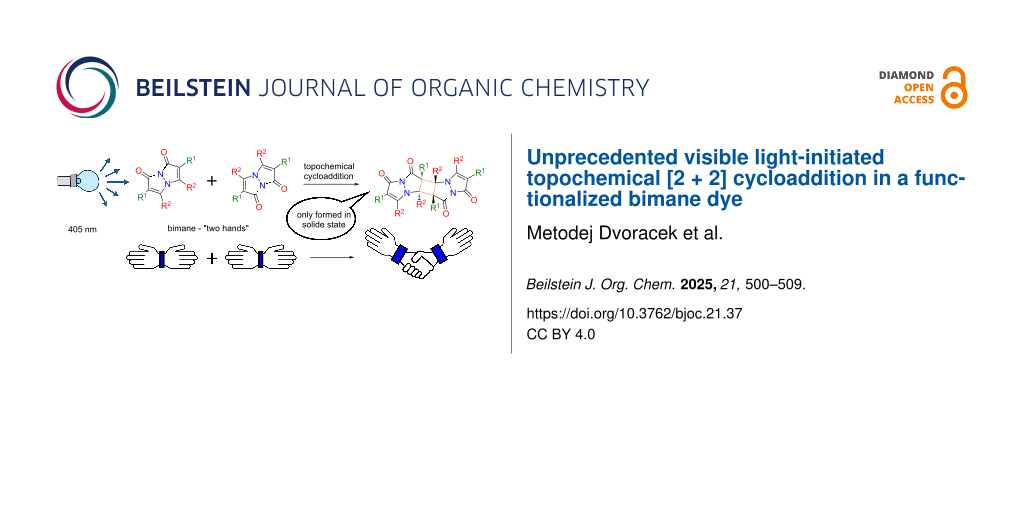
Abstract
Bimanes, a class of molecules based on the 1H,7H-pyrazolo[1,2-a]pyrazole-1,7-dione scaffold, were first introduced by E. M. Kosower in 1978. In this study, we report the topochemical cycloaddition of diethyl 2,6-dichloro-1,7-dioxo-1H,7H-pyrazolo[1,2-a]pyrazole-3,5-dicarboxylate (Cl2B), initiated by visible light. Crystal structure analysis confirmed that the reactive double bonds are parallel and coplanar, in line with the Schmidt criteria for topochemical cycloaddition. Additionally, two other bimane derivatives with different substitution patterns were synthesized and investigated. Our findings suggest that functionalizing bimanes to redshift their absorption maxima into the visible-light spectrum provides a promising strategy for synthesizing substituted cyclobutanes without the need for ultraviolet irradiation.

Graphical Abstract
Introduction
Topochemical polymerizations refer to polymerization reactions occurring in the solid state, which are highly dependent on molecular packing. The concept of topochemical reactions was first introduced by Kohlschütter in 1919 [1], and topochemical polymerizations were systematically studied by Schmidt and co-workers in the 1960s and 1970s [2-4]. Schmidt's pioneering work primarily focused on [2 + 2] photocycloaddition in the solid state, making it one of the earliest known examples of topochemical polymerization and a valuable method for synthesizing substituted cyclobutanes [5]. These solid-state reactions are highly influenced by the arrangement of monomers within the crystal lattice and are often reversible, requiring either high-energy light or heat for initiation [6-8]. Notably, topochemical polymerizations align with the principles of green chemistry, as they are solvent-free and do not involve toxic reagents [9].
The highly ordered and uniform nature of crystalline compounds significantly enhances the stereoselectivity and yield of topochemical reactions compared to those conducted in solution [10]. A key requirement for these reactions is the intermolecular distance between the two reactive double bonds. Schmidt’s rule, which governs topochemical [2 + 2] cycloaddition reactions, states that for the reaction to occur, the distance between the centers of the double bonds must be less than 4.2 Å, and the double bonds must be coplanar and parallel [11].
Designing topochemically active compounds is a challenging task, as the crystal packing mode must first be optimized, which can only be predicted computationally – a process that becomes increasingly complex with more intricate structures [12]. Since these reactions depend on both crystal packing and specific intermolecular distances, another key challenge lies in designing novel structures capable of undergoing topochemical reactions. Currently, topochemically active monomers are limited to large, planar aromatic molecules, aryl-substituted olefins, and polyenes, often complexed with metals. These compounds are frequently employed in the construction of metal-organic frameworks [13-15]. The limited range of compounds used thus far is likely due to reliance on their ability to undergo π-stacking interactions, which facilitate face-to-face packing. Therefore, the discovery of a new type of structure capable of undergoing this reaction, as reported here, represents a significant advancement, enabling the development of a novel scaffold for topochemically active monomers.
Bimanes
Bimanes, a class of bicyclic compounds first synthesized by Kosower and Pazhenchevsky in 1978 [16], are based on a 1H,7H-pyrazolo[1,2-a]pyrazole-1,7-dione scaffold. The general structure of a bimane is depicted in Figure 1a.
![[1860-5397-21-37-1]](/bjoc/content/figures/1860-5397-21-37-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of a) the unfunctionalized bimane scaffold and b) the two isomers of bimanes with their respective naming conventions.
