Institute of Organic Chemistry, Center for Molecular Biosciences Innsbruck (CMBI), Innrain 80-82, 6020 Innsbruck, Austria
Corresponding author email
Associate Editor: K. N. Allen Beilstein J. Org. Chem.2025,21, 483–489.https://doi.org/10.3762/bjoc.21.35 Received 06 Dec 2024,
Accepted 17 Feb 2025,
Published 04 Mar 2025
The preQ1 cIass-I riboswitch aptamer can utilize 7-aminomethyl-7-deazaguanine (preQ1) ligands that are equipped with an electrophilic handle for the covalent attachment of the ligand to the RNA. The simplicity of the underlying design of irreversibly bound ligand–RNA complexes has provided a new impetus in the fields of covalent RNA labeling and RNA drugging. Here, we present short and robust synthetic routes for such reactive preQ1 and (2,6-diamino-7-aminomethyl-7-deazapurine) DPQ1 ligands. The readily accessible key intermediates of preQ0 and DPQ0 (both bearing a nitrile moiety instead of the aminomethyl group) were reduced to the corresponding 7-formyl-7-deazapurine counterparts. These readily undergo reductive amination to form the hydroxyalkyl handles, which were further converted to the haloalkyl or mesyloxyalkyl-modified target compounds. In addition, we report hydrogenation conditions for preQ0 and DPQ0 that allow for cleaner and faster access to preQ1 compared to existing routes and provide the novel compound DPQ1.
Pre-queuosine 1 (preQ1) is a biosynthetic precursor of the hypermodified nucleoside queuosine (Q) that is found in the wobble position of bacterial as well as eukaryotic aspartyl-, asparaginyl-, histidyl- and tyrosyl-tRNA isoacceptors bearing the G34U35N36 anticodon motif [1]. Like other tRNA anticodon modifications, queuosine has been shown to increase translational fidelity and efficiency [2]. Structurally, queuosine and preQ1 (compound 1, Scheme 1A) belong to the 7-deazapurine family, which contain a pyrrolo[2,3-d]pyrimidine core. A rich pool of natural 7-deazapurine products has been identified, often (apparently) sharing a common biosynthetic pathway. Their functions are diverse; while some have been identified as having antifungal or antibiotic properties, others expand the chemical diversity and thus the functional sophistication of ribonucleic acids, as in the case of Q [3].
Scheme 1:
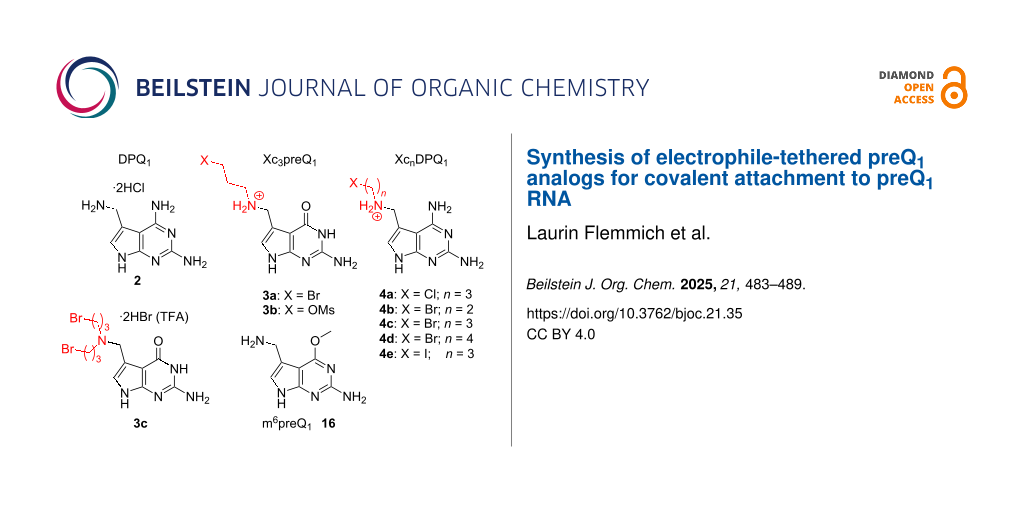
A) Chemical structures of hypermodified nucleobase queuine and nucleoside queuosine (Q) occurring as natural tRNA modifications. Purine ring numbering is indicated in grey. The synthetic targets of this study are highlighted in grey: Natural riboswitch ligand pre-queuosine 1 (preQ1), a novel preQ1 analog with altered base-pairing properties (2,6-diamino-pre-queuosine 2, DPQ1) and haloalkyl-modified preQ1 and DPQ1. B) The ligand classes of XcnpreQ1 and XcnDPQ1 allow specific formation of covalent small molecule–RNA complexes as has been recently demonstrated (see ref. [4]). Electrophile (E).
