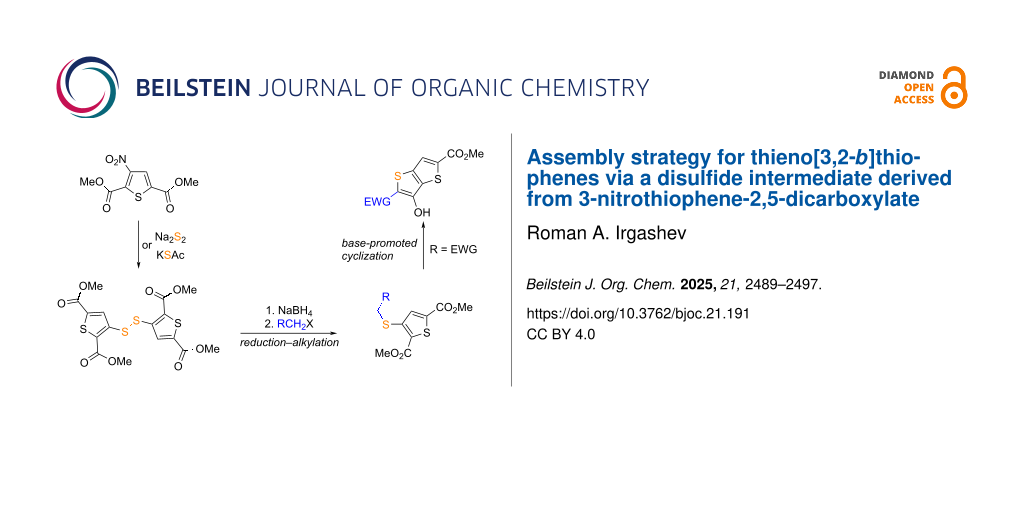
Abstract
A versatile synthetic route to thieno[3,2-b]thiophenes was elaborated from dimethyl 3-nitrothiophene-2,5-dicarboxylate. Nucleophilic substitution of the nitro group with sulfur nucleophiles, including thioacetate or disulfide anions as well as thioacetamide, yielded bis(thiophen-3-yl)disulfide and sulfide derivatives. The disulfide served as a suitable precursor for the preparation of 3-alkylthio-substituted thiophene-2,5-dicarboxylates by its one-pot reduction–alkylation using NaBH4 in DMF followed by an alkylating agent. Base-promoted cyclization of electron-deficient 3-alkylthio derivatives furnished 2-aryl-, 2-aroyl-, and 2-cyano-substituted thieno[3,2-b]thiophenes, bearing a 3-hydroxy group. This protocol broadens access to functionalized thieno[3,2-b]thiophenes with potential applications in pharmaceutical and materials chemistry.

Graphical Abstract
Introduction
Thieno[3,2-b]thiophene (TT) derivatives are a valuable class of fused heteroaromatic compounds characterized by rigid planar π-conjugated backbones that provide narrow band gaps, high charge carrier mobility, and intermolecular π–π stacking interactions, making them particularly attractive for the design of organic materials. Indeed, the use of TT frameworks for semiconductors has led to significant progress in organic field-effect transistors (OFETs), organic photovoltaics, organic light-emitting diodes, and chemical sensors [1-3]. For instance, TT-containing copolymers have shown high hole mobility, strong absorption in the visible region, and improved morphological stability [4-6]. Recent reports have shown that the development of alkyl-supported TT-based semiconductors has led to high-performance, air-stable OFETs [7], while integration of TT frameworks into donor–acceptor architectures has enabled the development of materials for various types of organic solar cells [8-13]. TT-extended phthalocyanines and related dyes have also been applied in gas-sensing devices, benefiting from their π-stacked alignment and electron-rich character [14].
On the other hand, the biological potential of TT molecules has attracted increasing interest as numerous TT-based compounds were proposed as promising candidates in medicinal chemistry due to their diverse pharmacological profiles including antitumor, antiviral, antimicrobial, enzymatic and phototherapeutic activities [15]. Among them, TT-2-carboxanilides exhibit notable DNA intercalation and antiproliferative effects [16,17], while TT–BODIPY derivatives were shown to efficiently generate singlet oxygen and demonstrate light-induced cytotoxicity, highlighting their promise as photodynamic therapeutic agents [18]. Near-infrared TT–DPP-based dyes have also been developed for photothermal and photodynamic therapy, where their π-conjugated frameworks contribute to both imaging and therapeutic functions [19]. Some other TT-based compounds have been reported to inhibit specific carbonic anhydrase isoforms [20], protein tyrosine phosphatase 1B [21], as well as to act as agonists of the GPR35 receptor [22], further underscoring the value of the TT scaffold in enzyme- and receptor-targeted drug discovery.