Figure 1: Structures of a) the unfunctionalized bimane scaffold and b) the two isomers of bimanes with their ...
The term "bimanes" is derived from the Latin ‘bi’ (two) and ‘manus’ (hand). syn-Bimanes are often characterized by high fluorescence quantum yields, ranging from 60% to 100% [17], while anti-isomers typically exhibit phosphorescence [18]. Figure 1b illustrates the two isomers along with their simplified naming conventions. Although these dyes have been known since 1978, their photochemistry has only been briefly explored. In this work, during the synthesis of a series of these dyes (Figure 2a), we observed an unprecedented intermolecular [2 + 2] cycloaddition of syn-(ethoxycarbonyl,chloro)bimane (Cl2B), seen in Figure 2b.
![[1860-5397-21-37-2]](/bjoc/content/figures/1860-5397-21-37-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: a) Structures of the bimanes studied and b) the reaction scheme of the [2 + 2] photocycloaddition of Cl2B.
Figure 2: a) Structures of the bimanes studied and b) the reaction scheme of the [2 + 2] photocycloaddition o...
We hypothesized that this reaction occurred in the solid state, and further investigations confirmed this. To validate our findings, we employed single-crystal X-ray diffraction, which revealed the packing mode in the crystal and identified any photoproducts formed. Two additional compounds, syn-(ethoxycarbonyl,methyl)bimane (Me2B) and syn-(methyl,methyl)bimane (Me4B), were also examined for this phenomenon but did not undergo the reaction.
Results and Discussion
Synthesis
Bimanes are typically synthesized through a three-step process, as outlined in Figure 3. First, a β-keto ester 1 reacts with hydrazine hydrate and is heated to reflux, forming a pyrazolinone 2. This product is then chlorinated at the α-carbon, introducing a leaving group, to yield 3. The parent pyrazolinone 3 is subsequently treated with a base (either K2CO3 or N,N-diisopropylethylamine) to form the bimane 4.
![[1860-5397-21-37-3]](/bjoc/content/figures/1860-5397-21-37-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
For the chlorination reactions on the synthetic path to Cl2B and Me2B, chlorine gas was generated by reacting MnO₂ with HCl and then passed through a stirred suspension of the pyrazolinone. In the case of Me4B, trichloroisocyanuric acid (TCICA) was used as the chlorinating agent as a safer alternative to chlorine gas, with the reaction conducted by stirring the pyrazolinone with TCICA in DCM for 18 hours.
Bimanes can be purified easily using column chromatography with silica gel as the stationary phase, with the compounds Cl2B, Me2B, and Me4B only requiring neat DCM as the eluent. After chromatographic purification, the 1H NMR of Cl2B showed 6% of an impurity, possibly the anti-isomer. Further purification of Cl2B reduced the impurity content to less than 3%.
Topochemical photocycloaddition
Upon examining the crystal structure of our first sample of Cl2B, Cl2B (A), we observed that the compound had undergone a [2 + 2] cycloaddition reaction, with approximately 20% yield of the dimer in the crystal.
This was particularly notable, given that the compound had spent minimal time in solution with limited exposure to visible light, and no exposure to ultraviolet light. To further explore this phenomenon, we included two additional bimane samples, Me2B and Me4B. These samples were crystallized from a MeOH–DCM 1:1 (v/v) mixture which was left to slowly evaporate over the course of several days at room temperature, followed by single-crystal X-ray diffraction to determine their structure (Figure 4) and crystal packing mode. Extra precautions were taken to shield the compounds from light during synthesis, purification, and storage.
![[1860-5397-21-37-4]](/bjoc/content/figures/1860-5397-21-37-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: View of the molecular structures in the crystal of the functionalized bimanes studied: a) Cl2B (B), representing 90% of the disordered asymmetric unit, b) Me2B, not disordered with dotted line representing an intramolecular hydrogen bond, and c) Me4B showing the majority occupied (55%) N–N moiety.
Figure 4: View of the molecular structures in the crystal of the functionalized bimanes studied: a) Cl2B (B),...
After crystallizing each bimane, a vial containing multiple crystals was selected for irradiation studies. Cl2B (B), which showed 5% presence of the [2 + 2] dimer after crystallography, was irradiated, resulting in Cl2B (C), which showed approximately 18% dimer formation, while no dimerization was observed in the Me2B and Me4B (C) samples (see experimental section for details). The initial presence of the [2 + 2] dimer in the Cl2B (A) sample can be attributed to improper shielding from light, and unavoidable light exposure during crystal selection, and the X-ray measurement itself. As an additional test, the compounds were prepared as NMR samples in DMSO-d6 and irradiated with 405 nm LED. Irradiation in the solution phase did not result in any dimerization; the respective ¹H NMR spectra remained unchanged compared to the neat compounds (Figures S1, S3, and S5, Supporting Information File 1), confirming that the reaction occurs selectively in the solid state. Cl2B was also irradiated in DCM with the same 405 nm LEDs, in a quartz fluorescence cuvette. A UV–vis spectrum was then taken at time intervals of 15–30 minutes for a total of 3 h. The UV–vis monitoring revealed that the absorbance decreases as irradiation continues, indicating photodegradation; however, no evidence of a [2 + 2] cycloaddition product was seen in the spectra (Supporting Information File 1, Figure S15). Following this, more 1H NMR irradiation studies were conducted, where each bimane was dissolved in dichloromethane-d2, and irradiated using the same 405 nm light source for 1.25 h. The NMR spectra showed that no [2 + 2] dimer yield was achieved (Supporting Information File 1, Figures S8–S10). Further details on this are available in the experimental section.
