forge the C2–C3’ linkage, but the approaches to prepare the C–H arylation precursors they employed differ completely. In the Tsukano synthesis, a Diels–Alder reaction and an intramolecular Heck reaction were used to build the key ring systems of complanadine A. In the Dai synthesis, they used an electron-rich and nucleophilic pyrrole as the precursor of the electron-deficient pyridine to enable a tandem sequence involving an intramolecular nucleophilic addition of the pyrrole to an iminium ion to form a key C–C bond. The pyrrole group was then converted to the desired pyridine via a single-atom skeletal editing using the 145-year-old Ciamician–Dennstedt rearrangement, completing its long overdue debut in total synthesis. Overall, each of these four total syntheses showcase innovative strategies and creative and enabling tactics including modern transition metal catalysis, C–H activation methods, biomimetic synthesis, classic rearrangements, skeletal editing logic, and others. In addition, these efforts enabled the identification of the potential cellular target of complanadine A, validation of its neurotrophic activity, establishment of preliminary structure activity relationships, and generation of synthetic analogs, all of which pave the way for further study and development of this unique natural product and/or its analogs.
![[1860-5397-21-178-i6]](/bjoc/content/inline/1860-5397-21-178-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Comparative summary of the four complanadine A total syntheses.
Scheme 6: Comparative summary of the four complanadine A total syntheses.
Data Availability Statement
Data sharing is not applicable as no new data was generated or analyzed in this study.
References
-
Nicolaou, K. C.; Hale, C. R. H. Natl. Sci. Rev. 2014, 1, 233–252. doi:10.1093/nsr/nwu001
Return to citation in text: [1] -
Ma, X.; Gang, D. R. Nat. Prod. Rep. 2004, 21, 752–772. doi:10.1039/b409720n
Return to citation in text: [1] -
Tun, M. K. M.; Herzon, S. B. J. Exp. Pharmacol. 2012, 4, 113–123. doi:10.2147/jep.s27084
Return to citation in text: [1] -
Kobayashi, J.; Hirasawa, Y.; Yoshida, N.; Morita, H. Tetrahedron Lett. 2000, 41, 9069–9073. doi:10.1016/s0040-4039(00)01630-0
Return to citation in text: [1] -
Morita, H.; Ishiuchi, K.; Haganuma, A.; Hoshino, T.; Obara, Y.; Nakahata, N.; Kobayashi, J. Tetrahedron 2005, 61, 1955–1960. doi:10.1016/j.tet.2005.01.011
Return to citation in text: [1] -
Ishiuchi, K.; Kubota, T.; Ishiyama, H.; Hayashi, S.; Shibata, T.; Mori, K.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2011, 19, 749–753. doi:10.1016/j.bmc.2010.12.025
Return to citation in text: [1] -
Ishiuchi, K.; Kubota, T.; Mikami, Y.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2007, 15, 413–417. doi:10.1016/j.bmc.2006.09.043
Return to citation in text: [1] -
Johnson, T.; Siegel, D. Bioorg. Med. Chem. Lett. 2014, 24, 3512–3515. doi:10.1016/j.bmcl.2014.05.060
Return to citation in text: [1] -
Heathcock, C. H.; Kleinman, E. F.; Binkley, E. S. J. Am. Chem. Soc. 1982, 104, 1054–1068. doi:10.1021/ja00368a024
