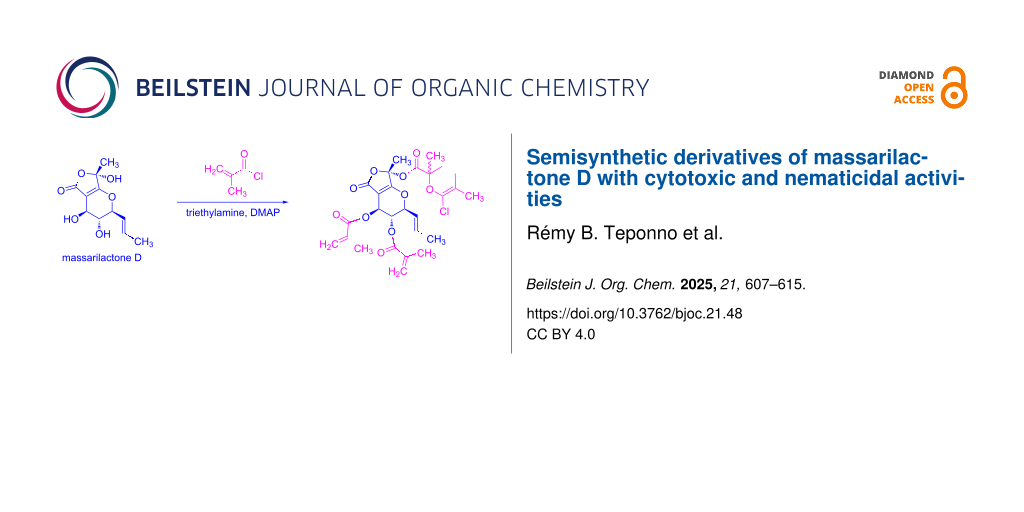
Abstract
Massarilactones constitute a rare class of polyketides produced mainly by endophytic fungi. Given that semisynthetic derivatives often exhibit biological activities greater than those of the substrates, seven previously unreported derivatives of massarilactone D, compounds 2–8, were synthetized by acylation with methacryloyl chloride, cinnamoyl chloride, 4-bromobenzoyl chloride, trans-2-methyl-2-butenoyl chloride, and crotonyl chloride. These compounds were evaluated for their cytotoxic activity against the murine fibroblasts L929, human cervix carcinoma KB-3-1, human lung carcinoma A549, human prostate cancer PC-3, epidermoid carcinoma A431, ovarian carcinoma SKOV-3, and breast cancer MCF-7 cell lines. Compounds 2 and 3 exhibited significant cytotoxicity against all the tested cells. Some of the semisynthetic derivatives were also tested for their nematicidal activity and compound 4 displayed significant and selective nematicidal activity with LD90 and LD50 of 100 and 12.5 µg/mL, respectively. Since the parent compound was not active, the present study supports the fact that the acylation reaction can improve bioactivities of some natural products.

Graphical Abstract
Introduction
Cancer continues to be responsible for morbidity and mortality all over the world. Endophytic fungi have been shown to be an important source of secondary metabolites endowed with interesting cytotoxic activities. However, resistance to cancer therapies is a persistent challenge in clinical practice. This resistance often leads to treatment failure and poor survival outcomes for patients [1]. Another ongoing problem is the excessive use of chemical pesticides such as methyl bromide, carbamates, and organophosphates to control plant-parasitic nematodes that has shown a negative impact on the environment and human health. Prolonged and widespread applications of these substances have also increased the development of nematode resistance to pesticides [2-4]. However, advancements in natural products chemistry have shown that chemical modifications of certain natural products can serve as effective scaffold for the design and synthesis of derivatives with improved biological activities [5-7].
Massarilactones are produced by marine and endophytic fungi and bear close biogenetic similarity to several other fungal PKS1-derived metabolites including rosigenin, the curvupallides, and the spirostaphylotrichins [8-10]. Massarilactones A and B were isolated for the first time from the freshwater aquatic fungus Massarina tunicata [8], massarilactones C and D from Coniothyrium sp. associated to the succulent plant Carpobrotus edulis [11], massarilactones E, F, and G from Coniothyrium sp. associated with the plant Artimisia maritima [12] while massarilactone H was first obtained from the marine-derived fungus Phoma herbarum [13]. Massarilactones D and H are also produced by the phytopathogenic fungus Kalmusia variispora associated with grapevine trunk diseases (GTDs) in Iran [14]. Massarilactones A and B were earlier shown to exhibit antimicrobial activity against Bacillus subtilis (ATCC 6051) and Staphylococcus aureus (ATCC 29213), affording zones of inhibition varying from 12 to 19 mm at 200 μg/disk [8], while massarilactone H displayed moderate cytotoxicity against three human cancer cell lines, namely A549, Hs683, and SKMEL-28 with IC50 of 32.9, 31.5, and 35.2 μM, respectively [15]. Recently, we discovered that massarilactone D was the main secondary metabolite produced during shake flasks fermentation in YMG medium (1.0% malt extract, 0.4% glucose, 0.4% yeast extract, pH 6.3) by the endophytic fungus Dendrothyrium variisporum. This fungus, isolated from the roots of the Algerian plant Globularia alypum, was explored for the first time for its potential to produce secondary metabolites [16]. Despite the abundant production of massarilactone D, it did not exhibit significant antimicrobial or cytotoxic activities upon testing [16]. This led us to hypothesize that possible chemical modification might enhance its biological activity. Therefore, the present research work aimed to investigate whether structural modifications could improve the biological activity of this polyketide. Seven analogues of massarilactone D, compounds 2–8, were synthetized through acylation modifications aimed at enhancing the compound’s interactions with biological targets (Scheme 1). These acylated analogues were subsequently screened for their cytotoxicity against a panel of human cancer cells and evaluated for their nematicidal activity.
![[1860-5397-21-48-i1]](/bjoc/content/inline/1860-5397-21-48-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of massarilactone D derivatives 2–8. Reagents and reactions conditions: a) (CH3CH2)3N/DMAP and methacryloyl chloride in CH2Cl2; b) (CH3CH2)3N/DMAP and cinnamoyl chloride in CH2Cl2; c) (CH3CH2)3N/DMAP and 4-bromobenzoyl chloride in CH2Cl2; d) (CH3CH2)3N/DMAP and trans-2-methyl-2-butenoyl chloride in CH2Cl2; e) (CH3CH2)3N/DMAP and crotonyl chloride in CH2Cl2.
Scheme 1: Preparation of massarilactone D derivatives 2–8. Reagents and reactions conditions: a) (CH3CH2)3N/D...
Results and Discussion
Semisynthesis of massarilactone D derivatives
The reaction of massarilactone D with methacryloyl chloride in triethylamine in the presence of catalytic amounts of 4-dimethylaminopyridine afforded compound 2 (11% yield) obtained as a white powder. Its molecular formula C27H31ClO10 was deduced from the HRESIMS spectrum (Figure S2, Supporting Information File 1), which exhibited the sodium adduct at m/z 573.1495 (calcd. for C27H31ClO10Na+: 573.1498) and NMR analysis. In addition to resonances attributed to massarilactone D, signals of two methacryloyl moieties were depicted in its 1H NMR spectrum at δH 1.92 (s, Me-2''), 1.89 (s, Me-2'''), 6.24 (dq, J = 2.1, 1.0 Hz, Ha-3''), 6.18 (dq, J = 2.2, 1.0 Hz, Ha-3'''), 5.80 (p, J = 1.6 Hz, Hb-3'''), and 5.73 (p, J = 1.6 Hz, Hb-3'') [17]. In the 13C NMR spectrum, the presence of these groups was evidenced by resonances observed at δC 166.4 (C-1''), 164.8 (C-1'''), 136.4 (C-2'', C-2'''), 128.4 (C-3'''), 128.0 (C-3''), 18.3 (Me-2''), and 18.1 (Me-2'''). The HMBC correlations from H-3 (δH 5.23, t, J = 3.4 Hz) and H-4 (δH 5.75, dd, J = 3.3, 1.3 Hz) to carbons at δC 164.8 (C-1''') and 166.4 (C-1''), respectively, revealed that the two methacryloyl moieties were linked at C-2 and C-3. The other salient feature of the 13C NMR spectrum was the presence of some signals at δC 171.7 (C-1'), 82.3 (C-2'), 31.4 (C-3'), 26.0 (C-4'), 105.1 (C-5'), 137.0 (C-6'), 25.1 (Me-2'), and 18.2 (Me-5') characteristic of a 6-chloro-3,4-dihydro-2,5-dimethyl-2H-pyran-2-carbonyl moiety [18]. The chemical shift of C-1' (δC 171.7) in compound 2 compared to 6-chloro-3,4-dihydro-2,5-dimethyl-2H-pyran-2-carbonyl chloride (δC 176.8) indicated that the 6-chloro-3,4-dihydro-2,5-dimethyl-2H-pyran-2-carbonyl group was linked through an ester bond and the only hydroxy group available for this esterification was the one at C-7. This compound was finally elucidated as massarilactone D 3,4-di-O-methacryloyl-7-O-(6-chloro-3,4-dihydro-2,5-dimethyl-2H-pyran-2-carbonyl).