Scheme 1:
A) Chemical structures of hypermodified nucleobase queuine and nucleoside queuosine (Q) occurring a...
In most bacteria, Q biosynthesis is tightly regulated by riboswitches, which are highly structured RNA elements located mostly in the 5’-leader of messenger RNA. PreQ1 riboswitches sense the cellular concentration of preQ1 and regulate the expression of downstream located genes associated with the biosynthesis or transport of Q in a feedback-like manner. Binding of PreQ1 to the mRNA causes the riboswitch to commit an altered folding pathway, which affects the transcription or translation of the mRNA and results in altered transcript and/or protein levels [5].
In recent years, there has been a growing interest in the modification of preQ1. preQ1 derivatives, preQ1 analogs and mimics of preQ1 have greatly expanded our understanding of preQ1-binding biomolecules, such as riboswitches [6-8] or the queuosine biosynthetic enzyme machinery [9-11]. Recently, even the self-methylation activity of a preQ1 riboswitch has been discovered with a methylated preQ1 derivative acting as a ribozyme cofactor [12]. Moreover, these analogs have found utility in several biotechnological applications, including the identification of queuosinylation sites in cellular RNA [13], RNA and DNA labeling [14,15], and mRNA photocaging [16]. The latter applications rely on the promiscuity of the tRNA-modifying enzyme tRNA-guanine transglycosylase (TGT), which can incorporate functionalized preQ1 congeners into oligonucleotide strands at specific recognition sites.
In addition, the potential of modified preQ1 for protein enzyme-independent RNA labeling has also been demonstrated [12,17]. In a recent study [4], sequence-specific RNA–small molecule crosslinking (Scheme 1B) was achieved in vitro and in living cells using rationally designed electrophile-tethered derivatives of preQ1 (1) and its Watson–Crick diamino-faced counterpart DPQ1 (2, Scheme 1A). These ligands (compound classes 3 and 4, Scheme 1A) were tailored to target a conserved guanine nucleobase within a natural preQ1-binding mRNA domain, namely the preQ1 class-I riboswitch (preQ1-I) from Thermoanaerobacter tengcongensis. By rigorously analyzing the high-resolution structures available for this ligand–RNA complex, the approach exploits the natural, sequence-inherent reactivity hotspots of RNA and thus avoids the use of highly electrophilic warheads otherwise typically employed in RNA-small molecule crosslinking [18-21]. Instead, primary alkyl halides (or mesylates, Scheme 1, in particular compounds 3a, 3b and 4c) were found to be potent yet mild alkylators that minimize off-target reactivity, while providing reasonably fast labeling kinetics and up to quantitative conversion under quasi-physiological conditions [4].
Obviously, the rapid dissemination and widespread acceptance of such labeling methods depend on fast and simple access to the small molecule probes. Here, we report efficient synthetic routes to haloalkyl- and mesylate-modified preQ13a and 3b, the corresponding variants with different Watson–Crick face of DPQ1 (4a–e), as well as to the nucleobase precursors preQ1 (1) and DPQ1 (2), respectively.