This suggests that the reaction is topochemical, with the bimane’s reactivity dependent on its crystal packing, since no [2 + 2] cycloaddition product was seen in the spectrum. While the reaction may also depend on the reactivities of the different bimane molecules, discussing the crystal packing may bring insights into identifying other structures which may undergo this reaction also.
It should be noted that all three Cl2B structures are, by necessity, modelled as highly disordered, and the whole molecule is disordered for Cl2B (A). Crystallographic bond length restraints and constraints were used in each model and bond lengths and angle accuracy are affected by the disorder model (see Supporting Information File 1 for further details). In the case of Cl2B (all crystals, Figure 5), the torsion angle between the two double bonds in the bimane is 0(0°), indicating that the reacting double bonds are coplanar and parallel. Furthermore, the intermolecular distances between the reacting carbons are identical. The distance between two carbon double bonds in Cl2B (B) was determined to be 3.487(4) Å (Table 1).
![[1860-5397-21-37-5]](/bjoc/content/figures/1860-5397-21-37-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: View of the molecular structure in the crystal of a) symmetry generated by inversion bimanes Cl2B (B). Each bimane is only 90% occupied and b) Cl2B (B) topochemical [2 + 2] cycloaddition product, symmetry generated also by inversion. This moiety is 5% occupied in total. Hydrogen atoms omitted for clarity. Both disordered moieties were modelled using bond length restraints and displacement parameter restraints.
Figure 5: View of the molecular structure in the crystal of a) symmetry generated by inversion bimanes Cl2B (...
The key question is why did Cl2B react and Me2B did not. One possible explanation lies in the light source used for irradiation. The emission wavelength (405 nm) is within 2 nm of the λmax of Cl2B (as a solution in DCM), allowing it to absorb the initiation light with much greater intensity. Additionally, Cl2B has a higher molar extinction coefficient (6100 M−1 cm−1) compared to Me2B (4700 M−1 cm−1) at their λmax values of 403 nm and 394 nm in DCM, respectively [19]. This difference in light absorption is further exacerbated by the fact that the irradiation wavelength is not aligned with the λmax of Me2B. Calculating from the reported value, the extinction coefficient of Me2B at 405 nm is approximately 4500 M−1 cm−1. As a result, initiating the reaction in Me2B is not as efficient, and may require a shorter wavelength of higher intensity. However, as reported by Kosower et al. [18], bimanes are prone to photoreactions under UV light irradiation, undergoing rearrangement and photodegradation, so there may be a risk of degradation if irradiation using UV light is tried. Alternatively, the lack of a [2 + 2] cycloaddition of Me2B, may be due to its photophysical properties not allowing for a reaction to occur, rather than the irradiation conditions. Inefficient intersystem crossing (or complete lack thereof) and short singlet-state lifetimes can both affect the efficiency of a photoreaction [20].
Crystal packing
Similar to Cl2B, the potentially reactive double bonds in Me2B are coplanar and parallel, with a torsion angle of 0(0°) (crystallographic unit cell can be seen in Figure 6a). However, in Me2B, the bonds are further apart, with an intermolecular separation of 3.862(2) Å. Although this is still within the range for a potential reaction, no reaction was observed. In contrast, Me4B lacks the proper alignment of the reactive double bonds and is therefore not expected to undergo this topochemical reaction (Figure 6b). Examination of the reported structure of 9,10-dioxa-syn(carboethoxy,methyl)bimane in the CCDC database (BESGAH) [19,21], shows that it crystallizes in the monoclinic (P2(1)/c space group, unlike Me2B which crystallizes in P-1 and the molecules pack in a different arrangement, where the double bonds are neither coplanar nor parallel. This product was obtained from propan-2-ol, which may have influenced the crystallization kinetics. The reactive packing mode of Cl2B is enhanced by the hydrogen bonding where the ester groups play a role by increasing the number of hydrogen bonds (C–H···O, 3.18(5)–3.62(6) Å) The intermolecular distances between the C–H groups and the chlorine atoms in Cl2B (A) are C13A–H13···Cl1A, 3.69(5), and C18A–H18C···Cl2A, 3.69(3) Å, indicating further molecular attraction [22].
A previously reported bimane, syn-(H,Cl)bimane (BIYGUL), reveals a ribbon-like packing pattern, which arises from a hydrogen-bonding network [23], which also features a coplanar and parallel arrangement of the potentially reactive double bonds. Due to these characteristics, the reaction site can be represented by a parallelogram (Table 1), allowing for a more detailed comparison of the bond alignment.
Table 1: Parallelogram representation data of the reactive double bonds in a) all Cl2B structures, Me2B, and also syn-(H,Cl)bimane.