For the formation of compound 2, an oxa-Diels–Alder reaction between two methacryloyl chloride molecules could have taken place to yield 6-chloro-3,4-dihydro-2,5-dimethyl-2H-pyran-2-carbonyl chloride as previously described [18] before esterification of the hydroxy group at C-7.
Compounds 3 (6% yield) and 4 (90% yield) were obtained each as white powder after HPLC purification of the mixture formed from reaction of massarilactone D with cinnamoyl chloride in triethylamine in the presence of catalytic amounts of 4-dimethylaminopyridine. The ESIMS of compound 3 exhibited the sodium adducts at m/z 507.20 [M + Na]+ and 991.39 [2M + Na]+ corresponding to the molecular formula C29H24O7. The presence of two cinnamoyl moieties was supported by resonances shown at δH 7.77 (d, J = 16.0 Hz, H-3'''), 7.76 (d, 16.0, H-3''), 7.72–7.70 (o, H-5'', H-9'', H-5''', H-9'''), 7.46–7.44 (o, H-6'', H-7'', H-8'', H-6''', H-7''', H-8'''), 6.60 (d, J = 16.0 Hz, H-2'''), and 6.58 (d, J = 16.0 Hz, H-2''). In the 13C NMR spectrum, the cinnamoyl moieties were evidenced by signals observed at δC 165.9 (C-1''), 165.7 (C-1'''), 147.4 (C-3''), 147.0 (C-3'''), 135.3 (C-4''), 135.2 (C-4'''), 131.7 (C-7'', C-7'''), 130.0 (C-6'', C-8'', C-6''', C-8'''), 129.4 (C-5'', C-9''), 129.3 (C-5''', C-9'''), 118.0 (C-2''), and 117.8 (C-2''') [19,20]. The HMBC correlations were observed from H-3 (δH 5.43, t, J = 3.6 Hz) to C-1''' (δC 165.7) and from H-4 (δH 5.90, o) to C-1'' (δC 165.9). The 1H NMR spectrum of compound 3 revealed the presence of two signals at δH 5.28 (d, J = 3.0 Hz, Ha-11) and 5.23 (d, J = 3.0 Hz, Hb-11) characteristic of exo-methylene protons as in massarilactone H [13,21], suggesting an exo-dehydration of the C-7 hydroxy group. This was further supported by resonances depicted at δC 148.8 (C-7) and 94.3 (C-11). Since compound 3 is massarilactone H 3,4-di-O-trans-cinnamoyl, these results suggested that massarilactone H could be an artifact formed during the extraction/isolation of massarilactone D.
The molecular formula of compound 4 was deduced to be C38H32O9 from its HRESIMS spectrum (Figure S19, Supporting Information File 1) which showed the protonated molecular ion at m/z 633.2077 [M + H]+ (calcd. for C38H32O9+: 633.2119). This implies that compound 4 has 390 more atomic mass units than massarilactone D, suggesting the presence of three additional cinnamoyl groups. The presence of three cinnamoyl moieties was supported by carbon signals observed at δC 165.8 (C-1''), 165.3 (C-1'''), 164.6 (C-1'), 148.0 (C-3'), 147.3 (C-3'''), 146.9 (C-3''), 135.3 (C-4'''), 135.2 (C-4''), 134.8 (C-4'), 131.9 (C-7''), 131.6 (C-7', C-7'''), 130.3 (C-6'', C-8''), 130.0 (C-6''', C-8'''), 129.9 (C-6', C-8'), 129.4 (C-5'', C-9'', C-5''', C-9'''), 129.3 (C-5', C-9') and 117.9 (C-2', C-2'', C-2''') [19,20]. Careful examination of 1H, 13C, 1H-1H COSY, HSQC, and HMBC spectra revealed compound 4 to be massarilactone D 3,4,7-tri-O-trans-cinnamoyl.
The molecular formula of compound 5 (product of the reaction of massarilactone D with 4-bromobenzoyl chloride in triethylamine in the presence of catalytic amounts of 4-dimethylaminopyridine, 82% yield) was proposed as C32H23Br3O9 based on the molecular ion cluster sodium adducts [M + Na]+ depicted at m/z 810.96, 812.94, 814.92, and 816.90 characteristic for the presence of three bromine atoms [22]. In addition to signals ascribed to the parent compound, the 1H NMR spectrum exhibited some resonances at δH 8.02 (d, J = 8.5 Hz, H-3'', H-7''), 7.87 (d, J = 8.9 Hz, H-3''', H-7'''), 7.83 (d, J = 8.5 Hz, H-3', H-7'), 7.61 (d, J = 8.5 Hz, H-4', H-6'), 7.60 (d, J = 8.9 Hz, H-4''', H-6'''), and 7.58 (d, J = 8.5 Hz, H-4'', H-6'') ascribed to three bromobenzoyl moieties [23]. Furthermore, signals of three carbonyl groups were observed at δC 164.3 (C-1''), 164.2 (C-1'''), and 162.6 (C-1') in the 13C NMR spectrum. On the basis of the above spectroscopic data and by interpretation of the 2D spectra, compound 5 was identified as massarilactone D 3,4,7-tri-O-bromobenzoyl.
Compound 6 was obtained in 38% yield from the reaction of massarilactone D and trans-2-methyl-2-butenoyl chloride in triethylamine with catalytic amounts of 4-dimethylaminopyridine. Its HRESIMS (Figure S36, Supporting Information File 1) displayed two sodium adducts at m/z 347.1098 [M + Na]+ and 671.2314 [2M + Na]+ corresponding to the molecular formula C16H20O7 (calcd. for C16H20O7Na+: 347.1101). The NMR data of compound 6 showed the presence of a trans-2-methyl-2-butenoyl moiety when compared to those of the parent compound [24]. This was revealed by proton signals observed at δH 6.92 (dddd, J = 8.5, 7.1, 5.6, 1.5 Hz, H-3'), 1.81 (dq, J = 7.1, 1.2 Hz, 3H-4'), and 1.77 (q, J = 1.2 Hz, 3H-5') as well as carbon resonances depicted at δC 165.6 (C-1'), 141.0 (C-3'), 128.8 (C-2'), 14.7 (C-4'), and 12.0 (C-5'). Due to the upfield shifts of H-3 (δH 3.82, dd, J = 5.1, 4.4 Hz) and H-4 (δH 4.38, dd, J = 4.3, 0.9 Hz, H-4), the trans-2-methyl-2-butenoyl moiety was placed at C-7. The above spectroscopic data combined to the analysis of 2D experiments led to the elucidation of the structure of compound 6 as massarilactone D 7-O-trans-2-methyl-2-butenoyl.