Results and Discussion
Synthesis of preQ1 and DPQ1
Several synthetic strategies towards preQ1 and its derivatives have been reported [22-26]. Among these reports, various silylation and protection strategies have been employed to address the poor solubility of preQ1 (and analogs) in organic solvents [9,22,23,25-29]. Herein, we report an optimized three-step protocol, free of protecting groups and time-consuming purification steps, that provides preQ1 in 43% overall yield with a purity of >98% (Scheme 2). The approach is based on the cyclocondensation reaction between 2-chloro-3-cyanopropan-1-al (6) (itself obtained from chloroacetonitrile and methyl formate) and 2,6-diaminopyrimidin-4(3H)-one to afford preQ0 (7), as originally reported by Townsend et al. [30]. The next step, namely the reduction of the nitrile moiety by hydrogenation is critical and notoriously difficult due to the low reactivity of this group in preQ0[26]. We solved this problem by applying strongly acidic protic conditions [31] together with a 7-fold increase in hydrogenation pressure (30 bar); this resulted in an almost quantitative conversion and pure preQ1 (1) in the form of its dihydrochloride salt which was isolated after a simple filtration step.
Scheme 2:
Three-step syntheses of preQ1 (1) and DPQ1 (2). For the synthesis of m6preQ1 (16) see Supporting Information File 1.
Scheme 2:
Three-step syntheses of preQ1 (1) and DPQ1 (2). For the synthesis of m6preQ1 (16) see Supporting Information File 1.
Using the same approach, we were able to prepare the novel 2,6-diamino preQ1 analog 2 (DPQ1) by hydrogenation of 8 (DPQ0[32]) (Scheme 2). In this case, however, a final purification step (by reversed-phase chromatography) was required. Notably, using the hydrogenation conditions described here, we were also able to streamline our previously reported 7-step synthesis of O6-methyl preQ1 (16, m6preQ1) [28]. The direct reduction of the precursor O6-methyl preQ0 (15, m6preQ0) was possible, eliminating the need for the previously introduced protection/solubility concept, which shortened the synthetic route to only four steps (Supporting Information File 1).
Synthesis of preQ1 and DPQ1 derivatives with electrophilic handles
For the synthesis of haloalkyl- and mesyloxyalkyl-modified preQ1 and DPQ1 ligands 3a,b and 4a–e (for target structures see Scheme 1), a divergent synthetic route was sought that provided flexibility with respect to linker length and nature of the electrophile. We thus identified aldehydes 9 and 10 as suitable branching points, which were easily derivatized to their aminomethyl-modified preQ1 analogs by reductive amination (Scheme 3). Their syntheses by Raney-Ni reduction of nitriles 7 and 8, previously described by Gangjee and co-workers [33], proceeded cleanly in our hands.
Scheme 3:
Syntheses of haloalkyl- and mesyloxyalkyl-modified preQ1 as and DPQ1 ligands.
Scheme 3:
Syntheses of haloalkyl- and mesyloxyalkyl-modified preQ1 as and DPQ1 ligands.
In the case of compound 4a, the chloropropyl moiety was directly installed by reductive amination of 9 with 3-chloropropylamine hydrochloride under basic conditions. A two-step reaction sequence, however, was necessary to generate derivatives 4b–e and 3a. First, the alkyl handles bearing a primary hydroxy group were introduced and then converted into the electrophile of choice. More specifically, to furnish compounds 4b–d, precursor 9 was treated with the corresponding amino alcohols in the presence of a desiccant. The imines formed were subjected to mild reduction with methanolic sodium borohydride. Upon purification by reversed-phase chromatography using aqueous hydrobromic acid as eluent (0.5% in H2O), a considerable fraction of the alcohols was already converted to the desired bromides 4c,d. Only in the case of 4b, no deoxygenative bromination was observed, and the alcohol intermediate was isolated in pure form. In both cases, quantitative bromination was achieved by heating the compounds in concentrated aqueous hydrobromic acid, which after evaporation afforded the pure compounds 4b–d.
To generate iodide 4e, alcohol 11 was isolated and subjected to Appel conditions in DMF, using elemental iodine as the halogen source. Notably, we were not able to efficiently generate the corresponding bromides with the same strategy.