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-37-i2.svg?max-width=637&scale=1.0)
|
X | Y | A | B | α | β | % bimane |
| Cl2B(A) | C6a | C7a | 1.346(7) | 3.556(8) | 78.5(3) | 101.4(3) | 58 |
| Cl2B(B) | C3a | C4a | 1.354(4) | 3.487(4) | 77.5(2) | 102.5(2) | 90 |
| Cl2B(C) | C6a | C7a | 1.350(8) | 3.489(14) | 77.9(4) | 102.1(4) | 63 |
| Me2B | C6 | C7 | 1.3592(18) | 3.8622(18) | 63.92(8) | 116.08(8) | 100 |
| syn-(H,Cl)bimane | C2 | C3 | 1.345 | 3.635 | 73.0 | 106.9 | 100 |
As shown in Table 1, while the crystallographic center between the bonds meets the Schmidt criteria, the reactive carbons in Cl2B are better aligned compared to the two unreactive bimanes. The packing arrangement in Me2B and Me4B is shown in Figure 6, indicating the alignment of the bimanes in the solid state.
![[1860-5397-21-37-6]](/bjoc/content/figures/1860-5397-21-37-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: View of the packing of the unit cells of a) Me2B viewed normal to the c-axis and b) Me4B viewed normal to (110). Only the majority occupied moiety of the N–N bridge is shown in Me4B.
Figure 6: View of the packing of the unit cells of a) Me2B viewed normal to the c-axis and b) Me4B viewed nor...
This reactive packing arrangement is also observed in an anti-bimane previously synthesized by Blenderman et al. In anti-(Me,Br)bimane (FOSREK), coplanar and parallel double bonds are present; however, the distance between these bonds is outside the acceptable range (4.2 Å) [24]. This finding, along with the re-examination of several previously synthesized bimane compounds, suggests that substituting the bimane chromophore with groups that sufficiently redshift the absorption maximum could result in a wide range of compounds capable of undergoing topochemical [2 + 2] cycloaddition reactions initiated by visible light.
A number of related bimane structures have been reported. These include anti-(Ph,Cl)bimane (CPDZBO10) where the Ph residues impart a bowl-shaped bimane distortion and syn-(Ph,Cl)bimane [25], anti-(H,Me)bimane (DMOCDO10) and syn-(Me,Me)bimane (DXABIM10) [26], a room temperature structure of Me4B (TNZBCO10) [27], and a planar syn-(H,ethynyl)bimane (WAYHEJ) [28].
Optical properties
The optical properties of the three bimanes were measured to investigate differences in their excited state properties, as shown in Table 2. These properties help to explain the variation in photoreactivity. Quantum yields were measured using Me2B as an internal reference, with a value of 33% in acetonitrile, as initially determined by Kosower [19]. While tentative, the low fluorescence quantum yields of these compounds may suggest potential triplet-state formation, which is involved in the proposed mechanism for the cycloaddition reaction. Although Me4B could theoretically form triplet states, given its fluorescence quantum yield of 51.7–67.3%, its crystallographic structure is not conducive to this reaction, regardless of its photophysical properties.
Table 2: Steady-state spectroscopic data from Me4B, Me2B, and Cl2B recorded in acetonitrile (ACN), dichloromethane (DCM), and toluene (TOL). Fluorescence quantum yields (Φfl) calculated with respect to Me2B in acetonitrile (Φfl = 33%) [19]. Shoulders (sh) on emission spectrum are indicated in parentheses.
| solvent | λabs (nm) | λem (nm) | Φfl | |
![[Graphic 2]](/bjoc/content/inline/1860-5397-21-37-i3.svg?max-width=637&scale=1.0)
Me4B |
TOL | 365 | 417 | 53.4% |
| DCM | 369 | 428 | 67.3% | |
| ACN | 368 | 433 | 51.7% | |
![[Graphic 3]](/bjoc/content/inline/1860-5397-21-37-i4.svg?max-width=637&scale=1.0)
Me2B |
TOL | 398 | 462 (492 sh) | 49.5% |
| DCM | 394 | 462 (502 sh) | 63.4% | |
| ACN | 391 | 463 (495 sh) | 33.0% | |
![[Graphic 4]](/bjoc/content/inline/1860-5397-21-37-i5.svg?max-width=637&scale=1.0)
Cl2B |
TOL | 414 | 474 (502 sh) | 40.4% |
| DCM | 404 | 473 (505 sh) | 47.6% | |
| ACN | 401 | 476 (505 sh) | 20.2% | |
While a pure sample of the dimer has not yet been isolated, a UV–vis spectrum of Cl2B after 1.75 hours of irradiation in DCM was recorded. The peak corresponding to the remaining monomer was observed, along with a new peak blueshifted by 47 nm compared to the parent bimane (Figure 7). The hypsochromic shift in this reaction means that provided correct irradiation wavelength is used, a 100% yield of the dimer could theoretically be achieved.
![[1860-5397-21-37-7]](/bjoc/content/figures/1860-5397-21-37-7.png?scale=2.3&max-width=1024&background=FFFFFF)
Figure 7: UV–vis spectrum of Cl2B after irradiation in DCM.