The reaction of massarilactone D and crotonyl chloride in triethylamine with catalytic amounts of 4-dimethylaminopyridine afforded compounds 7 (16% yield) and 8 (32% yield). Compound 7 was obtained as white powder and its molecular formula C16H20O7 was deduced from its positive-ion mode HRESIMS (Figure S45, Supporting Information File 1) which showed the protonated molecular ion peak and the sodium adduct at m/z 325.1281 [M + H]+ (calcd. for C16H21O7+: 325.1282) and 347.1103 (calcd. for C16H20O7Na+: 347.1101), respectively. Its 1HNMR spectrum displayed in addition to signals ascribed to massarilactone D, those of a crotonyl moiety at δH 6.97 (dq, J = 15.5, 7.1 Hz, H-3''), 5.84 (dq, J = 15.5, 1.7 Hz, H-2''), and 1.88 (dd, J = 6.9, 1.7 Hz, 3H-4'') and a singlet at δH 3.20 (s, OMe) attributable to a methoxy group. In the 13C NMR spectrum, the presence of these groups was revealed by resonances observed at δC 165.6 (C-1''), 146.8 (C-3''), 122.9 (C-2''), 51.5 (OMe), and 18.0 (C-4'') [25]. The HMBC correlations observed from H-4 (δH 5.55, dd, J = 3.2, 1.4 Hz) to C-1'' (δC 165.6) and from the OMe (δH 3.20, s) to C-7 (δC 104.4) evidenced the linkage of the methoxy and the crotonyl groups at C-7 and C-4, respectively. Based on the above data, compound 7 was found to be massarilactone D 4-O-crotonyl-7-O-methyl.
The HRESIMS of the reaction product 8 (Figure S54, Supporting Information File 1) revealed a protonated molecular ion peak at m/z 379.1383 [M + H]+ and a sodium adduct ion peak at m/z 401.1203 [M + Na]+ corresponding to the molecular formula C19H22O8 (calcd. for C19H23O8+: 379.1387 and for C19H22O8Na+: 401.1207). Apart from resonances attributable to the parent compound, the 1H NMR spectrum showed duplicated signals at δH 7.04 (dq, J = 15.5, 6.9, Hz, H-3'), 6.98 (dq, J = 15.5, 6.9, Hz, H-3''), 5.85 (dq, J = 15.5, 1.7 Hz, H-2', H-2''), 1.90 (dd, J = 6.9, 1.7 Hz, 3H-4'), and 1.88 (dd, J = 6.9, 1.7 Hz, 3H-4'') ascribed to two crotonyl moieties. In the 13C NMR spectrum, the signals depicted at δC 165.7 (C-1''), 163.9 (C-1'), 149.1 (C-3'), 146.7 (C-3''), 122.9 (C-2''), 122.3 (C-2'), and 18.1 (C-4', C-4'') further confirmed the presence of these groups. The HMBC correlation from H-4 (δH 5.65, dd, J = 4.4, 1.0 Hz) to C-1'' (δC 165.7) and the upfield shift of H-3 (δH 3.98, dd, J = 5.1, 4.4 Hz) compared to the same proton in compounds 2–5 evidenced that the crotonyl moieties were linked at C-4 and C-7. Compound 8 was then elucidated as massarilactone D 4,7-di-O-crotonyl.
Cytotoxic and nematicidal activities
The antiproliferative properties of massarilactone D and the newly synthesized analogues were evaluated against the murine fibroblasts L929, human cervix carcinoma KB3-1, human lung carcinoma A549, human prostate cancer PC-3, epidermoid carcinoma A431, ovarian carcinoma SKOV-3, and breast cancer MCF-7 cell lines. Although the parent compound was not active as previously described [15,16], compounds 2 and 3 exhibited a significant cytotoxic activity against all the tested cells lines with IC50 values ranging from 3.51 to 32.73 μM. Furthermore, compounds 7 and 8 displayed cytotoxicity against the L929 and KB3-1 cell lines with IC50 values ranging from 18.50 to 61.73 μM (Table 1). Even if compounds 4–6 were not active, the present investigation revealed that the acylation reaction can improve the cytotoxic activity of natural products by increasing the hydrophobicity, enhancing cell membrane permeability and binding affinity with intracellular targets [26]. Structure–activity relationships analysis of both hemisynthetic products 2 and 3 revealed a shared conjugated methylene olefinic function that might be key to their cytotoxic effects. Additionally, compound 3, which features the exo-methylene group at C-7, exhibited greater potency than compound 2, with the highest cytotoxic activity observed against the MCF-7 cell lines, with IC50 values of 3.51 µM for compound 3 compared to 7.09 µM for compound 2. This suggests that the presence of the exo-methylene group is a critical structural feature that influences the cytotoxic activity, as outlined earlier [15]. These data also demonstrate possible selective effects of acylated massarilactone D in biological systems.
Table 1: Cytotoxic effect (IC50) of compounds 1–8 against some cell lines.
| Samples | IC50 (µM)a | ||||||
| L929 | KB3.1 | A549 | PC-3 | A431 | SKOV-3 | MCF-7 | |
| 1 | na | na | na | na | na | na | na |
| 2 | 5.09 | 13.09 | 32.73 | 11.82 | 4.00 | 11.82 | 7.09 |
| 3 | 5.58 | 7.85 | 19.63 | 7.64 | 4.34 | 13.22 | 3.51 |
| 4 | na | na | na | na | na | na | na |
| 5 | na | na | na | na | na | na | na |
| 6 | na | na | na | na | na | na | na |
| 7 | 61.73 | 55.56 | na | na | na | na | na |
| 8 | 50.26 | 18.52 | na | na | na | na | na |
| epothilone B | 2.2 × 10−3 | 7.5 × 10‒5 | 5.3 × 10‒5 | 9.1 × 10−5 | 5.9 × 10‒5 | 9.1 × 10‒5 | 7.5 × 10−5 |
ana: not active.
Massarilactone D 3,4,7-tri-O-bromobenzoyl (4) also showed significant and selective nematicidal activity with LD90 and LD50 of 100 and 12.5 µg/mL, respectively. In the case of compound 5, the LD90 was not obtained but the LD50 was determined to be 100 µg/mL. Compounds 1, 6, 7, and 8 showed some mortality at concentrations of 50 and 100 μg/mL, but their LD50 values were not obtained (Table 2). Interestingly, the derivative massarilactone H 3,4-di-O-trans-cinnamoyl (3), which contains only two cinnamoyl groups, did not exhibit nematicidal activity. In contrast, massarilactone D 3,4,7-tri-O-trans-cinnamoyl (4), featuring three cinnamoyl groups, including an additional substitution on C-7, showed potent activity against nematodes. These acyl substituents, particularly the third cinnamoyl group on C-7, may significantly enhance the biological activity. This modification likely represents a key structural feature influencing its activity, as indicated by structure–activity relationship analysis.
Conclusion
In the present study, the use of various acylating reagents to modify massarilactone D introduces distinct functional groups, each with unique chemical properties. This approach led to the synthesis of seven previously undescribed derivatives 2–8. The preliminary characterization of these products, in contrast to the parent compound, showed that both massarilactone D 3,4-di-O-methacryloyl-7-O-(6-chloro-3,4-dihydro-2,5-dimethyl-2H-pyran-2-carbonyl) (2) and massarilactone H 3,4-di-O-trans-cinnamoyl (3) exhibited a significant cytotoxic activity against all tested cell lines with IC50 values ranging from 3.51 to 32.73 μM. Furthermore, massarilactone D 3,4,7-tri-O-trans-cinnamoyl (4) also displayed a good nematicidal activity against Caenorhabditis elegans. This work once again confirms that some hemisynthetic derivatives can be more active than their natural substrates, thus expanding their therapeutic potential for cancer and parasitic infections. However, the effect of sequence variation on activity remains unclear. Additionally, the presence of the massarilactone H core in compound 3 formed from the reaction of massarilactone D with cinnamoyl chloride suggests that these two polyketides can interconvert from each other and could be artifacts and vice versa formed during extraction and isolation. This finding suggests that, to improve the antiproliferative efficacy of these derivatives, the exo-methylene group should be preserved, with chemical modifications focusing on massarilactone H rather than massarilactone D. These modifications should target other regions of the molecule, such as the number and position of acyl substituents. Future research should investigate the stability and selectivity of these compounds, as well as detailed structure–activity relationships, to optimize massarilactone hemisynthetic derivatives for specific biological applications.