Return to citation in text: [1] -
Azuma, M.; Yoshikawa, T.; Kogure, N.; Kitajima, M.; Takayama, H. J. Am. Chem. Soc. 2014, 136, 11618–11621. doi:10.1021/ja507016g
Return to citation in text: [1] -
Yeap, J. S.-Y.; Lim, K.-H.; Yong, K.-T.; Lim, S.-H.; Kam, T.-S.; Low, Y.-Y. J. Nat. Prod. 2019, 82, 324–329. doi:10.1021/acs.jnatprod.8b00754
Return to citation in text: [1] -
Wang, J.; Zhang, Z.-K.; Jiang, F.-F.; Qi, B.-W.; Ding, N.; Hnin, S. Y. Y.; Liu, X.; Li, J.; Wang, X.-h.; Tu, P.-F.; Abe, I.; Morita, H.; Shi, S.-P. Org. Lett. 2020, 22, 8725–8729. doi:10.1021/acs.orglett.0c03339
Return to citation in text: [1] -
Martin, B. S.; Ma, D.; Saito, T.; Gallagher, K. S.; Dai, M. Synthesis 2024, 56, 107–117. doi:10.1055/a-2107-5159
Return to citation in text: [1] [2] -
Yuan, C.; Chang, C.-T.; Axelrod, A.; Siegel, D. J. Am. Chem. Soc. 2010, 132, 5924–5925. doi:10.1021/ja101956x
Return to citation in text: [1] -
Yuan, C.; Chang, C.-T.; Siegel, D. J. Org. Chem. 2013, 78, 5647–5668. doi:10.1021/jo400695c
Return to citation in text: [1] -
Fischer, D. F.; Sarpong, R. J. Am. Chem. Soc. 2010, 132, 5926–5927. doi:10.1021/ja101893b
Return to citation in text: [1] -
Zhao, L.; Tsukano, C.; Kwon, E.; Takemoto, Y.; Hirama, M. Angew. Chem., Int. Ed. 2013, 52, 1722–1725. doi:10.1002/anie.201208297
Return to citation in text: [1] -
Ma, D.; Martin, B. S.; Gallagher, K. S.; Saito, T.; Dai, M. J. Am. Chem. Soc. 2021, 143, 16383–16387. doi:10.1021/jacs.1c08626
Return to citation in text: [1] -
Uosis-Martin, M.; Pantoş, G. D.; Mahon, M. F.; Lewis, S. E. J. Org. Chem. 2013, 78, 6253–6263. doi:10.1021/jo401014n
Return to citation in text: [1] -
Newhouse, T.; Baran, P. S.; Hoffmann, R. W. Chem. Soc. Rev. 2009, 38, 3010–3021. doi:10.1039/b821200g
Return to citation in text: [1] -
Varela, J. A.; Saá, C. Chem. Rev. 2003, 103, 3787–3802. doi:10.1021/cr030677f
Return to citation in text: [1] -
Varela, J. A.; Castedo, L.; Saá, C. J. Am. Chem. Soc. 1998, 120, 12147–12148. doi:10.1021/ja982832r
Return to citation in text: [1] -
de la Torre, M. C.; Sierra, M. A. Angew. Chem., Int. Ed. 2004, 43, 160–181. doi:10.1002/anie.200200545
Return to citation in text: [1] -
Razzak, M.; De Brabander, J. K. Nat. Chem. Biol. 2011, 7, 865–875. doi:10.1038/nchembio.709
Return to citation in text: [1] -
Bao, R.; Zhang, H.; Tang, Y. Acc. Chem. Res. 2021, 54, 3720–3733. doi:10.1021/acs.accounts.1c00459
Return to citation in text: [1] -
Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 390–391. doi:10.1021/ja0173019
Return to citation in text: [1] -
Cho, J.-Y.; Tse, M. K.; Holmes, D.; Maleczka, R. E., Jr.; Smith, M. R., III. Science 2002, 295, 305–308. doi:10.1126/science.1067074
Return to citation in text: [1] -
Newton, J. N.; Fischer, D. F.; Sarpong, R. Angew. Chem., Int. Ed. 2013, 52, 1726–1730. doi:10.1002/anie.201208571
Return to citation in text: [1] -
Haley, H. M. S.; Payer, S. E.; Papidocha, S. M.; Clemens, S.; Nyenhuis, J.; Sarpong, R. J. Am. Chem. Soc. 2021, 143, 4732–4740. doi:10.1021/jacs.1c00457
Return to citation in text: [1] -