Figure 7: UV–vis spectrum of Cl2B after irradiation in DCM.
Tentative mechanism
The observed reaction likely follows a five-step mechanism, as suggested by previous studies [29,30]. The proposed pathway begins with the excitation of a bimane molecule to its singlet-excited state, followed by intersystem crossing (ISC) to the triplet state. The triplet state then forms an excimer complex with a neighboring bimane molecule, leading to the formation of a single bond and generating a triplet diradical. This triplet diradical undergoes another ISC, returning to the singlet state, which then forms a second single bond (Figure 8). This mechanism, involving both singlet-excited states and triplet states via ISC, is consistent with the relatively low fluorescence quantum yields observed for Cl2B.
![[1860-5397-21-37-8]](/bjoc/content/figures/1860-5397-21-37-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Proposed mechanism for the topochemical [2 + 2] photocycloaddition of Cl2B.
Figure 8: Proposed mechanism for the topochemical [2 + 2] photocycloaddition of Cl2B.
Conclusion
Following the observation of the topochemical cycloaddition of Cl2B, two other bimane dyes, Me2B and Me4B, were synthesized and investigated for their ability to undergo this reaction when initiated by visible light. A notable feature of this reaction is that it occurs only in the solid state, not in solution. Although Me2B had the correct alignment of double bonds, meeting the Schmidt criteria, the reaction did not proceed under 405 nm excitation. This may be due to steric effects, a lack of molecular attraction, or an insufficient molar extinction coefficient at that wavelength. Me4B did not have the correct packing mode for a topochemical [2 + 2] cycloaddition to occur, and as such no reaction was observed. Synthesizing bimanes with functional groups that redshift the absorption maximum into the visible-light spectrum could yield a variety of structures capable of undergoing this reaction.
Experimental
Synthesis
The studied bimanes were synthesized following the approach reported by Kosower and co-workers [17,19], and an alternative chlorination method developed by Neogi et al. [31]. The synthetic details can be found in Supporting Information File 1. Compounds were crystallized from a MeOH–DCM 1:1 (v/v) solution which was left to evaporate at room temperature over the course of a few days.
Photophysical properties
Fluorescence spectra were measured on a Horiba FluoroMax Plus spectrofluorimeter. UV–vis spectra were measured on a Shimadzu UV-1900i UV–vis spectrophotometer. Absorption and fluorescence spectra are presented in Supporting Information File 1 (Figures S12–S14).
For the fluorescence quantum yield measurements, solutions of each bimane were prepared, and the concentration was adjusted so that the absorbance at the right shoulder of the band at 360 nm falls within the range of 0.03–0.08. A fluorescence spectrum was then recorded, exciting at 370 nm, using 1 nm excitation and emission slits. The emission was recorded in the range of 370–850 nm. This was done for each solvent: DCM, acetonitrile, and toluene. The quantum yields were calculated by using Me2B in acetonitrile as the reference (Φfl = 33%) [19].
Quantum yields were calculated using the following formula (Equation 1):
![[1860-5397-21-37-i1]](/bjoc/content/inline/1860-5397-21-37-i1.svg?max-width=590&scale=1.18182)
where Φs is the fluorescence quantum yield of the analyte and Φref is the fluorescence quantum yield of the reference, Abs is the absorbance at the excitation wavelength, Area is the area under the fluorescence spectrum and η is the refractive index of the solvent.
Single crystal X-ray diffraction studies
Crystals of Me4B, Cl2B (A), (B), (C), and Me2B were mounted on a MiTeGen micromount with NVH immersion oil. Data were collected from a shock-cooled single crystal at 100(2) K on a Bruker Apex Kappa Duo Imus Cu Kα Kappa diffractometer with a microfocus sealed X-ray tube using mirror optics as a monochromator and an APEX2 detector. The diffractometer was equipped with an Oxford Cobra low temperature device and used Cu Kα radiation (λ = 1.54178 Å). All data were integrated with SAINT and a multi-scan absorption correction using SADABS was applied [32,33]. Structures were solved by dual methods using SHELXT and refined by full-matrix least-squares methods against F2 by SHELXL using Olex2 [34,35]. All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were refined isotropic on calculated positions using a riding model with their Uiso values constrained to 1.5 times the Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. Disordered moieties were refined using bond length restraints and displacement parameter restraints [21]. Full crystal and refinement details are given in Supporting Information File 1, section 4).
Irradiation tests
The bimanes were crystallized from a MeOH–DCM 1:1 (v/v) solution and were allowed to evaporate at room temperature over the course of several days. In a typical experiment, the crystals were then irradiated for variable periods of time using 405 nm LED (75 mW cm−2) in an Elegoo Mercury Plus Wash and Cure (turntable chamber). Samples were placed approximately 8 cm far from the light source. The average light power measured at this distance was 5 mW cm−2. During the irradiation, the crystals changed their appearance from light yellow to dark orange within 15 minutes.
An alternative method for solid-state irradiation involved dissolving 5–10 mg of the bimane in a minimal amount of acetone, which was then evaporated in a 25 mL round-bottomed flask, forming a seemingly amorphous solid spread across the flask. The entire flask was then irradiated using 405 nm LEDs in the turntable chamber.