Experimental
General experimental procedures
UV–vis spectra were recorded on a Shimadzu UV/Vis 2450 spectrophotometer. Optical rotations were obtained from an Anton Paar MCP-150 Polarimeter with sodium D line at 589 nm and 100 mm path length. HRESIMS mass spectra were measured with a maXis ESI TOF mass spectrometer (Bruker Daltonics) [scan range m/z 100–2500, rate 2 Hz, capillary voltage 4500 V, dry temperature 200 °C], coupled to an Agilent 1200 series HPLC-UV system [column 2.1 × 50 mm, 1.7 μm, C18 Acquity UPLC BEH (Waters), solvent A: H2O + 0.1% formic acid; solvent B: ACN + 0.1% formic acid, gradient: 5% B for 0.5 min, increasing to 100% B in 19.5 min, maintaining 100% B for 5 min, RF = 0.6 mL/min, UV–vis detection 200–600 nm]. NMR spectra were recorded in deuterated solvents (CDCl3 and acetone-d6) with an Avance III 700 (Bruker, Billerica, MA, USA) (1H: 700 MHz, 13C: 175 MHz) and an Avance III 500 (Bruker, Bremen, Germany) (1H: 500 MHz, 13C: 125 MHz) spectrometers. Chemical shifts are given in parts per million (ppm), and coupling constants in hertz (Hz). HPLC-DAD-MS analysis was performed using an amaZon speed ETD ion trap mass spectrometer (Bruker Daltonics) in positive and negative ionization modes. The mass spectrometer was coupled to a DIONEX UltiMate 3000 diode array detector [column 2.1 × 50 mm, 1.7 μm, C18 Acquity UPLC BEH (Waters), solvent A: H2O + 0.1% formic acid; solvent B: acetonitrile (ACN) + 0.1% formic acid, gradient: 5% B for 0.5 min, increasing to 100% B in 20 min, maintaining isocratic conditions at 100% B for 10 min, flow = 0.6 mL/min, UV–vis detection 190–600 nm]. Preparative HPLC was achieved at room temperature on an Agilent 1100 series preparative HPLC system [ChemStation software (Rev. B.04.03 SP1); binary pump system; column: Kinetex 5u RP C18, dimensions 250 × 21.20 mm; mobile phase: ACN + 0.05% trifluoroacetic acid (TFA) and water + 0.05% TFA; flow rate: 20 mL/min; diode array UV detector; 226 fraction collector].
Semisynthesis of massarilactone D derivatives
Preparation of compound 2
Massarilactone D (1, 15 mg, 0.062 mmol) was dissolved in CH2Cl2 (10 mL). Triethylamine (20 μL) and methacryloyl chloride (30 μL, 0.31 mmol) were added to the solution. After adding a catalytic amount of 4-dimethylaminopyridine, the reaction mixture was stirred at room temperature overnight. The reaction mixture was suspended in water (25 mL) and extracted with CH2Cl2 (2 × 25 mL). The combined organic layer was evaporated to dryness to give a residue, which was dissolved in methanol (300 μL) and purified by preparative HPLC [ChemStation software (Rev. B.04.03 SP1); binary pump system; column: Kinetex 5u RP C18, dimensions 250 × 21.20 mm; mobile phase: ACN + 0.05% trifluoroacetic acid (TFA) and water + 0.05% TFA; flow rate 20 mL/min; diode array UV detector; 226 fraction collector. A gradient from 47 to 72% solvent B in 50 min was used]. Compound 2 was obtained in 11% yield (3.50 mg, tR = 39.28 min). White powder; [α]D25 − 38.8 (c 0.0006, acetone); UV (c 0.075 mg/mL, EtOH) λmax 241 nm (3.84); 1H NMR (500 MHz, acetone-d6) δH 6.24 (dq, J = 2.1, 1.0 Hz, Ha-3''), 6.18 (dq, J = 2.2, 1.0 Hz, Ha-3'''), 6.12–6.08 (m, H-9), 5.93 (ddq, J = 15.2, 8.7, 1.6 Hz, H-8), 5.80 (p, J = 1.6 Hz, Hb-3'''), 5.75 (dd, J = 3.3, 1.3 Hz, H-4), 5.73 (p, J = 1.6 Hz, Hb-3''), 5.27 (dd, J = 8.7, 3.4 Hz, H-2), 5.23 (t, J = 3.4 Hz, H-3), 2.31 (ddd, J = 13.7, 6.6, 1.9 Hz, Ha-3'), 2.07 (o, Ha-4'), 2.00 (o, Hb-4'), 1.92 (s, Me-2''), 1.89 (s, Me-2'''), 1.83 (s, 3H-11), 1.83–182 (m, Hb-3'),1.80–178 (m, 3H-10), 1.61 (s, Me-5'), 1.50 (s, Me-2'); 13C NMR (175 MHz, acetone-d6) δC 175.8 (C-7a), 171.7 (C-1'), 166.4 (C-1''), 166.0 (C-5), 164.8 (C-1'''), 137.0 (C-6'), 136.4 (C-2'', C-2'''), 135.2 (C-9), 128.4 (C-3'''), 128.0 (C-3''), 124.1 (C-8), 105.1 (C-5'), 100.8 (C-7), 99.2 (C-4a), 83.7 (C-2), 82.3 (C-2'), 69.9 (C-3), 62.3 (C-4), 31.4 (C-3'), 26.0 (C-4'), 25.1 (Me-2'), 23.5 (C-11), 18.3 (Me-2''), 18.2 (Me-5'), 18.1 (C-10, Me-2'''); HRESIMS (m/z): [M + Na]+ calcd for C27H31O10NaCl+, 573.1498; found, 573.1495.
Preparation of compounds 3 and 4
Massarilactone D (1, 15 mg, 0.062 mmol) was dissolved in CH2Cl2 (10 mL). Triethylamine (20 μL) and cinnamoyl chloride (50 mg, 0.30 mmol) were added to the solution. After adding a catalytic amount of 4-dimethylaminopyridine, the reaction mixture was stirred at room temperature overnight. The reaction mixture was suspended in water (25 mL) and extracted with CH2Cl2 (2 × 25 mL). The combined organic layer was evaporated to dryness to give a residue, which was dissolved in methanol (300 μL) and purified by preparative HPLC (gradient from 60 to 85% solvent B in 50 min) to yield compounds 3 in 6% yield (1.72 mg, tR = 25.24 min) and 4 in 90% yield (35.30 mg, tR = 36.34 min).