Given the established practical significance of thieno[3,2-b]thiophene compounds, the construction of molecules featuring the TT core remains a key objective in organic synthesis. A widely adopted strategy for building the TT scaffold involves the annulation of a second thiophene ring onto a suitably functionalized thiophene precursor. Most commonly, this approach starts from halogenated thiophenes such as 3-bromo- or 3-chlorothiophenes, which undergo subsequent cyclization to furnish the TT framework [2,23,24]. Despite the wide structural diversity of TT derivatives, 3-hydroxy-substituted analogues remain rare, with only a few synthetic strategies described for their preparation, as illustrated in Scheme 1. One such route involves sulfur insertion into a thiophen-3-yl lithium derivative, followed by electrophilic trapping of the thiolate and base-induced cyclization to afford the 3-hydroxy-TT [25]. Route II is represented by the single example of a one-pot reaction of 2-methylquinoline and 3-bromothiophene-2-carbaldehyde in the presence of elemental S and K2CO3 in DMSO to give the desired product [26]. Route III, previously elaborated in our group, utilizes the nucleophilic substitution of the Cl atom in 3-chlorothiophene-2-carboxylates by methyl thioglycolate in the presence of KOt-Bu, followed by KOt-Bu-mediated cyclization to the 3-hydroxy-TTs [27]. In route IV, cleavage of the ethyl xanthate group in the starting substrate by NaOMe generates a thiolate intermediate, which undergoes S-alkylation and subsequent NaOMe-promoted cyclization to afford the 3-hydroxy-TT [28]. In our recent works, it was presented an effective strategy for synthesizing TTs from the activated 3-nitrosubstituted thiophenes [29,30]. This strategy provides an effective access to the 3-hydroxy-TTs via the nucleophilic substitution of the nitro group in 3-nitrothiophene precursors using S-nucleophiles, such as alkyl thioglycolates or mercaptoacetone (Scheme 1, route V).
![[1860-5397-21-191-i1]](/bjoc/content/inline/1860-5397-21-191-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The synthetic routes to 3-hydroxy-substituted TT derivatives.
Scheme 1: The synthetic routes to 3-hydroxy-substituted TT derivatives.
Herein, we wish to report a modified strategy based on route IV involving the stepwise construction of the TT framework. This approach is accomplished via the nucleophilic substitution of the nitro group with a S-nucleophile to obtain the thiophene-3-thiolate or its equivalent, followed by further S-alkylation and base-promoted cyclization to form the 3-hydroxy-TT molecules (Scheme 2).
![[1860-5397-21-191-i2]](/bjoc/content/inline/1860-5397-21-191-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The present retrosynthetic plan for constructing TT molecules.
Scheme 2: The present retrosynthetic plan for constructing TT molecules.
Results and Discussion
We began our study by investigating the reaction of dimethyl 3-nitrothiophene-2,5-dicarboxylate (1) with Na2S, inspired by Beck’s reported synthesis of 2-substituted-3-aminobenzo[b]thiophenes via the nucleophilic substitution of the nitro group in 2-nitrobenzonitriles using Na2S in a DMF/water medium, followed by alkylation of the resulting 2-cyanophenylthiolates and subsequent cyclization [31]. However, in our case, the reaction of ester 1 with Na2S in either acetone or a DMF/water mixture predominantly led to degradation of the starting material. Subsequent treatment with iodomethane gave a complex mixture of compounds, in which only trace amounts of the expected derivative 1-SMe were detected by GC–MS analysis (Scheme 3). In this regard, the reaction of 3-nitrothiophene 1 with Na2S turned out to be an unsuitable route for nucleophilic substitution of its nitro group.
![[1860-5397-21-191-i3]](/bjoc/content/inline/1860-5397-21-191-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: An attempt to nucleophilically substitute the NO2 group in ester 1.
Scheme 3: An attempt to nucleophilically substitute the NO2 group in ester 1.
Next, we explored an alternative route using potassium thioacetate (KSAc) as a softer nucleophile. Unexpectedly, the reaction of ester 1 with KSAc in acetone gave a mixture of products other than the desired 3-AcS-substituted thiophene, promoting further study of these structures and reaction mechanism. 1H NMR spectroscopy of the obtained mixture provided the first key information about its nature, namely the presence of two major products 2 and 3, both of which showed three characteristic singlets with signal integration 1:3:3. For compound 2, these signals appeared at 7.40, 3.91, and 3.89 ppm, whereas for compound 3, the signals were observed at 7.83, 3.97, and 3.87 ppm. These data strongly suggest the absence of a proton at the C-3 position of the thiophene ring and the presence of two methoxycarbonyl groups. Further isolation of compounds 2 and 3 in analytically pure form allowed their structure to be established thanks to elemental analysis and HRMS. It was found that compounds 2 and 3 are dimeric derivatives of the starting thiophene, linked by sulfur bridges located at the C-3 position instead of the nitro group. Compound 2 was identified as a bis(thiophen-3-yl)sulfide derivative and compound 3 as a bis(thiophen-3-yl)disulfide derivative, both with methoxycarbonyl groups at the C-2 and C-5 positions of each thiophene ring (Scheme 4).
![[1860-5397-21-191-i4]](/bjoc/content/inline/1860-5397-21-191-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The reaction of ester 1 with potassium thioacetate.
Scheme 4: The reaction of ester 1 with potassium thioacetate.