Solution-phase irradiation tests
For solution-phase irradiations, 10 mg of each compound was dissolved in DMSO-d6 and placed into NMR tubes. The samples were irradiated directly inside the tubes, using the same light source as for the solid-state samples.
Another series of tests involved dissolving 5–15 mg of each bimane in dichloromethane-d2 in an NMR tube. The tubes were irradiated for 1.25 h using the same light source as for the solid-state samples.
Cl2B irradiation was also monitored using UV–vis spectroscopy, where the concentration was adjusted such that the absorbance is 2.164 at the λmax. The solution was added into a quartz fluorescence cuvette, and irradiated for 15 minutes at a time using the same 405 nm LEDs in a turntable chamber.
Supporting Information
| Supporting Information File 1: Spectroscopic and crystallographic information as well as synthetic details. | ||
| Format: PDF | Size: 3.0 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Kohlschütter, V.; Haenni, P. Z. Anorg. Allg. Chem. 1919, 105, 121–144. doi:10.1002/zaac.19191050109
Return to citation in text: [1] -
Cohen, M. D.; Elgavi, A.; Green, B. S.; Ludmer, Z.; Schmidt, G. M. J. J. Am. Chem. Soc. 1972, 94, 6776–6779. doi:10.1021/ja00774a034
Return to citation in text: [1] -
Green, B. S.; Schmidt, G. M. J. Tetrahedron Lett. 1970, 11, 4249–4252. doi:10.1016/s0040-4039(00)89456-3
Return to citation in text: [1] -
Green, B. S.; Lahav, M.; Schmidt, G. M. J. J. Chem. Soc. B 1971, 1552. doi:10.1039/j29710001552
Return to citation in text: [1] -
Hema, K.; Ravi, A.; Raju, C.; Pathan, J. R.; Rai, R.; Sureshan, K. M. Chem. Soc. Rev. 2021, 50, 4062–4099. doi:10.1039/d0cs00840k
Return to citation in text: [1] -
Medishetty, R.; Bai, Z.; Yang, H.; Wong, M. W.; Vittal, J. J. Cryst. Growth Des. 2015, 15, 4055–4061. doi:10.1021/acs.cgd.5b00664
Return to citation in text: [1] -
Johnston, P.; Braybrook, C.; Saito, K. Chem. Sci. 2012, 3, 2301–2306. doi:10.1039/c2sc20380d
Return to citation in text: [1] -
Marschner, D. E.; Frisch, H.; Offenloch, J. T.; Tuten, B. T.; Becer, C. R.; Walther, A.; Goldmann, A. S.; Tzvetkova, P.; Barner-Kowollik, C. Macromolecules 2018, 51, 3802–3807. doi:10.1021/acs.macromol.8b00613
Return to citation in text: [1] -
Basics of Green Chemistry. US Environmental Protection Agency: Washington, D.C., USA, 2024; https://www.epa.gov/greenchemistry/basics-green-chemistry (accessed Oct 26, 2024).
Return to citation in text: [1] -
Khan, S.; Dutta, B.; Mir, M. H. Dalton Trans. 2020, 49, 9556–9563. doi:10.1039/d0dt01534b
Return to citation in text: [1] -
Schmidt, G. M. J. Pure Appl. Chem. 1971, 27, 647–678. doi:10.1351/pac197127040647
Return to citation in text: [1] -
Beran, G. J. O. Chem. Sci. 2023, 14, 13290–13312. doi:10.1039/d3sc03903j
Return to citation in text: [1] -
Yu, J.-G.; Gan, M.-M.; Bai, S.; Han, Y.-F. CrystEngComm 2019, 21, 4673–4683. doi:10.1039/c9ce00971j
Return to citation in text: [1] -
Yadava, K.; Vittal, J. J. Chem. – Eur. J. 2019, 25, 10394–10399. doi:10.1002/chem.201901237
Return to citation in text: [1] -
Medishetty, R.; Park, I.-H.; Lee, S. S.; Vittal, J. J. Chem. Commun. 2016, 52, 3989–4001. doi:10.1039/c5cc08374e
Return to citation in text: [1] -
Kosower, E. M.; Pazhenchevsky, B.; Hershkowitz, E. J. Am. Chem. Soc. 1978, 100, 6516–6518. doi:10.1021/ja00488a050
Return to citation in text: [1] -
Kosower, E. M.; Pazhenchevsky, B. J. Am. Chem. Soc. 1980, 102, 4983–4993. doi:10.1021/ja00535a028
Return to citation in text: [1] [2] -
Kosower, E. M.; Kanety, H.; Dodiuk, H. J. Photochem. 1983, 21, 171–182. doi:10.1016/0047-2670(83)80020-3
Return to citation in text: [1] [2] -
Kosower, E. M.; Faust, D.; Ben-Shoshan, M.; Goldberg, I. J. Org. Chem. 1982, 47, 214–221. doi:10.1021/jo00341a007
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Poplata, S.; Tröster, A.; Zou, Y.-Q.; Bach, T. Chem. Rev. 2016, 116, 9748–9815. doi:10.1021/acs.chemrev.5b00723
Return to citation in text: [1] -
Groom, C. R.; Bruno, I. J.; Lightfoot, M. P.; Ward, S. C. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, 72, 171–179. doi:10.1107/s2052520616003954
Return to citation in text: [1] [2] -
Sarma, J. A. R. P.; Desiraju, G. R. Chem. Phys. Lett. 1985, 117, 160–164. doi:10.1016/0009-2614(85)85227-1
Return to citation in text: [1] -
Goldberg, I.; Kosower, E. M. J. Phys. Chem. 1982, 86, 332–335. doi:10.1021/j100392a011
Return to citation in text: [1] -
Blenderman, W. G.; Carroll, P. J.; Joullie', M. M.; Ulatowski, T. G.; Nemeroff, N. H. J. Prakt. Chem. 1986, 328, 648–650. doi:10.1002/prac.19863280428
Return to citation in text: [1] -
Bernstein, J.; Goldstein, E.; Goldberg, I. Cryst. Struct. Commun. 1980, 9, 295–299.