The preQ1 derivative 3a was synthesized in a 2-step reaction sequence analogous to the DPQ1 derivative 4b, while four steps were required to obtain the corresponding mesylate 3b. Similar to 11, compound 12 was isolated as its trifluoroacetate salt. Selective Boc protection of the aliphatic amine gave 13, which was selectively O-mesylated to give compound 14. Compound 14 was found to slowly undergo intramolecular cyclization by displacement of the mesyl group to give a six-membered cyclic carbamate, a reactivity that has been described earlier [34]. Thus, care was taken to quickly isolate compound 14 and use it immediately in the next step. Deblocking of the secondary amine by treatment with trifluoroacetic acid afforded 3b in almost quantitative yield.
The bis(3-bromopropyl)-modified ligand 3c was generated by heating preQ1 together with bis(3-hydroxypropyl)amine. It is noteworthy that the amine exchange reaction is thought to proceed via a purine methide intermediate [11]. Subsequent treatment of the diol with aqueous hydrobromic acid provided 3c.
Conclusion
We have developed a divergent synthesis of 7-aminomethyl-7-deazapurines (preQ1 and DPQ1) with various electrophilic handles extending the aminomethyl moiety. These derivatives are capable of covalent tethering to the preQ1-I RNA aptamer. This aptamer occurs naturally in mRNA riboswitches in bacteria and is involved in ligand-dependent gene regulation. Therefore, this riboswitch (like others) has become an attractive target for drug design.
To date, most known RNA–small molecule binders interact in a non-covalent manner. The compounds presented here are part of our research program to tailor non-covalent RNA–small molecule ligands to their covalent counterparts. While “covalent drugs” have become a leading principle in medicinal chemistry in the “protein world” [35,36] – approximately 30% of all FDA-approved drugs form a covalent bond with their target protein – this concept is underexplored in the field of RNA drugging [37]. Recent studies suggest that the validation of RNA–small molecule interactions [38-40], drug efficacy or the identification of off-target effects of approved drugs on the transcriptome [41,42] could greatly benefit from covalency. We believe that these exciting new research directions will be furthered by the efficient synthetic routes to covalent RNA binders presented here.
Experimental
General procedure for reductive aminations (compounds 3a, 4b–d, 11, and 12). Aldehyde 9 or 10 (50.0 mg, 282 µmol) was suspended in methanol (1.3 mL). Anhydrous magnesium sulfate (340 mg, 2.82 mmol, 10 equiv) and the respective amino alcohol (2.82 mmol, 10 equiv) were added. The mixture was sonicated for 30 minutes and subsequently stirred at room temperature for 16 h. After cooling to 0 °C, sodium borohydride (96.1 mg, 2.54 mmol, 9 equiv) was added in portions over the course of 1 h. The mixture was stirred for additional 2.5 h at room temperature. Afterwards, the volatiles were removed under reduced pressure and the residue was taken up in dilute aqueous acid (for composition see individual experiments in Supporting Information File 1; compounds 3a, 4b–d, 11, and 12). If insolubles were present after pH 1–2 was reached, the suspension was filtered. Purification is described in Supporting Information File 1 for the individual compounds 3a, 4b–d, 11, and 12.
Supporting Information
Supporting Information File 1:
Experimental part, HPLC analysis of preQ1 and NMR spectra.
We thank Clemens Eichler (University of Innsbruck) for synthetic contributions.
Funding
This work was funded in part by the Austrian Science Fund FWF [P31691, F8011-B] and the Austrian Research Promotion Agency FFG [West Austrian Bio NMR 858017].