The formation of these unexpected products, sulfide 2 and disulfide 3, necessitates consideration of a reaction mechanism beyond straightforward nucleophilic substitution of the nitro group in ester 1. Although the initial reaction probably involves a nucleophilic attack by the S-nucleophile on the activated thiophene ring, resulting in displacement of the nitro group, the subsequent fate of the intermediate appears to be critical. Based on our observations and literature data, a probable reaction mechanism was proposed. The initial nucleophilic substitution of the nitro group in ester 1 by the AcS− anion yields the S-acetyl derivative (intermediate A) and NO2− anion. The key step leading to the observed products is the reaction of the S-acetyl intermediate A with the generated NO2− anion, resulting in the formation of a thiophene-3-thiolate species (intermediate B) and acetyl nitrite. Thiolate B then acts as a nucleophile toward starting ester 1, resulting in the formation of sulfide 2. Simultaneously, thiolate B reacts with acetyl nitrite to form the S-nitrosothiol intermediate C and the acetate anion. S-Nitrosothiol C subsequently undergoes decomposition, losing nitrogen oxide to give disulfide 3 (Scheme 5). This proposed mechanism, in particular the conversion of the S-acetyl compound to the thiolate and then to the disulfide with the participation of nitrite, is consistent with the previously reported synthesis of bis(per-O-acetylated glycosyl)disulfides from S-acetyl glycosides and KNO2, providing a literature precedent for similar transformations [32].
![[1860-5397-21-191-i5]](/bjoc/content/inline/1860-5397-21-191-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: A probable mechanism for the formation of compounds 2 and 3.
Scheme 5: A probable mechanism for the formation of compounds 2 and 3.
In addition to potassium thioacetate, the reactivity of ester 1 was investigated with other related S-nucleophiles, such as potassium ethyl xanthate (KSC(S)OEt), sodium diethyldithiocarbamate (NaSC(S)NEt2) as well as thioacetamide in the presence of K2CO3 and Na2S2 (Table 1, entries 2–5). In each case, the reaction resulted in the formation of a mixture of sulfide 2 and disulfide 3, although the ratio of products varied considerably depending on the nature of the used S-nucleophile. Thus, the reaction with KSAc gave a 2/3 ratio of 1:5.82 (Table 1, entry 1), which greatly favored disulfide formation. The use of KSC(S)OEt and NaSC(S)NEt2 gave a 2/3 ratio of 1:0.92 and 1:0.8, respectively, indicating almost equivalent amounts of both products (Table 1, entries 2 and 3). The reaction with thioacetamide promoted the formation of sulfide 2 in a 2/3 ratio of 1:0.28 (Table 1, entry 4).
Table 1: The reaction of ester 1 with S-nucleophiles to form sulfide 2 and disulfide 3.
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-191-i8.svg?max-width=637&scale=1.0)
|
|||
| entry | S-nucleophile | 2/3 ratioa | yield |
| 1 | KSAc (1.1 equiv) | 1:5.82 | 3, 75%b |
| 2 | KSC(S)OEt (1.1 equiv) | 1:0.92 | 2 + 3, 33%/2, 15%b |
| 3 | NaSC(S)NEt2 (1.1 equiv) | 1:0.80 | 2 + 3, 27% |
| 4 | MeC(S)NH2 (1.2 equiv)/K2CO3 | 1:0.28 | 2, 51%b |
| 5c | Na2S2 (0.527 equiv) | 1:19.91 | 3, 71%b |
aAccording to 1H NMR spectroscopy. bThe yield of product isolated in analytically pure form. cA mixture of DMF/water/acetone was used in this experiment.
The most pronounced preference for disulfide formation was observed in the reaction with Na2S2, where the 2/3 ratio reached 1:19.91, indicating the almost exclusive formation of disulfide 3 (Table 1, entry 5). This result is consistent with a mechanism in which the nucleophilic substitution of the nitro group in ester 1 occurs via attack by the disulfide anion, wherein each sulfur atom reacts with one molecule 1, resulting in the formation of product 3. The minor presence of sulfide 2 in the product mixture can be attributed to partial decomposition of an intermediate aryl disulfide anion, which can fragment under the reaction conditions to release thiolate species capable of reacting separately with ester 1 to form compound 2.