Compound 3: White powder; [α]D25 +189.6 (c 0.00058, acetone); UV (c 0.01875 mg/mL, EtOH) λmax 232 nm (4.27), 284 nm (4.59); 1H NMR (700 MHz, acetone-d6) δH 7.77 (d, J = 16.0 Hz, H-3'''), 7.76 (d, J = 16.0 Hz, H-3''), 7.72–7.70 (o, H-5'', H-9'', H-5''', H-9'''), 7.46–7.44 (o, H-6'', H-7'', H-8'', H-6''', H-7''', H-8'''), 6.60 (d, J = 16.0 Hz, H-2'''), 6.58 (d, J = 16.0 Hz, H-2''), 6.07 (dqd, J = 15.3, 6.5, 1.1 Hz, H-9), 5.90 (o, H-4, H-8), 5.43 (t, J = 3.6 Hz, H-3), 5.28 (d, J = 3.0 Hz, Ha-11), 5.23 (d, J = 3.0 Hz, Hb-11), 1.78 (ddd, J = 6.6, 1.6, 0.6 Hz, 3H-10); 13C NMR (125 MH2, acetone-d6) δC 165.9 (C-5, C-1''), 165.7 (C-7a, C-1'''), 148.8 (C-7), 147.4 (C-3''), 147.0 (C-3'''), 135.3 (C-4''), 135.2 (C-4'''), 134.1 (C-9), 131.7 (C-7'', C-7'''), 130.0 (C-6'', C-8'', C-6''', C-8'''), 129.4 (C-5'', C-9''), 129.3 (C-5''', C-9'''), 124.6 (C-8), 118.0 (C-2''), 117.8 (C-2'''), 100.1 (C-4a), 94.3 (C-11), 82.9 (C-2), 70.0 (C-3), 62.1 (C-4), 18.1 (C-10); ESIMS m/z: [M + Na]+ 507.20, [2M + Na]+ 991.39
Compound 4: White powder, [α]D25 +252.2 (c 0.0029, acetone); UV (c 0.009375 mg/mL, EtOH) λmax 234 nm (4.69), 284 nm (5.15); 1H NMR (700 MHz, acetone-d6) δH 7.90 (d, J = 16.1 Hz, H-3'''), 7.76 (d, J = 16.0 Hz, H-3''), 7.76 (d, J = 16.0, H-3'), 7.73–7.69 (o, H-5', H-9', H-5''', H-9'''), 7.57–7.56 (m, H-5'', H-9''), 7.47–7.41 (o, H-6', H-7', H-8', H-6'', H-7'', H-8'', H-6''', H-7''', H-8'''), 6.58 (d, J = 16.0 Hz, H-2''), 6.57 (d, J = 16.0 Hz, H-2'), 6.55 (d, J = 16.1 Hz, H-2'''), 6.09 (dqd, J = 15.3, 6.5, 1.1 Hz, H-9), 5.94 (dqd, J = 15.3, 8.0, 1.6 Hz, H-8), 5.89 (dd, J = 3.2, 1.4 Hz, H-4), 5.43 (t, J = 3.3 Hz, H-3), 5.36 (ddt, J = 6.9, 3.3, 1.2 Hz, H-2), 1.79 (ddd, J = 6.5, 1.7, 0.8 Hz, 3H-10),1.91 (s, 3H-11); 13C NMR (125 MHz, acetone-d6) δC 175.4 (C-7a), 167.0 (C-5), 165.8 (C-1''), 165.3 (C-1'''), 164.6 (C-1'), 148.0 (C-3'), 147.3 (C-3'''), 146.9 (C-3''), 135.3 (C-4'''), 135.2 (C-4''), 134.8 (C-4'), 134.2 (C-9), 131.9 (C-7''), 131.6 (C-7', C-7'''), 130.3 (C-6'', C-8''), 130.0 (C-6''', C-8'''), 129.9 (C-6', C-8'), 129.4 (C-5'', C-9'', C-5''', C-9'''), 129.3 (C-5', C-9'), 124.2 (C-8), 117.9 (C-2', C-2'', C-2'''), 100.9 (C-7), 99.3 (C-4a), 83.2 (C-2), 69.7 (C-3), 62.2 (C-4), 23.6 (C-11), 18.2 (C-10); HRESIMS (m/z): [M + H]+ calcd for C38H32O9+, 633.2119; found, 633.2077.
Preparation of compound 5
Massarilactone D (1, 15 mg, 0.062 mmol) was dissolved in CH2Cl2 (10 mL). Triethylamine (20 μL) and 4-bromobenzoyl chloride (70 mg, 0.32 mmol) were added to the solution. After adding a catalytic amount of 4-dimethylaminopyridine, the reaction mixture was stirred at room temperature overnight. The reaction mixture was suspended in water (25 mL) and extracted with CH2Cl2 (2 × 25 mL). The combined organic layer was evaporated to dryness to give a residue, which was dissolved in methanol (300 μL) and purified by preparative HPLC (gradient from 60 to 100% solvent B in 50 min) to yield compound 5 in 82% yield (39.79 mg, tR = 22.50 min). White powder; [α]D25 +38.79 (c 0.0099, acetone); UV (c 0.0375 mg/mL, EtOH) λmax 252 nm (4.80); 1H NMR (500 MHz, CDCl3) δH 8.02 (d, J = 8.5 Hz, H-3'', H-7''), 7.87 (d, J = 8.9 Hz, H-3''', H-7'''), 7.83 (d, J = 8.5 Hz, H-3', H-7'), 7.61 (d, J = 8.5 Hz, H-4', H-6'), 7.60 (d, J = 8.9 Hz, H-4''', H-6'''), 7.58 (d, J = 8.5 Hz, H-4'', H-6''), 6.07 (dd, J = 2.8, 1.4 Hz, H-4), 5.98 (dqd, J = 14.2, 6.6, 1.1 Hz, H-9), 5.73 (dqd, J = 15.3, 7.8, 1.5 Hz, H-8), 5.56 (t, J = 2.9 Hz, H-3), 5.25 (ddt, J = 7.8, 2.9, 1.2 Hz, H-2), 1.74 (ddd, J = 6.6, 1.6, 0.9 Hz, 3H-10), 1.99 (s, 3H-11); 13C NMR (125 MHz, CDCl3) δC 174.8 (C-7a), 166.2 (C-5), 164.3 (C-1''), 164.2 (C-1'''), 162.6 (C-1'), 133.6 (C-9), 132.0 (C-4', C-6'), 131.8 (C-3'', C-4'', C-6'', C-7'', C-4''', C-6'''), 131.3 (C-3', C-7', C-3''', C-7'''), 129.3 (C-5'), 129.0 (C-5''), 128.8 (C-5'''), 128.0 (C-2'''), 127.6 (C-2', C-2''), 122.4 (C-8), 100.2 (C-7), 98.2 (C-4a), 82.1 (C-2), 69.2 (C-3), 61.8 (C-4), 23.2 (C-11), 18.0 (C-10). ESIMS m/z: [M + Na]+ 810.96
Preparation of compound 6
Massarilactone D (1, 15 mg, 0.062 mmol) was dissolved in CH2Cl2 (10 mL). Triethylamine (20 μL) and trans-2-methyl-2-butenoyl chloride (34 μL, 0.30 mmol) were added to the solution. After adding a catalytic amount of 4-dimethylaminopyridine, the reaction mixture was stirred at room temperature overnight. The reaction mixture was suspended in water (25 mL) and extracted with CH2Cl2 (2 × 25 mL). The combined organic layer was evaporated to dryness to give a residue, which was dissolved in methanol (300 μL) and purified by preparative HPLC (gradient from 20 to 54% solvent B in 50 min) to yield compound 6 in 38% yield (7.52 mg, tR = 25.92 min). White powder; [α]D25 −98.37 (c 0.00123, acetone); UV (c 0.15 mg/mL, EtOH) λmax 247 nm (3.84); 1H NMR (500 MHz, acetone-d6) δH 6.92 (dddd, J = 8.5, 7.1, 5.6, 1.5 Hz, H-3'), 5.92 (m, H-9), 5.85 (ddq, J = 15.3, 8.1, 1.3 Hz, H-8), 4.81 (dd, J = 7.6, 5.2 Hz, H-2), 4.38 (dd, J = 4.3, 0.9 Hz, H-4), 3.82 (dd, J = 5.1, 4.4 Hz, H-3), 1.81 (dq, J = 7.1, 1.2 Hz, 3H-4'), 1.77 (q, J = 1.2 Hz, 3H-5'), 1.76 (s, 3H-11), 1.72 (m, 3H-10); 13C NMR (125 MHz, acetone-d6) δC 173.8 (C-7a), 168.0 (C-5), 165.6 (C-1'), 141.0 (C-3'), 132.8 (C-9), 128.8 (C-2'), 126.6 (C-8), 100.4 (C-7), 102.9 (C-4a), 86.3 (C-2), 72.8 (C-3), 64.7 (C-4), 23.3 (C-11), 18.1 (C-10), 14.7 (C-4'), 12.0 (C-5'); HRESIMS (m/z): [M + Na]+ calcd for C16H20O7Na+, 347.1101; found, 347.1098
Preparation of compounds 7 and 8
Massarilactone D (1, 15 mg, 0.062 mmol) was dissolved in CH2Cl2 (10 mL). Triethylamine (20 μL) and crotonyl chloride (25 μL, 0.26 mmol) were added to the solution. After adding a catalytic amount of 4-dimethylaminopyridine, the reaction mixture was stirred at room temperature overnight. The reaction mixture was suspended in water (25 mL) and extracted with CH2Cl2 (2 × 25 mL). The combined organic layers were evaporated to dryness to give a residue, which was dissolved in methanol (300 μL) and purified by preparative HPLC (gradient from 26 to 50% solvent B in 50 min) to yield compounds 7 in 16% yield (3.2 mg, tR = 31.50 min) and 8 in 32% yield (7.3 mg, tR = 46.23 min).