Campeau, L.-C.; Rousseaux, S.; Fagnou, K. J. Am. Chem. Soc. 2005, 127, 18020–18021. doi:10.1021/ja056800x
Return to citation in text: [1] -
Zhao, L.; Tsukano, C.; Kwon, E.; Shirakawa, H.; Kaneko, S.; Takemoto, Y.; Hirama, M. Chem. – Eur. J. 2017, 23, 802–812. doi:10.1002/chem.201604647
Return to citation in text: [1] -
Al-Ahmad, R.; Dai, M. Acc. Chem. Res. 2025, 58, 1392–1406. doi:10.1021/acs.accounts.5c00030
Return to citation in text: [1] -
Jurczyk, J.; Woo, J.; Kim, S. F.; Dherange, B. D.; Sarpong, R.; Levin, M. D. Nat. Synth. 2022, 1, 352–364. doi:10.1038/s44160-022-00052-1
Return to citation in text: [1] -
Li, H.; Shen, S.-J.; Zhu, C.-L.; Xu, H. J. Am. Chem. Soc. 2019, 141, 9415–9421. doi:10.1021/jacs.9b04381
Return to citation in text: [1] -
Ciamician, G. L.; Dennstedt, M. Ber. Dtsch. Chem. Ges. 1881, 14, 1153–1163. doi:10.1002/cber.188101401240
Return to citation in text: [1] -
Welin, E. R.; Ngamnithiporn, A.; Klatte, M.; Lapointe, G.; Pototschnig, G. M.; McDermott, M. S. J.; Conklin, D.; Gilmore, C. D.; Tadross, P. M.; Haley, C. K.; Negoro, K.; Glibstrup, E.; Grünanger, C. U.; Allan, K. M.; Virgil, S. C.; Slamon, D. J.; Stoltz, B. M. Science 2019, 363, 270–275. doi:10.1126/science.aav3421
Return to citation in text: [1]
| 13. | Martin, B. S.; Ma, D.; Saito, T.; Gallagher, K. S.; Dai, M. Synthesis 2024, 56, 107–117. doi:10.1055/a-2107-5159 |
| 1. | Nicolaou, K. C.; Hale, C. R. H. Natl. Sci. Rev. 2014, 1, 233–252. doi:10.1093/nsr/nwu001 |
| 8. | Johnson, T.; Siegel, D. Bioorg. Med. Chem. Lett. 2014, 24, 3512–3515. doi:10.1016/j.bmcl.2014.05.060 |
| 20. | Newhouse, T.; Baran, P. S.; Hoffmann, R. W. Chem. Soc. Rev. 2009, 38, 3010–3021. doi:10.1039/b821200g |
| 4. | Kobayashi, J.; Hirasawa, Y.; Yoshida, N.; Morita, H. Tetrahedron Lett. 2000, 41, 9069–9073. doi:10.1016/s0040-4039(00)01630-0 |
| 5. | Morita, H.; Ishiuchi, K.; Haganuma, A.; Hoshino, T.; Obara, Y.; Nakahata, N.; Kobayashi, J. Tetrahedron 2005, 61, 1955–1960. doi:10.1016/j.tet.2005.01.011 |
| 6. | Ishiuchi, K.; Kubota, T.; Ishiyama, H.; Hayashi, S.; Shibata, T.; Mori, K.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2011, 19, 749–753. doi:10.1016/j.bmc.2010.12.025 |
| 7. | Ishiuchi, K.; Kubota, T.; Mikami, Y.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2007, 15, 413–417. doi:10.1016/j.bmc.2006.09.043 |
| 21. | Varela, J. A.; Saá, C. Chem. Rev. 2003, 103, 3787–3802. doi:10.1021/cr030677f |
| 3. | Tun, M. K. M.; Herzon, S. B. J. Exp. Pharmacol. 2012, 4, 113–123. doi:10.2147/jep.s27084 |
| 18. | Ma, D.; Martin, B. S.; Gallagher, K. S.; Saito, T.; Dai, M. J. Am. Chem. Soc. 2021, 143, 16383–16387. doi:10.1021/jacs.1c08626 |
| 19. | Uosis-Martin, M.; Pantoş, G. D.; Mahon, M. F.; Lewis, S. E. J. Org. Chem. 2013, 78, 6253–6263. doi:10.1021/jo401014n |
| 13. | Martin, B. S.; Ma, D.; Saito, T.; Gallagher, K. S.; Dai, M. Synthesis 2024, 56, 107–117. doi:10.1055/a-2107-5159 |
| 16. | Fischer, D. F.; Sarpong, R. J. Am. Chem. Soc. 2010, 132, 5926–5927. doi:10.1021/ja101893b |