Disulfide 3 was found to be an accessible and stable precursor of dimethyl 3-mercaptothiophene-2,5-dicarboxylate, a molecule that is suitable for S-alkylation. In this regard, reductive cleavage of the S–S bond in disulfide 3 followed by in situ alkylation was investigated for the one-pot synthesis of 3-alkylthio-substituted thiophene-2,5-dicarboxylates. We first focused our efforts on optimizing the conditions of this two-step process to determine suitable reducing agents and solvents (Table 2). Thus, 4-(chloromethyl)benzonitrile was used as a model alkylating agent, NaBH4 and Na2S2O4 were used as mild and accessible reducing agents, and K2CO3 was originally considered as a base for the alkylation step. In our first experiment, NaBH4 in methanol was used to reduce disulfide 3 (Table 2, entry 1). However, methanol proved to be an ineffective solvent due to the rapid decomposition of NaBH4 and poor solubility of substrate 3. Most of the starting material was recovered unchanged after complete decomposing NaBH4. In contrast, when ethanol (Table 2, entry 2) or isopropanol (Table 2, entry 3) was used as solvent, the reduction of disulfide 3 with NaBH4 proceeded more efficiently at reflux for 2 h. The reaction mixture was then treated with K2CO3 and the alkylating agent at room temperature. The product was identified as the desired 3-benzylthio-substituted thiophene-2,5-dicarboxylate, but it was obtained as a mixture of ester forms, namely dimethyl and diethyl esters in ethanol, and dimethyl and diisopropyl esters in isopropanol. These results indicate that the reaction with alcohol solvents resulted in partial transesterification of the methoxycarbonyl groups, while reductive cleavage of the S–S bond and the subsequent S-alkylation reaction were successful. To suppress the side reaction and improve reduction efficiency, we next employed DMF as a polar aprotic solvent. In Table 2, entry 4, reduction of disulfide 3 in DMF at 75 °C with NaBH4 was complete within 15 min. The excess reductant was quenched with methanol, and the reaction mixture was then treated with K2CO3 and the alkylating agent for 20 min, which allowed to obtain the desired product in 45% yield. A partial saponification of the ester groups was observed under these conditions, which contributed to the reduced yield. To address this, in a subsequent experiment (Table 2, entry 5), the alkylation step was performed without K2CO3, while DMF was retained as a solvent. The reaction proceeded cleanly, and product 4a was isolated in 88% yield. This result confirms that under these conditions, base-promoted side reactions, probably involving ester hydrolysis or alkylating agent decomposition, can be avoided by omitting the external base. In addition, Na2S2O4 was investigated as a reductant in a DMF/water 9:1 (v/v) mixture at room temperature (Table 2, entry 6). In the presence of both K2CO3 and the alkylating agent, the reaction proceeded smoothly over 24 h to afford the desired product in 60% yield. In contrast, when the reaction was performed without K2CO3 (Table 2, entry 7), no product was obtained, indicating that a basic medium is essential not only for alkylation but also to enable effective S–S bond reduction by Na2S2O4 under these conditions. These experiments show that the optimal conditions for the one-pot synthesis of 3-(alkylthio)thiophene-2,5-dicarboxylates involve NaBH4-mediated reduction of substrate 3 in DMF, and next alkylation in the absence of added base (Table 2, entry 5).
Table 2: Optimization of the reaction conditions for the reduction–alkylation of disulfide 3.
![[Graphic 2]](/bjoc/content/inline/1860-5397-21-191-i9.svg?max-width=637&scale=1.0)
|
||||||
| entrya | reductant | solvent | temp. | time | base | yield |
| 1 | NaBH4 | MeOH | rt | 30 min | – | nr |
| 2 | NaBH4 | EtOH | reflux | 2 h | K2CO3 | R = Me / R = Etb |
| 3 | NaBH4 | iPrOH | reflux | 2 h | K2CO3 | R = Me / R = iPrb |
| 4c | NaBH4 | DMF/MeOH | 75 °C | 15 min | K2CO3 | 4a, R = Me, 45%d |
| 5c | NaBH4 | DMF/MeOH | 75 °C | 15 min | – | 4a, R = Me, 88%d |
| 6 | Na2S2O4 | DMF/H2O | rt | 24 h | K2CO3 | 4a, R = Me, 60%d |
| 7 | Na2S2O4 | DMF/H2O | rt | 24 h | – | nr |
aSulfide 3 (0.4 mmol), NaBH4 (2.5 equiv) or Na2S2O4 (3 equiv) and 4-(chloromethyl)benzonitrile (2.2 equiv) were used in these experiments. bA mixture of 3-RS-substituted dialkyl thiophene-2,5-dicarboxylates was obtained. cThe oil bath temperature is indicated. dThe yield of product isolated in analytically pure form.
A number of benzyl halides and other electrophiles were used under optimal reaction conditions to obtain various 3-(alkylthio)thiophene-2,5-dicarboxylates. Among them, compounds 4b–g, bearing benzylthio substituents, were prepared using different benzyl-type alkylating agents. It was found that the reaction tolerated both electron-donating and electron-withdrawing groups on the aromatic ring, and the yields for this series ranged from 76% to 92%. Piperonyl mesylate was successfully used in the synthesis of compound 4g (92% yield), while thiophene-2-ylmethyl mesylate was used for the preparation of compound 4h (77% yield). For product 4b, methyl 4-(bromomethyl)benzoate was employed as the alkylating agent. In all other cases, benzyl chlorides were used for the alkylation.
The alkylation with chloroacetonitrile afforded 3-(cyanomethyl)thio derivative 5a in 48% yield, shown in parenthesis in Scheme 6, while bromoacetonitrile significantly improved the result, allowing product 5a to be obtained in 91% yield. Furthermore, we extended our approach to phenacyl-type alkylators to access 3-(phenacyl)thio compounds 6a and 6b, as well as their 3-[(thiophen-2-yl)carbonylmethyl]thio analog 6c. The corresponding phenacyl chlorides reacted smoothly to afford products 6a and 6b in 62% and 69% yields, whereas compound 6c was prepared in 76% yield using 2-(chloroacetyl)thiophene as the alkylating agent. This shows that the reaction tolerates a broader class of electrophiles, making it suitable for accessing precursors of TT molecules (Scheme 6).
![[1860-5397-21-191-i6]](/bjoc/content/inline/1860-5397-21-191-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: The synthesis of 3-(alkylthio)thiophene-2,5-dicarboxylates 4–6, yields, and scope of products. *From BrCH2CN (from ClCH2CN).