Roth, A.; Winkler, W. C.; Regulski, E. E.; Lee, B. W. K.; Lim, J.; Jona, I.; Barrick, J. E.; Ritwik, A.; Kim, J. N.; Welz, R.; Iwata-Reuyl, D.; Breaker, R. R. Nat. Struct. Mol. Biol.2007,14, 308–317. doi:10.1038/nsmb1224
Return to citation in text:
[1]
Wu, M.-C.; Lowe, P. T.; Robinson, C. J.; Vincent, H. A.; Dixon, N.; Leigh, J.; Micklefield, J. J. Am. Chem. Soc.2015,137, 9015–9021. doi:10.1021/jacs.5b03405
Return to citation in text:
[1]
Neuner, E.; Frener, M.; Lusser, A.; Micura, R. RNA Biol.2018,15, 1376–1383. doi:10.1080/15476286.2018.1534526
Return to citation in text:
[1]
Wilding, B.; Winkler, M.; Petschacher, B.; Kratzer, R.; Egger, S.; Steinkellner, G.; Lyskowski, A.; Nidetzky, B.; Gruber, K.; Klempier, N. Chem. – Eur. J.2013,19, 7007–7012. doi:10.1002/chem.201300163
Return to citation in text:
[1]
[2]
Fergus, C.; Al-qasem, M.; Cotter, M.; McDonnell, C. M.; Sorrentino, E.; Chevot, F.; Hokamp, K.; Senge, M. O.; Southern, J. M.; Connon, S. J.; Kelly, V. P. Nucleic Acids Res.2021,49, 4877–4890. doi:10.1093/nar/gkab289
Return to citation in text:
[1]
Akimoto, H.; Nomura, H.; Yoshida, M.; Shindo-Okada, N.; Hoshi, A.; Nishimura, S. J. Med. Chem.1986,29, 1749–1753. doi:10.1021/jm00159a031
Return to citation in text:
[1]
[2]
Flemmich, L.; Heel, S.; Moreno, S.; Breuker, K.; Micura, R. Nat. Commun.2021,12, 3877. doi:10.1038/s41467-021-24193-7
Return to citation in text:
[1]
[2]
Bessler, L.; Kaur, N.; Vogt, L.-M.; Flemmich, L.; Siebenaller, C.; Winz, M.-L.; Tuorto, F.; Micura, R.; Ehrenhofer-Murray, A. E.; Helm, M. Nucleic Acids Res.2022,50, 10785–10800. doi:10.1093/nar/gkac822
Return to citation in text:
[1]
Tota, E. M.; Devaraj, N. K. J. Am. Chem. Soc.2023,145, 8099–8106. doi:10.1021/jacs.3c00861
Return to citation in text:
[1]
Alexander, S. C.; Busby, K. N.; Cole, C. M.; Zhou, C. Y.; Devaraj, N. K. J. Am. Chem. Soc.2015,137, 12756–12759. doi:10.1021/jacs.5b07286
Return to citation in text:
[1]
Zhang, D.; Zhou, C. Y.; Busby, K. N.; Alexander, S. C.; Devaraj, N. K. Angew. Chem., Int. Ed.2018,57, 2822–2826. doi:10.1002/anie.201710917
Return to citation in text:
[1]
Balaratnam, S.; Rhodes, C.; Bume, D. D.; Connelly, C.; Lai, C. C.; Kelley, J. A.; Yazdani, K.; Homan, P. J.; Incarnato, D.; Numata, T.; Schneekloth, J. S., Jr. Nat. Commun.2021,12, 5856. doi:10.1038/s41467-021-25973-x
Return to citation in text:
[1]
Krochmal, D.; Shao, Y.; Li, N.-S.; DasGupta, S.; Shelke, S. A.; Koirala, D.; Piccirilli, J. A. Nat. Chem. Biol.2022,18, 376–384. doi:10.1038/s41589-021-00950-z
Return to citation in text:
[1]
Ameta, S.; Jäschke, A. Chem. Sci.2013,4, 957–964. doi:10.1039/c2sc21588h
Return to citation in text:
[1]
Wilson, C.; Szostak, J. W. Nature1995,374, 777–782. doi:10.1038/374777a0
Return to citation in text:
[1]
McDonald, R. I.; Guilinger, J. P.; Mukherji, S.; Curtis, E. A.; Lee, W. I.; Liu, D. R. Nat. Chem. Biol.2014,10, 1049–1054. doi:10.1038/nchembio.1655