Compound 7: White powder; [α]D25 +25.97 (c 0.00077, acetone); UV (c 0.01875 mg/mL, EtOH) λmax 241 nm (4.41); 1H NMR (500 MHz, acetone-d6) δ 6.97 (dq, J = 15.5, 7.1 Hz, H-3''), 5.92 (m, H-9), 5.84 (dq, J = 15.5, 1.7 Hz, H-2''), 5.73 (ddq, J = 15.5, 7.7, 1.6 Hz, H-8), 5.55 (dd, J = 3.2, 1.4 Hz, H-4), 5.12 (ddt, J = 6.6, 3.3, 1.2 Hz, H-2), 4.06 (t, J = 3.2 Hz, H-3), 3.20 (s, OMe), 1.88 (dd, J = 6.9, 1.7 Hz, 3H-4''), 1.73 (ddd, J = 6.5, 1.6, 0.9 Hz, 3H-10), 1.63 (s, 3H-11); 13C NMR (125 MHz, acetone-d6) δC 174.1 (C-7a), 168.1 (C-5), 165.6 (C-1''), 146.8 (C-3''), 132.0 (C-9), 125.7 (C-8), 122.9 (C-2''), 104.4 (C-7), 100.3 (C-4a), 85.3 (C-2), 69.1 (C-3), 63.5 (C-4), 51.5 (OMe), 22.8 (C-11), 18.1 (C-10), 18.0 (C-4''); HRESIMS (m/z): [M + H]+ calcd for C16H21O7+, 325.1282; found, 325.1281; [M + Na]+ calcd for C16H20O7Na+, 347.1101; found, 347.1103.
Compound 8: White powder; [α]D25 −34.74 (c 0.0019, acetone); UV (c 0.075 mg/mL, EtOH) λmax 241 nm (4.00); 1H NMR (500 MHz, acetone-d6) δH 7.04 (dq, J = 15.5, 6.9, Hz, H-3'), 6.98 (dq, J = 15.5, 6.9, Hz, H-3''), 5.97 (m, H-9), 5.85 (dq, J = 15.5, 1.7 Hz, H-2', H-2''), 5.74 (ddq, J = 15.4, 7.9, 1.6 Hz, H-8), 5.65 (dd, J = 4.4, 1.0 Hz, H-4), 4.96 (ddq, J = 7.9, 5.1, 0.9 Hz, H-2), 3.98 (dd, J = 5.1, 4.4 Hz, H-3), 1.90 (dd, J = 6.9, 1.7 Hz, 3H-4'), 1.88 (dd, J = 6.9, 1.7 Hz, 3H-4''), 1.79 (s, 3H-11), 1.74 (ddd, J = 6.6, 1.7, 0.8 Hz, 3H-10), 1.79 (s, 3H-11); 13C NMR (125 MHz, acetone-d6) δC 175.4 (C-7a), 167.1 (C-5), 165.7 (C-1''), 163.9 (C-1'), 149.1(C-3'), 146.7 (C-3''), 133.2 (C-9), 125.8 (C-8), 122.9 (C-2''), 122.3 (C-2'), 100.5 (C-7), 99.4 (C-4a), 86.0 (C-2), 69.8 (C-3), 65.1 (C-4), 23.2 (C-11), 18.2 (C-10), 18.1 (C-4', C-4''); HRESIMS (m/z): [M + H]+ calcd for C19H23O8+, 379.1387; found, 379.1383.
Cytotoxic activity
The cytotoxicity against the murine fibroblasts L929, human cervix carcinoma KB-3-1, human lung carcinoma A549, human prostate cancer PC-3, epidermoid carcinoma A431, ovarian carcinoma SKOV-3, and breast cancer MCF-7 cell lines was determined by using the MTT (2-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) method in 96-well microplates as previously described [27-29]. Briefly, the cell lines were cultured in DMEM (Gibco), 60 µL aliquots of serial dilutions from an initial stock of 1 mg/mL in MeOH of the test compounds were added to 120 µL aliquots of a cell suspension (5 × 104 cells/mL) in 96-well microplates. After 5 days incubation, an MTT assay was performed, and the absorbance measured at 590 nm using an ELISA plate reader (Victor). The concentration at which the growth of cells was inhibited to 50% of the control (IC50) was obtained from the dose–response curves. Epothilone B was used as the positive control.
Nematicidal activity
Nematicidal activity was performed using Caenorhabditis elegans in a microtiter plate assay as described by Rupcic et al. (2018) [30]. The assay was performed in four concentrations (100, 50, 25 and 12.5 μg/mL). Ivermectin was used as a positive control at the same concentration ranges as the tested compounds and MeOH was used as a negative control. Percentages of mortality were calculated, then the results were expressed as a LD90 and LD50.
Supporting Information
| Supporting Information File 1: Copies of NMR, mass, and UV spectra for compounds 2–8. | ||
| Format: PDF | Size: 5.9 MB | Download |
Data Availability Statement
All data is available in the published article and/or the supporting information.
References
-
Lei, Z.-N.; Tian, Q.; Teng, Q.-X.; Wurpel, J. N. D.; Zeng, L.; Pan, Y.; Chen, Z.-S. MedComm 2023, 4, e265. doi:10.1002/mco2.265
Return to citation in text: [1] -
Seid, A.; Fininsa, C.; Mekete, T.; Decraemer, W.; Wesemael, W. M. L. Nematology 2015, 17, 995–1009. doi:10.1163/15685411-00002935
Return to citation in text: [1] -
Sun, H.; Li, H.; Wang, J.; Song, G. Chin. Chem. Lett. 2018, 29, 977–980. doi:10.1016/j.cclet.2017.10.015
Return to citation in text: [1] -
Ghareeb, R. Y.; Shams El-Din, N. G. E.-D.; Maghraby, D. M. E.; Ibrahim, D. S. S.; Abdel-Megeed, A.; Abdelsalam, N. R. Sci. Rep. 2022, 12, 3841. doi:10.1038/s41598-022-06600-1
Return to citation in text: [1] -
Dias, K. S. T.; Januário, J. P.; D’Dego, J. L.; Dias, A. L. T.; dos Santos, M. H.; Camps, I.; Coelho, L. F. L.; Viegas, C., Jr. Bioorg. Med. Chem. 2012, 20, 2713–2720. doi:10.1016/j.bmc.2012.02.023
Return to citation in text: [1] -
Teponno, R. B.; Dzoyem, J. P.; Nono, R. N.; Kauhl, U.; Sandjo, L. P.; Tapondjou, L. A.; Bakowsky, U.; Opatz, T. Rec. Nat. Prod. 2017, 11, 421–430. doi:10.25135/rnp.54.17.03.050
Return to citation in text: [1] -
Solipeta, D. R.; Bandi, S.; Vemulapalli, S. P. B.; Pallavi, P. M. C.; Vemireddy, S.; Balasubramania, S.; Mahabalarao, S. K. H.; Katragadda, S. B. J. Nat. Prod. 2019, 82, 2731–2743. doi:10.1021/acs.jnatprod.9b00346
Return to citation in text: [1] -
Oh, H.; Swenson, D. C.; Gloer, J. B.; Shearer, C. A. Tetrahedron Lett. 2001, 42, 975–977. doi:10.1016/s0040-4039(00)02160-2
Return to citation in text: [1] [2] [3] -
Keller, N. P.; Turner, G.; Bennett, J. W. Nat. Rev. Microbiol. 2005, 3, 937–947. doi:10.1038/nrmicro1286