Scheme 6: The synthesis of 3-(alkylthio)thiophene-2,5-dicarboxylates 4–6, yields, and scope of products. *Fro...
For the next synthesis of TT derivatives, the base-promoted cyclization of the obtained thiophene-2,5-dicarboxylates 4–6 was studied for those derivatives having electron-withdrawing groups in the alkylthio fragment to ensure the closure of the thiophene ring. To establish the optimal conditions for the cyclization of compounds 4–6, a series of exploratory experiments was carried out using various bases and solvents. Initial attempts involved the use of NaOMe in methanol, a simple base–solvent combination for such substrates. However, this system proved unsuitable due to the limited solubility of the starting materials in methanol, which prevented efficient conversion. Subsequently, the substrates were introduced as THF solutions into NaOMe in methanol. Under these conditions, the cyclization did occur to form the corresponding Na salts of 3-hydroxy-TTs. Upon aqueous work-up and acidification, however, the reaction afforded mixtures of the desired products and the corresponding carboxylic acids due to partial hydrolysis of the ester groups. This behavior was observed during cyclization of substrates 4a and 5a. In the case of substrate 6b, treatment with NaOMe resulted primarily in the formation of carboxylic acid 9bA, isolated in 71% yield (Scheme 7). In addition, an attempt to cyclize 6b in the presence of DBU without a solvent at 110 °C led to significant destruction of the substrate and product 9b was obtained with a yield of about 20%. Cyclization of substrate 4a with NaH in toluene also gave product 7a in about 25% yield, along with the formation of a mixture of byproducts. It was found that the most favorable conditions for cyclization of compounds 4a, 4b, and 5a were their treatment with excess LiH in a solution of dry DMF at room temperature for 24 h. In this way, products 7a,b, and 8a were obtained in 62%, 55%, and 45% yields, respectively. In turn, the suitable reaction conditions for cyclization of substrates 6a–c were their treatment with excess Mg(OMe)2 in a solution of methanol/toluene, which afforded products 9a–c in 66–73% yield. It should be noted that the bases LiH and Mg(OMe)2 chosen for the cyclization are convenient from the point of view of availability and work safety. Indeed, LiH is not a pyrophoric hydride compared to other alkali metal hydrides, NaH or KH, while Mg(OMe)2 is easily prepared by dissolving I2-activated Mg metal in dry methanol.
![[1860-5397-21-191-i7]](/bjoc/content/inline/1860-5397-21-191-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: The synthesis of TT derivatives, yields, and scope of products. Conditions: i) LiH (5 equiv), DMF, rt, 24 h. ii) Mg(OMe)2 (5 equiv), PhMe/MeOH, 90 °C, 2 h. *NaOMe (2 equiv), MeOH/THF, 75 °C, 2 h.
Scheme 7: The synthesis of TT derivatives, yields, and scope of products. Conditions: i) LiH (5 equiv), DMF, ...
Conclusion
In summary, this study reveals an unusual and previously poorly understood reactivity of 3-nitrothiophene-2,5-dicarboxylate toward sulfur-containing nucleophiles such as potassium thioacetate or thioacetamide, which resulted in the efficient formation of both bis(thiophen-3-yl) sulfide and disulfide derivatives via substitution of the nitro group. This reaction opens a new route to sulfur-containing derivatives of the thiophene nucleus. Beyond enabling an effective synthesis of 3-hydroxy-substituted TTs bearing electron-withdrawing groups at the C-2 position, this work also establishes a robust approach to a wide range of 3-substituted thiophene-2,5-dicarboxylates. These intermediates are of interest as valuable building blocks for the synthesis of more complex thiophene derivatives in both materials and medicinal chemistry contexts.