Return to citation in text:
[1]
Kondo, T.; Ohgi, T.; Goto, T. Chem. Lett.1983,12, 419–422. doi:10.1246/cl.1983.419
Return to citation in text:
[1]
[2]
Akimoto, H.; Imamiya, E.; Hitaka, T.; Nomura, H.; Nishimura, S. J. Chem. Soc., Perkin Trans. 11988, 1637–1644. doi:10.1039/p19880001637
Return to citation in text:
[1]
[2]
Gerber, H.-D.; Klebe, G. Org. Biomol. Chem.2012,10, 8660–8668. doi:10.1039/c2ob26387d
Return to citation in text:
[1]
Chen, Y.-C.; Brooks, A. F.; Goodenough-Lashua, D. M.; Kittendorf, J. D.; Showalter, H. D.; Garcia, G. A. Nucleic Acids Res.2011,39, 2834–2844. doi:10.1093/nar/gkq1188
Return to citation in text:
[1]
[2]
Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta2005,88, 2610–2616. doi:10.1002/hlca.200590201
Return to citation in text:
[1]
[2]
[3]
Brooks, A. F.; Garcia, G. A.; Showalter, H. D. H. Tetrahedron Lett.2010,51, 4163–4165. doi:10.1016/j.tetlet.2010.06.008
Return to citation in text:
[1]
Flemmich, L.; Moreno, S.; Micura, R. Beilstein J. Org. Chem.2021,17, 2295–2301. doi:10.3762/bjoc.17.147
Return to citation in text:
[1]
[2]
Anthony, N. G.; Baiget, J.; Berretta, G.; Boyd, M.; Breen, D.; Edwards, J.; Gamble, C.; Gray, A. I.; Harvey, A. L.; Hatziieremia, S.; Ho, K. H.; Huggan, J. K.; Lang, S.; Llona-Minguez, S.; Luo, J. L.; McIntosh, K.; Paul, A.; Plevin, R. J.; Robertson, M. N.; Scott, R.; Suckling, C. J.; Sutcliffe, O. B.; Young, L. C.; Mackay, S. P. J. Med. Chem.2017,60, 7043–7066. doi:10.1021/acs.jmedchem.7b00484
Return to citation in text:
[1]
Migawa, M. T.; Hinkley, J. M.; Hoops, G. C.; Townsend, L. B. Synth. Commun.1996,26, 3317–3322. doi:10.1080/00397919608004641
Return to citation in text:
[1]
Dincer, S.; Cetin, K. T.; Onay-Besikci, A.; Ölgen, S. J. Enzyme Inhib. Med. Chem.2013,28, 1080–1087. doi:10.3109/14756366.2012.715288
Return to citation in text:
[1]
Zhu, G.; Liu, Z.; Xu, Y.; Mao, Z. Heterocycles2008,75, 1631. doi:10.3987/com-08-11323
Return to citation in text:
[1]
Gangjee, A.; Mavandadi, F.; Queener, S. F.; McGuire, J. J. J. Med. Chem.1995,38, 2158–2165. doi:10.1021/jm00012a016
Return to citation in text:
[1]
Lee, J. W.; Lee, J. H.; Son, H. J.; Choi, Y. K.; Yoon, G. J.; Park, M. H. Synth. Commun.1996,26, 83–88. doi:10.1080/00397919608003865
Return to citation in text:
[1]
Sutanto, F.; Konstantinidou, M.; Dömling, A. RSC Med. Chem.2020,11, 876–884. doi:10.1039/d0md00154f
Return to citation in text:
[1]
Singh, J.; Petter, R. C.; Baillie, T. A.; Whitty, A. Nat. Rev. Drug Discovery2011,10, 307–317. doi:10.1038/nrd3410
Return to citation in text:
[1]
Connelly, C. M.; Moon, M. H.; Schneekloth, J. S., Jr. Cell Chem. Biol.2016,23, 1077–1090. doi:10.1016/j.chembiol.2016.05.021
Return to citation in text:
[1]
Guan, L.; Disney, M. D. Angew. Chem., Int. Ed.2013,52, 10010–10013. doi:10.1002/anie.201301639
Return to citation in text:
[1]