Return to citation in text: [1] -
Goldberg, I.; Bernstein, J.; Kosower, E. M. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1982, 38, 1990–2000. doi:10.1107/s0567740882007730
Return to citation in text: [1] -
Bernstein, J.; Goldstein, E.; Goldberg, I. Cryst. Struct. Commun. 1980, 9, 301–305.
Return to citation in text: [1] -
Kosower, E. M.; Ben-Shoshan, M.; Goldberg, I. J. Chem. Soc., Chem. Commun. 1993, 1644–1645. doi:10.1039/c39930001644
Return to citation in text: [1] -
Loutfy, R. O.; de Mayo, P. Can. J. Chem. 1972, 50, 3465–3471. doi:10.1139/v72-560
Return to citation in text: [1] -
Wei, H.; Zhu, H.; Li, Q.; Zheng, X. J. Mater. Chem. C 2024, 12, 2613–2622. doi:10.1039/d3tc03792d
Return to citation in text: [1] -
Neogi, I.; Grynszpan, F.; Das, P. J. Synlett 2018, 29, 1043–1046. doi:10.1055/s-0036-1591964
Return to citation in text: [1] -
SAINT; Bruker AXS Inc.: Madison, WI, USA.
Return to citation in text: [1] -
Krause, L.; Herbst-Irmer, R.; Sheldrick, G. M.; Stalke, D. J. Appl. Crystallogr. 2015, 48, 3–10. doi:10.1107/s1600576714022985
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1]
| 31. | Neogi, I.; Grynszpan, F.; Das, P. J. Synlett 2018, 29, 1043–1046. doi:10.1055/s-0036-1591964 |
| 19. | Kosower, E. M.; Faust, D.; Ben-Shoshan, M.; Goldberg, I. J. Org. Chem. 1982, 47, 214–221. doi:10.1021/jo00341a007 |
| 32. | SAINT; Bruker AXS Inc.: Madison, WI, USA. |
| 33. | Krause, L.; Herbst-Irmer, R.; Sheldrick, G. M.; Stalke, D. J. Appl. Crystallogr. 2015, 48, 3–10. doi:10.1107/s1600576714022985 |
| 1. | Kohlschütter, V.; Haenni, P. Z. Anorg. Allg. Chem. 1919, 105, 121–144. doi:10.1002/zaac.19191050109 |
| 9. | Basics of Green Chemistry. US Environmental Protection Agency: Washington, D.C., USA, 2024; https://www.epa.gov/greenchemistry/basics-green-chemistry (accessed Oct 26, 2024). |
| 20. | Poplata, S.; Tröster, A.; Zou, Y.-Q.; Bach, T. Chem. Rev. 2016, 116, 9748–9815. doi:10.1021/acs.chemrev.5b00723 |
| 6. | Medishetty, R.; Bai, Z.; Yang, H.; Wong, M. W.; Vittal, J. J. Cryst. Growth Des. 2015, 15, 4055–4061. doi:10.1021/acs.cgd.5b00664 |
| 7. | Johnston, P.; Braybrook, C.; Saito, K. Chem. Sci. 2012, 3, 2301–2306. doi:10.1039/c2sc20380d |
| 8. | Marschner, D. E.; Frisch, H.; Offenloch, J. T.; Tuten, B. T.; Becer, C. R.; Walther, A.; Goldmann, A. S.; Tzvetkova, P.; Barner-Kowollik, C. Macromolecules 2018, 51, 3802–3807. doi:10.1021/acs.macromol.8b00613 |
| 19. | Kosower, E. M.; Faust, D.; Ben-Shoshan, M.; Goldberg, I. J. Org. Chem. 1982, 47, 214–221. doi:10.1021/jo00341a007 |
| 21. | Groom, C. R.; Bruno, I. J.; Lightfoot, M. P.; Ward, S. C. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, 72, 171–179. doi:10.1107/s2052520616003954 |
| 5. | Hema, K.; Ravi, A.; Raju, C.; Pathan, J. R.; Rai, R.; Sureshan, K. M. Chem. Soc. Rev. 2021, 50, 4062–4099. doi:10.1039/d0cs00840k |
| 19. | Kosower, E. M.; Faust, D.; Ben-Shoshan, M.; Goldberg, I. J. Org. Chem. 1982, 47, 214–221. doi:10.1021/jo00341a007 |
| 2. | Cohen, M. D.; Elgavi, A.; Green, B. S.; Ludmer, Z.; Schmidt, G. M. J. J. Am. Chem. Soc. 1972, 94, 6776–6779. doi:10.1021/ja00774a034 |