Supporting Information
| Supporting Information File 1: Full experimental details, characterization data, copies of NMR spectra and HRMS for all new compounds. | ||
| Format: PDF | Size: 4.4 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z
Return to citation in text: [1] -
Cinar, M. E.; Ozturk, T. Chem. Rev. 2015, 115, 3036–3140. doi:10.1021/cr500271a
Return to citation in text: [1] [2] -
Podlesný, J.; Bureš, F. Organics 2022, 3, 446–469. doi:10.3390/org3040029
Return to citation in text: [1] -
Qi, F.; Song, J.; Xiong, W.; Huo, L.; Sun, X.; Sun, Y. Dyes Pigm. 2018, 155, 126–134. doi:10.1016/j.dyepig.2018.03.013
Return to citation in text: [1] -
Kim, H.-S.; Kim, Y.-H.; Kim, T.-H.; Noh, Y.-Y.; Pyo, S.; Yi, M. H.; Kim, D.-Y.; Kwon, S.-K. Chem. Mater. 2007, 19, 3561–3567. doi:10.1021/cm070053g
Return to citation in text: [1] -
Bronstein, H.; Chen, Z.; Ashraf, R. S.; Zhang, W.; Du, J.; Durrant, J. R.; Shakya Tuladhar, P.; Song, K.; Watkins, S. E.; Geerts, Y.; Wienk, M. M.; Janssen, R. A. J.; Anthopoulos, T.; Sirringhaus, H.; Heeney, M.; McCulloch, I. J. Am. Chem. Soc. 2011, 133, 3272–3275. doi:10.1021/ja110619k
Return to citation in text: [1] -
Wu, K.; Chen, P.; Cui, T.; Zhang, B.; Sun, C.-L.; Wang, J.; Shao, X.; Chen, L.; Chen, Y.; Liu, Z. Macromol. Rapid Commun. 2025, 46, 2401055. doi:10.1002/marc.202401055
Return to citation in text: [1] -
Wang, W.; Wang, T.; Wu, X.; Tong, H.; Wang, L. Mater. Chem. Front. 2020, 4, 3328–3337. doi:10.1039/d0qm00514b
Return to citation in text: [1] -
Zhang, P.; Chen, K.; Gao, X.; Zhang, J.; Zeng, Y.; Tang, R.; Wu, F.; Zhong, C.; Zhu, L. Dyes Pigm. 2023, 220, 111693. doi:10.1016/j.dyepig.2023.111693
Return to citation in text: [1] -
Isci, R.; Unal, M.; Yesil, T.; Ekici, A.; Sütay, B.; Zafer, C.; Ozturk, T. Front. Mater. 2023, 10, 1125462. doi:10.3389/fmats.2023.1125462
Return to citation in text: [1] -
Li, J.; Zhang, C.; Sun, X.; Wang, H.; Hu, H.; Wang, K.; Xiao, M. Nano Energy 2024, 125, 109542. doi:10.1016/j.nanoen.2024.109542
Return to citation in text: [1] -
Gao, Y.; Zhang, H.; Xie, Y.; Xu, X.; Liu, Y.; Lu, H.; Zhang, W.; Liu, Y.; Li, C.; Bo, Z. Chin. Chem. Lett. 2026, 37, 111622. doi:10.1016/j.cclet.2025.111622
Return to citation in text: [1] -
Fürk, P.; Mallick, S.; Rath, T.; Reinfelds, M.; Wu, M.; Spiecker, E.; Simic, N.; Haberfehlner, G.; Kothleitner, G.; Ressel, B.; Holler, S.; Schaubeder, J. B.; Materna, P.; Amenitsch, H.; Trimmel, G. J. Mater. Chem. C 2023, 11, 8393–8404. doi:10.1039/d3tc01112g
Return to citation in text: [1] -
Isci, R.; Yavuz, O.; Faraji, S.; Gunturkun, D.; Eroglu, M.; Majewski, L. A.; Yilmaz, I.; Ozturk, T. J. Mater. Chem. C 2024, 13, 472–483. doi:10.1039/d4tc03208j
Return to citation in text: [1] -
Rafiq, A.; Aslam, S.; Ahmad, M.; Nazir, M. S.; Farooq, A.; Sultan, S. Mol. Diversity 2024, 28, 1793–1821. doi:10.1007/s11030-023-10647-1
Return to citation in text: [1] -
Jarak, I.; Kralj, M.; Piantanida, I.; Šuman, L.; Žinić, M.; Pavelić, K.; Karminski-Zamola, G. Bioorg. Med. Chem. 2006, 14, 2859–2868. doi:10.1016/j.bmc.2005.12.004
Return to citation in text: [1] -
Aleksić, M.; Bertoša, B.; Nhili, R.; Uzelac, L.; Jarak, I.; Depauw, S.; David-Cordonnier, M.-H.; Kralj, M.; Tomić, S.; Karminski-Zamola, G. J. Med. Chem. 2012, 55, 5044–5060. doi:10.1021/jm300505h
Return to citation in text: [1] -
Cao, N.; Jiang, Y.; Song, Z.-B.; Chen, D.; Wu, D.; Chen, Z.-L.; Yan, Y.-J. Eur. J. Med. Chem. 2024, 264, 116012. doi:10.1016/j.ejmech.2023.116012
Return to citation in text: [1] -