Return to citation in text: [1] -
Yokoyama, M.; Hirayama, Y.; Yamamoto, T.; Kishimoto, S.; Tsunematsu, Y.; Watanabe, K. Org. Lett. 2017, 19, 2002–2005. doi:10.1021/acs.orglett.7b00559
Return to citation in text: [1] -
Kock, I.; Krohn, K.; Egold, H.; Draeger, S.; Schulz, B.; Rheinheimer, J. Eur. J. Org. Chem. 2007, 2186–2190. doi:10.1002/ejoc.200600987
Return to citation in text: [1] -
Krohn, K.; Zia‐Ullah; Hussain, H.; Flörke, U.; Schulz, B.; Draeger, S.; Pescitelli, G.; Salvadori, P.; Antus, S.; Kurtán, T. Chirality 2007, 19, 464–470. doi:10.1002/chir.20402
Return to citation in text: [1] -
Zhang, G. F.; Han, W. B.; Cui, J. T.; Ng, S. W.; Guo, Z. K.; Tan, R. X.; Ge, H. M. Planta Med. 2012, 78, 76–78. doi:10.1055/s-0031-1280251
Return to citation in text: [1] [2] -
Cimmino, A.; Bahmani, Z.; Masi, M.; Di Lecce, R.; Amini, J.; Abdollahzadeh, J.; Tuzi, A.; Evidente, A. Nat. Prod. Res. 2021, 35, 5192–5198. doi:10.1080/14786419.2020.1791116
Return to citation in text: [1] -
Mathieu, V.; Superchi, S.; Masi, M.; Scafato, P.; Kornienko, A.; Evidente, A. Toxins 2022, 14, 517. doi:10.3390/toxins14080517
Return to citation in text: [1] [2] [3] -
Teponno, R. B.; Noumeur, S. R.; Helaly, S. E.; Hüttel, S.; Harzallah, D.; Stadler, M. Molecules 2017, 22, 1674. doi:10.3390/molecules22101674
Return to citation in text: [1] [2] [3] -
Li, M.-Y.; Yang, X.-B.; Pan, J.-Y.; Feng, G.; Xiao, Q.; Sinkkonen, J.; Satyanandamurty, T.; Wu, J. J. Nat. Prod. 2009, 72, 2110–2114. doi:10.1021/np900625w
Return to citation in text: [1] -
Warneke, J.; Wang, Z.; Zeller, M.; Leibfritz, D.; Plaumann, M.; Azov, V. A. Tetrahedron 2014, 70, 6515–6521. doi:10.1016/j.tet.2014.07.019
Return to citation in text: [1] [2] -
Nguyen Ngoc, H.; Alilou, M.; Stonig, M.; Nghiem, D. T.; Kim, L. T.; Gostner, J. M.; Stuppner, H.; Ganzera, M. J. Nat. Prod. 2019, 82, 2941–2952. doi:10.1021/acs.jnatprod.9b00208
Return to citation in text: [1] [2] -
Chen, F.-Y.; Li, C.-J.; Ma, J.; Zhou, J.; Li, L.; Zhang, Z.; Chen, N.-H.; Zhang, D.-M. J. Nat. Prod. 2018, 81, 270–278. doi:10.1021/acs.jnatprod.7b00615
Return to citation in text: [1] [2] -
Alliot, J.; Gravel, E.; Larquetoux, L.; Nicolas, M.; Doris, E. J. Nat. Prod. 2013, 76, 2346–2349. doi:10.1021/np400869z
Return to citation in text: [1] -
El-Gamal, A. A.; Wang, W.-L.; Duh, C.-Y. J. Nat. Prod. 2005, 68, 815–817. doi:10.1021/np058001y
Return to citation in text: [1] -
Maqbool, T.; Younas, H.; Bilal, M.; Rasool, N.; Bajaber, M. A.; Mubarik, A.; Parveen, B.; Ahmad, G.; Shah, S. A. A. ACS Omega 2023, 8, 30306–30314. doi:10.1021/acsomega.3c03183
Return to citation in text: [1] -
Shen, C.-C.; Wei, W.-C.; Lin, L.-C. J. Nat. Prod. 2019, 82, 3181–3185. doi:10.1021/acs.jnatprod.9b00674
Return to citation in text: [1] -
Lee, S.-S.; Chen, W.-C.; Chen, C.-H. J. Nat. Prod. 2000, 63, 1580–1583. doi:10.1021/np000225n
Return to citation in text: [1] -
Yang, Y.; Zhang, H.; Wanyan, Y.; Liu, K.; Lv, T.; Li, M.; Chen, Y. ACS Omega 2020, 5, 21513–21523. doi:10.1021/acsomega.0c02093
Return to citation in text: [1] -
Wittstein, K.; Rascher, M.; Rupcic, Z.; Löwen, E.; Winter, B.; Köster, R. W.; Stadler, M. J. Nat. Prod. 2016, 79, 2264–2269. doi:10.1021/acs.jnatprod.6b00371
Return to citation in text: [1] -
Helaly, S. E.; Ashrafi, S.; Teponno, R. B.; Bernecker, S.; Dababat, A. A.; Maier, W.; Stadler, M. J. Nat. Prod. 2018, 81, 2228–2234. doi:10.1021/acs.jnatprod.8b00486
Return to citation in text: [1] -
Noumeur, S. R.; Teponno, R. B.; Helaly, S. E.; Wang, X.-W.; Harzallah, D.; Houbraken, J.; Crous, P. W.; Stadler, M. Mycol. Prog. 2020, 19, 589–603. doi:10.1007/s11557-020-01581-9
Return to citation in text: [1] -
Rupcic, Z.; Chepkirui, C.; Hernández-Restrepo, M.; Crous, P. W.; Luangsa-ard, J. J.; Stadler, M. MycoKeys 2018, 33, 1–23. doi:10.3897/mycokeys.33.23341
Return to citation in text: [1]
| 30. | Rupcic, Z.; Chepkirui, C.; Hernández-Restrepo, M.; Crous, P. W.; Luangsa-ard, J. J.; Stadler, M. MycoKeys 2018, 33, 1–23. doi:10.3897/mycokeys.33.23341 |
| 1. | Lei, Z.-N.; Tian, Q.; Teng, Q.-X.; Wurpel, J. N. D.; Zeng, L.; Pan, Y.; Chen, Z.-S. MedComm 2023, 4, e265. doi:10.1002/mco2.265 |
| 8. | Oh, H.; Swenson, D. C.; Gloer, J. B.; Shearer, C. A. Tetrahedron Lett. 2001, 42, 975–977. doi:10.1016/s0040-4039(00)02160-2 |
| 18. | Warneke, J.; Wang, Z.; Zeller, M.; Leibfritz, D.; Plaumann, M.; Azov, V. A. Tetrahedron 2014, 70, 6515–6521. doi:10.1016/j.tet.2014.07.019 |
| 8. | Oh, H.; Swenson, D. C.; Gloer, J. B.; Shearer, C. A. Tetrahedron Lett. 2001, 42, 975–977. doi:10.1016/s0040-4039(00)02160-2 |
| 9. | Keller, N. P.; Turner, G.; Bennett, J. W. Nat. Rev. Microbiol. 2005, 3, 937–947. doi:10.1038/nrmicro1286 |
| 10. | Yokoyama, M.; Hirayama, Y.; Yamamoto, T.; Kishimoto, S.; Tsunematsu, Y.; Watanabe, K. Org. Lett. 2017, 19, 2002–2005. doi:10.1021/acs.orglett.7b00559 |
| 18. | Warneke, J.; Wang, Z.; Zeller, M.; Leibfritz, D.; Plaumann, M.; Azov, V. A. Tetrahedron 2014, 70, 6515–6521. doi:10.1016/j.tet.2014.07.019 |