| 12. | Wang, J.; Zhang, Z.-K.; Jiang, F.-F.; Qi, B.-W.; Ding, N.; Hnin, S. Y. Y.; Liu, X.; Li, J.; Wang, X.-h.; Tu, P.-F.; Abe, I.; Morita, H.; Shi, S.-P. Org. Lett. 2020, 22, 8725–8729. doi:10.1021/acs.orglett.0c03339 |
| 17. | Zhao, L.; Tsukano, C.; Kwon, E.; Takemoto, Y.; Hirama, M. Angew. Chem., Int. Ed. 2013, 52, 1722–1725. doi:10.1002/anie.201208297 |
| 11. | Yeap, J. S.-Y.; Lim, K.-H.; Yong, K.-T.; Lim, S.-H.; Kam, T.-S.; Low, Y.-Y. J. Nat. Prod. 2019, 82, 324–329. doi:10.1021/acs.jnatprod.8b00754 |
| 9. | Heathcock, C. H.; Kleinman, E. F.; Binkley, E. S. J. Am. Chem. Soc. 1982, 104, 1054–1068. doi:10.1021/ja00368a024 |
| 10. | Azuma, M.; Yoshikawa, T.; Kogure, N.; Kitajima, M.; Takayama, H. J. Am. Chem. Soc. 2014, 136, 11618–11621. doi:10.1021/ja507016g |
| 14. | Yuan, C.; Chang, C.-T.; Axelrod, A.; Siegel, D. J. Am. Chem. Soc. 2010, 132, 5924–5925. doi:10.1021/ja101956x |
| 15. | Yuan, C.; Chang, C.-T.; Siegel, D. J. Org. Chem. 2013, 78, 5647–5668. doi:10.1021/jo400695c |
| 26. | Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 390–391. doi:10.1021/ja0173019 |
| 27. | Cho, J.-Y.; Tse, M. K.; Holmes, D.; Maleczka, R. E., Jr.; Smith, M. R., III. Science 2002, 295, 305–308. doi:10.1126/science.1067074 |
| 22. | Varela, J. A.; Castedo, L.; Saá, C. J. Am. Chem. Soc. 1998, 120, 12147–12148. doi:10.1021/ja982832r |
| 23. | de la Torre, M. C.; Sierra, M. A. Angew. Chem., Int. Ed. 2004, 43, 160–181. doi:10.1002/anie.200200545 |
| 24. | Razzak, M.; De Brabander, J. K. Nat. Chem. Biol. 2011, 7, 865–875. doi:10.1038/nchembio.709 |
| 25. | Bao, R.; Zhang, H.; Tang, Y. Acc. Chem. Res. 2021, 54, 3720–3733. doi:10.1021/acs.accounts.1c00459 |
| 35. | Ciamician, G. L.; Dennstedt, M. Ber. Dtsch. Chem. Ges. 1881, 14, 1153–1163. doi:10.1002/cber.188101401240 |
| 36. | Welin, E. R.; Ngamnithiporn, A.; Klatte, M.; Lapointe, G.; Pototschnig, G. M.; McDermott, M. S. J.; Conklin, D.; Gilmore, C. D.; Tadross, P. M.; Haley, C. K.; Negoro, K.; Glibstrup, E.; Grünanger, C. U.; Allan, K. M.; Virgil, S. C.; Slamon, D. J.; Stoltz, B. M. Science 2019, 363, 270–275. doi:10.1126/science.aav3421 |
| 32. | Al-Ahmad, R.; Dai, M. Acc. Chem. Res. 2025, 58, 1392–1406. doi:10.1021/acs.accounts.5c00030 |
| 33. | Jurczyk, J.; Woo, J.; Kim, S. F.; Dherange, B. D.; Sarpong, R.; Levin, M. D. Nat. Synth. 2022, 1, 352–364. doi:10.1038/s44160-022-00052-1 |
| 34. | Li, H.; Shen, S.-J.; Zhu, C.-L.; Xu, H. J. Am. Chem. Soc. 2019, 141, 9415–9421. doi:10.1021/jacs.9b04381 |
| 30. | Campeau, L.-C.; Rousseaux, S.; Fagnou, K. J. Am. Chem. Soc. 2005, 127, 18020–18021. doi:10.1021/ja056800x |
| 31. | Zhao, L.; Tsukano, C.; Kwon, E.; Shirakawa, H.; Kaneko, S.; Takemoto, Y.; Hirama, M. Chem. – Eur. J. 2017, 23, 802–812. doi:10.1002/chem.201604647 |
| 28. | Newton, J. N.; Fischer, D. F.; Sarpong, R. Angew. Chem., Int. Ed. 2013, 52, 1726–1730. doi:10.1002/anie.201208571 |
| 29. | Haley, H. M. S.; Payer, S. E.; Papidocha, S. M.; Clemens, S.; Nyenhuis, J.; Sarpong, R. J. Am. Chem. Soc. 2021, 143, 4732–4740. doi:10.1021/jacs.1c00457 |
© 2025 Al-Ahmad and Dai; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.