| 3. | Green, B. S.; Schmidt, G. M. J. Tetrahedron Lett. 1970, 11, 4249–4252. doi:10.1016/s0040-4039(00)89456-3 |
| 4. | Green, B. S.; Lahav, M.; Schmidt, G. M. J. J. Chem. Soc. B 1971, 1552. doi:10.1039/j29710001552 |
| 18. | Kosower, E. M.; Kanety, H.; Dodiuk, H. J. Photochem. 1983, 21, 171–182. doi:10.1016/0047-2670(83)80020-3 |
| 13. | Yu, J.-G.; Gan, M.-M.; Bai, S.; Han, Y.-F. CrystEngComm 2019, 21, 4673–4683. doi:10.1039/c9ce00971j |
| 14. | Yadava, K.; Vittal, J. J. Chem. – Eur. J. 2019, 25, 10394–10399. doi:10.1002/chem.201901237 |
| 15. | Medishetty, R.; Park, I.-H.; Lee, S. S.; Vittal, J. J. Chem. Commun. 2016, 52, 3989–4001. doi:10.1039/c5cc08374e |
| 17. | Kosower, E. M.; Pazhenchevsky, B. J. Am. Chem. Soc. 1980, 102, 4983–4993. doi:10.1021/ja00535a028 |
| 18. | Kosower, E. M.; Kanety, H.; Dodiuk, H. J. Photochem. 1983, 21, 171–182. doi:10.1016/0047-2670(83)80020-3 |
| 11. | Schmidt, G. M. J. Pure Appl. Chem. 1971, 27, 647–678. doi:10.1351/pac197127040647 |
| 34. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
| 35. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 10. | Khan, S.; Dutta, B.; Mir, M. H. Dalton Trans. 2020, 49, 9556–9563. doi:10.1039/d0dt01534b |
| 16. | Kosower, E. M.; Pazhenchevsky, B.; Hershkowitz, E. J. Am. Chem. Soc. 1978, 100, 6516–6518. doi:10.1021/ja00488a050 |
| 21. | Groom, C. R.; Bruno, I. J.; Lightfoot, M. P.; Ward, S. C. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, 72, 171–179. doi:10.1107/s2052520616003954 |
| 24. | Blenderman, W. G.; Carroll, P. J.; Joullie', M. M.; Ulatowski, T. G.; Nemeroff, N. H. J. Prakt. Chem. 1986, 328, 648–650. doi:10.1002/prac.19863280428 |
| 22. | Sarma, J. A. R. P.; Desiraju, G. R. Chem. Phys. Lett. 1985, 117, 160–164. doi:10.1016/0009-2614(85)85227-1 |
| 23. | Goldberg, I.; Kosower, E. M. J. Phys. Chem. 1982, 86, 332–335. doi:10.1021/j100392a011 |
| 29. | Loutfy, R. O.; de Mayo, P. Can. J. Chem. 1972, 50, 3465–3471. doi:10.1139/v72-560 |
| 30. | Wei, H.; Zhu, H.; Li, Q.; Zheng, X. J. Mater. Chem. C 2024, 12, 2613–2622. doi:10.1039/d3tc03792d |
| 17. | Kosower, E. M.; Pazhenchevsky, B. J. Am. Chem. Soc. 1980, 102, 4983–4993. doi:10.1021/ja00535a028 |
| 19. | Kosower, E. M.; Faust, D.; Ben-Shoshan, M.; Goldberg, I. J. Org. Chem. 1982, 47, 214–221. doi:10.1021/jo00341a007 |
| 19. | Kosower, E. M.; Faust, D.; Ben-Shoshan, M.; Goldberg, I. J. Org. Chem. 1982, 47, 214–221. doi:10.1021/jo00341a007 |
| 19. | Kosower, E. M.; Faust, D.; Ben-Shoshan, M.; Goldberg, I. J. Org. Chem. 1982, 47, 214–221. doi:10.1021/jo00341a007 |
| 27. | Bernstein, J.; Goldstein, E.; Goldberg, I. Cryst. Struct. Commun. 1980, 9, 301–305. |
| 28. | Kosower, E. M.; Ben-Shoshan, M.; Goldberg, I. J. Chem. Soc., Chem. Commun. 1993, 1644–1645. doi:10.1039/c39930001644 |
| 25. | Bernstein, J.; Goldstein, E.; Goldberg, I. Cryst. Struct. Commun. 1980, 9, 295–299. |
| 26. | Goldberg, I.; Bernstein, J.; Kosower, E. M. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1982, 38, 1990–2000. doi:10.1107/s0567740882007730 |
© 2025 Dvoracek et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.