Childs-Disney, J. L.; Yang, X.; Gibaut, Q. M. R.; Tong, Y.; Batey, R. T.; Disney, M. D. Nat. Rev. Drug Discovery2022,21, 736–762. doi:10.1038/s41573-022-00521-4
Return to citation in text:
[1]
Velagapudi, S. P.; Li, Y.; Disney, M. D. Bioorg. Med. Chem. Lett.2019,29, 1532–1536. doi:10.1016/j.bmcl.2019.04.001
Return to citation in text:
[1]
Fang, L.; Velema, W. A.; Lee, Y.; Xiao, L.; Mohsen, M. G.; Kietrys, A. M.; Kool, E. T. Nat. Chem.2023,15, 1374–1383. doi:10.1038/s41557-023-01309-8
Return to citation in text:
[1]
Fullenkamp, C. R.; Schneekloth, J. S., Jr. Nat. Chem.2023,15, 1329–1331. doi:10.1038/s41557-023-01330-x
Return to citation in text:
[1]
Childs-Disney, J. L.; Yang, X.; Gibaut, Q. M. R.; Tong, Y.; Batey, R. T.; Disney, M. D. Nat. Rev. Drug Discovery2022,21, 736–762. doi:10.1038/s41573-022-00521-4
Chen, Y.-C.; Brooks, A. F.; Goodenough-Lashua, D. M.; Kittendorf, J. D.; Showalter, H. D.; Garcia, G. A. Nucleic Acids Res.2011,39, 2834–2844. doi:10.1093/nar/gkq1188
Fergus, C.; Al-qasem, M.; Cotter, M.; McDonnell, C. M.; Sorrentino, E.; Chevot, F.; Hokamp, K.; Senge, M. O.; Southern, J. M.; Connon, S. J.; Kelly, V. P. Nucleic Acids Res.2021,49, 4877–4890. doi:10.1093/nar/gkab289
11.
Akimoto, H.; Nomura, H.; Yoshida, M.; Shindo-Okada, N.; Hoshi, A.; Nishimura, S. J. Med. Chem.1986,29, 1749–1753. doi:10.1021/jm00159a031
Roth, A.; Winkler, W. C.; Regulski, E. E.; Lee, B. W. K.; Lim, J.; Jona, I.; Barrick, J. E.; Ritwik, A.; Kim, J. N.; Welz, R.; Iwata-Reuyl, D.; Breaker, R. R. Nat. Struct. Mol. Biol.2007,14, 308–317. doi:10.1038/nsmb1224
7.
Wu, M.-C.; Lowe, P. T.; Robinson, C. J.; Vincent, H. A.; Dixon, N.; Leigh, J.; Micklefield, J. J. Am. Chem. Soc.2015,137, 9015–9021. doi:10.1021/jacs.5b03405
Akimoto, H.; Imamiya, E.; Hitaka, T.; Nomura, H.; Nishimura, S. J. Chem. Soc., Perkin Trans. 11988, 1637–1644. doi:10.1039/p19880001637
25.
Chen, Y.-C.; Brooks, A. F.; Goodenough-Lashua, D. M.; Kittendorf, J. D.; Showalter, H. D.; Garcia, G. A. Nucleic Acids Res.2011,39, 2834–2844. doi:10.1093/nar/gkq1188
Flemmich, L.; Moreno, S.; Micura, R. Beilstein J. Org. Chem.2021,17, 2295–2301. doi:10.3762/bjoc.17.147
29.
Anthony, N. G.; Baiget, J.; Berretta, G.; Boyd, M.; Breen, D.; Edwards, J.; Gamble, C.; Gray, A. I.; Harvey, A. L.; Hatziieremia, S.; Ho, K. H.; Huggan, J. K.; Lang, S.; Llona-Minguez, S.; Luo, J. L.; McIntosh, K.; Paul, A.; Plevin, R. J.; Robertson, M. N.; Scott, R.; Suckling, C. J.; Sutcliffe, O. B.; Young, L. C.; Mackay, S. P. J. Med. Chem.2017,60, 7043–7066. doi:10.1021/acs.jmedchem.7b00484



![[1860-5397-21-35-i1]](/bjoc/content/inline/1860-5397-21-35-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-21-35-i2]](/bjoc/content/inline/1860-5397-21-35-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-21-35-i3]](/bjoc/content/inline/1860-5397-21-35-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)