| 5. | Dias, K. S. T.; Januário, J. P.; D’Dego, J. L.; Dias, A. L. T.; dos Santos, M. H.; Camps, I.; Coelho, L. F. L.; Viegas, C., Jr. Bioorg. Med. Chem. 2012, 20, 2713–2720. doi:10.1016/j.bmc.2012.02.023 |
| 6. | Teponno, R. B.; Dzoyem, J. P.; Nono, R. N.; Kauhl, U.; Sandjo, L. P.; Tapondjou, L. A.; Bakowsky, U.; Opatz, T. Rec. Nat. Prod. 2017, 11, 421–430. doi:10.25135/rnp.54.17.03.050 |
| 7. | Solipeta, D. R.; Bandi, S.; Vemulapalli, S. P. B.; Pallavi, P. M. C.; Vemireddy, S.; Balasubramania, S.; Mahabalarao, S. K. H.; Katragadda, S. B. J. Nat. Prod. 2019, 82, 2731–2743. doi:10.1021/acs.jnatprod.9b00346 |
| 16. | Teponno, R. B.; Noumeur, S. R.; Helaly, S. E.; Hüttel, S.; Harzallah, D.; Stadler, M. Molecules 2017, 22, 1674. doi:10.3390/molecules22101674 |
| 2. | Seid, A.; Fininsa, C.; Mekete, T.; Decraemer, W.; Wesemael, W. M. L. Nematology 2015, 17, 995–1009. doi:10.1163/15685411-00002935 |
| 3. | Sun, H.; Li, H.; Wang, J.; Song, G. Chin. Chem. Lett. 2018, 29, 977–980. doi:10.1016/j.cclet.2017.10.015 |
| 4. | Ghareeb, R. Y.; Shams El-Din, N. G. E.-D.; Maghraby, D. M. E.; Ibrahim, D. S. S.; Abdel-Megeed, A.; Abdelsalam, N. R. Sci. Rep. 2022, 12, 3841. doi:10.1038/s41598-022-06600-1 |
| 17. | Li, M.-Y.; Yang, X.-B.; Pan, J.-Y.; Feng, G.; Xiao, Q.; Sinkkonen, J.; Satyanandamurty, T.; Wu, J. J. Nat. Prod. 2009, 72, 2110–2114. doi:10.1021/np900625w |
| 14. | Cimmino, A.; Bahmani, Z.; Masi, M.; Di Lecce, R.; Amini, J.; Abdollahzadeh, J.; Tuzi, A.; Evidente, A. Nat. Prod. Res. 2021, 35, 5192–5198. doi:10.1080/14786419.2020.1791116 |
| 15. | Mathieu, V.; Superchi, S.; Masi, M.; Scafato, P.; Kornienko, A.; Evidente, A. Toxins 2022, 14, 517. doi:10.3390/toxins14080517 |
| 13. | Zhang, G. F.; Han, W. B.; Cui, J. T.; Ng, S. W.; Guo, Z. K.; Tan, R. X.; Ge, H. M. Planta Med. 2012, 78, 76–78. doi:10.1055/s-0031-1280251 |
| 16. | Teponno, R. B.; Noumeur, S. R.; Helaly, S. E.; Hüttel, S.; Harzallah, D.; Stadler, M. Molecules 2017, 22, 1674. doi:10.3390/molecules22101674 |
| 12. | Krohn, K.; Zia‐Ullah; Hussain, H.; Flörke, U.; Schulz, B.; Draeger, S.; Pescitelli, G.; Salvadori, P.; Antus, S.; Kurtán, T. Chirality 2007, 19, 464–470. doi:10.1002/chir.20402 |
| 11. | Kock, I.; Krohn, K.; Egold, H.; Draeger, S.; Schulz, B.; Rheinheimer, J. Eur. J. Org. Chem. 2007, 2186–2190. doi:10.1002/ejoc.200600987 |
| 8. | Oh, H.; Swenson, D. C.; Gloer, J. B.; Shearer, C. A. Tetrahedron Lett. 2001, 42, 975–977. doi:10.1016/s0040-4039(00)02160-2 |
| 19. | Nguyen Ngoc, H.; Alilou, M.; Stonig, M.; Nghiem, D. T.; Kim, L. T.; Gostner, J. M.; Stuppner, H.; Ganzera, M. J. Nat. Prod. 2019, 82, 2941–2952. doi:10.1021/acs.jnatprod.9b00208 |
| 20. | Chen, F.-Y.; Li, C.-J.; Ma, J.; Zhou, J.; Li, L.; Zhang, Z.; Chen, N.-H.; Zhang, D.-M. J. Nat. Prod. 2018, 81, 270–278. doi:10.1021/acs.jnatprod.7b00615 |
| 19. | Nguyen Ngoc, H.; Alilou, M.; Stonig, M.; Nghiem, D. T.; Kim, L. T.; Gostner, J. M.; Stuppner, H.; Ganzera, M. J. Nat. Prod. 2019, 82, 2941–2952. doi:10.1021/acs.jnatprod.9b00208 |
| 20. | Chen, F.-Y.; Li, C.-J.; Ma, J.; Zhou, J.; Li, L.; Zhang, Z.; Chen, N.-H.; Zhang, D.-M. J. Nat. Prod. 2018, 81, 270–278. doi:10.1021/acs.jnatprod.7b00615 |
| 13. | Zhang, G. F.; Han, W. B.; Cui, J. T.; Ng, S. W.; Guo, Z. K.; Tan, R. X.; Ge, H. M. Planta Med. 2012, 78, 76–78. doi:10.1055/s-0031-1280251 |
| 21. | Alliot, J.; Gravel, E.; Larquetoux, L.; Nicolas, M.; Doris, E. J. Nat. Prod. 2013, 76, 2346–2349. doi:10.1021/np400869z |
| 15. | Mathieu, V.; Superchi, S.; Masi, M.; Scafato, P.; Kornienko, A.; Evidente, A. Toxins 2022, 14, 517. doi:10.3390/toxins14080517 |
| 27. | Wittstein, K.; Rascher, M.; Rupcic, Z.; Löwen, E.; Winter, B.; Köster, R. W.; Stadler, M. J. Nat. Prod. 2016, 79, 2264–2269. doi:10.1021/acs.jnatprod.6b00371 |
| 28. | Helaly, S. E.; Ashrafi, S.; Teponno, R. B.; Bernecker, S.; Dababat, A. A.; Maier, W.; Stadler, M. J. Nat. Prod. 2018, 81, 2228–2234. doi:10.1021/acs.jnatprod.8b00486 |
| 29. | Noumeur, S. R.; Teponno, R. B.; Helaly, S. E.; Wang, X.-W.; Harzallah, D.; Houbraken, J.; Crous, P. W.; Stadler, M. Mycol. Prog. 2020, 19, 589–603. doi:10.1007/s11557-020-01581-9 |
| 15. | Mathieu, V.; Superchi, S.; Masi, M.; Scafato, P.; Kornienko, A.; Evidente, A. Toxins 2022, 14, 517. doi:10.3390/toxins14080517 |
| 16. | Teponno, R. B.; Noumeur, S. R.; Helaly, S. E.; Hüttel, S.; Harzallah, D.; Stadler, M. Molecules 2017, 22, 1674. doi:10.3390/molecules22101674 |
| 26. | Yang, Y.; Zhang, H.; Wanyan, Y.; Liu, K.; Lv, T.; Li, M.; Chen, Y. ACS Omega 2020, 5, 21513–21523. doi:10.1021/acsomega.0c02093 |
| 24. | Shen, C.-C.; Wei, W.-C.; Lin, L.-C. J. Nat. Prod. 2019, 82, 3181–3185. doi:10.1021/acs.jnatprod.9b00674 |
| 25. | Lee, S.-S.; Chen, W.-C.; Chen, C.-H. J. Nat. Prod. 2000, 63, 1580–1583. doi:10.1021/np000225n |
| 22. | El-Gamal, A. A.; Wang, W.-L.; Duh, C.-Y. J. Nat. Prod. 2005, 68, 815–817. doi:10.1021/np058001y |
| 23. | Maqbool, T.; Younas, H.; Bilal, M.; Rasool, N.; Bajaber, M. A.; Mubarik, A.; Parveen, B.; Ahmad, G.; Shah, S. A. A. ACS Omega 2023, 8, 30306–30314. doi:10.1021/acsomega.3c03183 |
© 2025 Teponno et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.