Yang, X.; Yu, Q.; Yang, N.; Xue, L.; Shao, J.; Li, B.; Shao, J.; Dong, X. J. Mater. Chem. B 2019, 7, 2454–2462. doi:10.1039/c8tb03185a
Return to citation in text: [1] -
Prugh, J. D.; Hartman, G. D.; Mallorga, P. J.; McKeever, B. M.; Michelson, S. R.; Murcko, M. A.; Schwam, H.; Smith, R. L.; Sondey, J. M.; Springer, J. P.; Sugrue, M. F. J. Med. Chem. 1991, 34, 1805–1818. doi:10.1021/jm00110a008
Return to citation in text: [1] -
Moretto, A. F.; Kirincich, S. J.; Xu, W. X.; Smith, M. J.; Wan, Z.-K.; Wilson, D. P.; Follows, B. C.; Binnun, E.; Joseph-McCarthy, D.; Foreman, K.; Erbe, D. V.; Zhang, Y. L.; Tam, S. K.; Tam, S. Y.; Lee, J. Bioorg. Med. Chem. 2006, 14, 2162–2177. doi:10.1016/j.bmc.2005.11.005
Return to citation in text: [1] -
Deng, H.; Hu, H.; He, M.; Hu, J.; Niu, W.; Ferrie, A. M.; Fang, Y. J. Med. Chem. 2011, 54, 7385–7396. doi:10.1021/jm200999f
Return to citation in text: [1] -
Litvinov, V. P. Adv. Heterocycl. Chem. 2006, 90, 125–203. doi:10.1016/s0065-2725(05)90002-0
Return to citation in text: [1] -
Ilhan, K. T.; Topal, S.; Eroglu, M. S.; Ozturk, T. RSC Adv. 2019, 9, 38407–38413. doi:10.1039/c9ra08023f
Return to citation in text: [1] -
Hergué, N.; Frère, P.; Roncali, J. Org. Biomol. Chem. 2011, 9, 588–595. doi:10.1039/c0ob00585a
Return to citation in text: [1] -
Wang, D.; Xiao, F.; Zhang, F.; Deng, G.-J. Chin. J. Chem. 2021, 39, 2483–2488. doi:10.1002/cjoc.202100238
Return to citation in text: [1] -
Demina, N. S.; Kazin, N. A.; Rasputin, N. A.; Irgashev, R. A.; Rusinov, G. L. Beilstein J. Org. Chem. 2019, 15, 2678–2683. doi:10.3762/bjoc.15.261
Return to citation in text: [1] -
Abdallah, S.; Darias, V.; Donoso, R.; de Urríes, P. J.; Lissavetzky, J. Arch. Pharm. (Weinheim, Ger.) 1996, 329, 216–222. doi:10.1002/ardp.19963290408
Return to citation in text: [1] -
Irgashev, R. A.; Kazin, N. A. Organics 2024, 5, 507–519. doi:10.3390/org5040027
Return to citation in text: [1] -
Irgashev, R.; Steparuk, A.; Kazin, N. ARKIVOC 2025, No. 5, 202512401. doi:10.24820/ark.5550190.p012.401
Return to citation in text: [1] -
Beck, J. R.; Yahner, J. A. J. Org. Chem. 1974, 39, 3440–3441. doi:10.1021/jo00937a040
Return to citation in text: [1] -
Ge, J.-T.; Zhang, L.-F.; Pu, L.; Zhang, Y.; Pei, Z.-C.; Dong, H. ChemistrySelect 2020, 5, 14549–14553. doi:10.1002/slct.202003566
Return to citation in text: [1]
| 31. | Beck, J. R.; Yahner, J. A. J. Org. Chem. 1974, 39, 3440–3441. doi:10.1021/jo00937a040 |
| 28. | Abdallah, S.; Darias, V.; Donoso, R.; de Urríes, P. J.; Lissavetzky, J. Arch. Pharm. (Weinheim, Ger.) 1996, 329, 216–222. doi:10.1002/ardp.19963290408 |
| 29. | Irgashev, R. A.; Kazin, N. A. Organics 2024, 5, 507–519. doi:10.3390/org5040027 |
| 30. | Irgashev, R.; Steparuk, A.; Kazin, N. ARKIVOC 2025, No. 5, 202512401. doi:10.24820/ark.5550190.p012.401 |
| 1. | Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z |
| 2. | Cinar, M. E.; Ozturk, T. Chem. Rev. 2015, 115, 3036–3140. doi:10.1021/cr500271a |
| 3. | Podlesný, J.; Bureš, F. Organics 2022, 3, 446–469. doi:10.3390/org3040029 |
| 14. | Isci, R.; Yavuz, O.; Faraji, S.; Gunturkun, D.; Eroglu, M.; Majewski, L. A.; Yilmaz, I.; Ozturk, T. J. Mater. Chem. C 2024, 13, 472–483. doi:10.1039/d4tc03208j |
| 26. | Wang, D.; Xiao, F.; Zhang, F.; Deng, G.-J. Chin. J. Chem. 2021, 39, 2483–2488. doi:10.1002/cjoc.202100238 |
| 8. | Wang, W.; Wang, T.; Wu, X.; Tong, H.; Wang, L. Mater. Chem. Front. 2020, 4, 3328–3337. doi:10.1039/d0qm00514b |
| 9. | Zhang, P.; Chen, K.; Gao, X.; Zhang, J.; Zeng, Y.; Tang, R.; Wu, F.; Zhong, C.; Zhu, L. Dyes Pigm. 2023, 220, 111693. doi:10.1016/j.dyepig.2023.111693 |
| 10. | Isci, R.; Unal, M.; Yesil, T.; Ekici, A.; Sütay, B.; Zafer, C.; Ozturk, T. Front. Mater. 2023, 10, 1125462. doi:10.3389/fmats.2023.1125462 |
| 11. | Li, J.; Zhang, C.; Sun, X.; Wang, H.; Hu, H.; Wang, K.; Xiao, M. Nano Energy 2024, 125, 109542. doi:10.1016/j.nanoen.2024.109542 |
| 12. | Gao, Y.; Zhang, H.; Xie, Y.; Xu, X.; Liu, Y.; Lu, H.; Zhang, W.; Liu, Y.; Li, C.; Bo, Z. Chin. Chem. Lett. 2026, 37, 111622. doi:10.1016/j.cclet.2025.111622 |
| 13. | Fürk, P.; Mallick, S.; Rath, T.; Reinfelds, M.; Wu, M.; Spiecker, E.; Simic, N.; Haberfehlner, G.; Kothleitner, G.; Ressel, B.; Holler, S.; Schaubeder, J. B.; Materna, P.; Amenitsch, H.; Trimmel, G. J. Mater. Chem. C 2023, 11, 8393–8404. doi:10.1039/d3tc01112g |
| 27. | Demina, N. S.; Kazin, N. A.; Rasputin, N. A.; Irgashev, R. A.; Rusinov, G. L. Beilstein J. Org. Chem. 2019, 15, 2678–2683. doi:10.3762/bjoc.15.261 |
| 7. | Wu, K.; Chen, P.; Cui, T.; Zhang, B.; Sun, C.-L.; Wang, J.; Shao, X.; Chen, L.; Chen, Y.; Liu, Z. Macromol. Rapid Commun. 2025, 46, 2401055. doi:10.1002/marc.202401055 |
| 2. | Cinar, M. E.; Ozturk, T. Chem. Rev. 2015, 115, 3036–3140. doi:10.1021/cr500271a |
| 23. | Litvinov, V. P. Adv. Heterocycl. Chem. 2006, 90, 125–203. doi:10.1016/s0065-2725(05)90002-0 |
| 24. | Ilhan, K. T.; Topal, S.; Eroglu, M. S.; Ozturk, T. RSC Adv. 2019, 9, 38407–38413. doi:10.1039/c9ra08023f |
| 4. | Qi, F.; Song, J.; Xiong, W.; Huo, L.; Sun, X.; Sun, Y. Dyes Pigm. 2018, 155, 126–134. doi:10.1016/j.dyepig.2018.03.013 |
| 5. | Kim, H.-S.; Kim, Y.-H.; Kim, T.-H.; Noh, Y.-Y.; Pyo, S.; Yi, M. H.; Kim, D.-Y.; Kwon, S.-K. Chem. Mater. 2007, 19, 3561–3567. doi:10.1021/cm070053g |
| 6. | Bronstein, H.; Chen, Z.; Ashraf, R. S.; Zhang, W.; Du, J.; Durrant, J. R.; Shakya Tuladhar, P.; Song, K.; Watkins, S. E.; Geerts, Y.; Wienk, M. M.; Janssen, R. A. J.; Anthopoulos, T.; Sirringhaus, H.; Heeney, M.; McCulloch, I. J. Am. Chem. Soc. 2011, 133, 3272–3275. doi:10.1021/ja110619k |
| 25. | Hergué, N.; Frère, P.; Roncali, J. Org. Biomol. Chem. 2011, 9, 588–595. doi:10.1039/c0ob00585a |
| 19. | Yang, X.; Yu, Q.; Yang, N.; Xue, L.; Shao, J.; Li, B.; Shao, J.; Dong, X. J. Mater. Chem. B 2019, 7, 2454–2462. doi:10.1039/c8tb03185a |
| 21. | Moretto, A. F.; Kirincich, S. J.; Xu, W. X.; Smith, M. J.; Wan, Z.-K.; Wilson, D. P.; Follows, B. C.; Binnun, E.; Joseph-McCarthy, D.; Foreman, K.; Erbe, D. V.; Zhang, Y. L.; Tam, S. K.; Tam, S. Y.; Lee, J. Bioorg. Med. Chem. 2006, 14, 2162–2177. doi:10.1016/j.bmc.2005.11.005 |
| 18. | Cao, N.; Jiang, Y.; Song, Z.-B.; Chen, D.; Wu, D.; Chen, Z.-L.; Yan, Y.-J. Eur. J. Med. Chem. 2024, 264, 116012. doi:10.1016/j.ejmech.2023.116012 |
| 22. | Deng, H.; Hu, H.; He, M.; Hu, J.; Niu, W.; Ferrie, A. M.; Fang, Y. J. Med. Chem. 2011, 54, 7385–7396. doi:10.1021/jm200999f |
| 16. | Jarak, I.; Kralj, M.; Piantanida, I.; Šuman, L.; Žinić, M.; Pavelić, K.; Karminski-Zamola, G. Bioorg. Med. Chem. 2006, 14, 2859–2868. doi:10.1016/j.bmc.2005.12.004 |
| 17. | Aleksić, M.; Bertoša, B.; Nhili, R.; Uzelac, L.; Jarak, I.; Depauw, S.; David-Cordonnier, M.-H.; Kralj, M.; Tomić, S.; Karminski-Zamola, G. J. Med. Chem. 2012, 55, 5044–5060. doi:10.1021/jm300505h |
| 32. | Ge, J.-T.; Zhang, L.-F.; Pu, L.; Zhang, Y.; Pei, Z.-C.; Dong, H. ChemistrySelect 2020, 5, 14549–14553. doi:10.1002/slct.202003566 |
| 15. | Rafiq, A.; Aslam, S.; Ahmad, M.; Nazir, M. S.; Farooq, A.; Sultan, S. Mol. Diversity 2024, 28, 1793–1821. doi:10.1007/s11030-023-10647-1 |
| 20. | Prugh, J. D.; Hartman, G. D.; Mallorga, P. J.; McKeever, B. M.; Michelson, S. R.; Murcko, M. A.; Schwam, H.; Smith, R. L.; Sondey, J. M.; Springer, J. P.; Sugrue, M. F. J. Med. Chem. 1991, 34, 1805–1818. doi:10.1021/jm00110a008 |
© 2025 Irgashev; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.