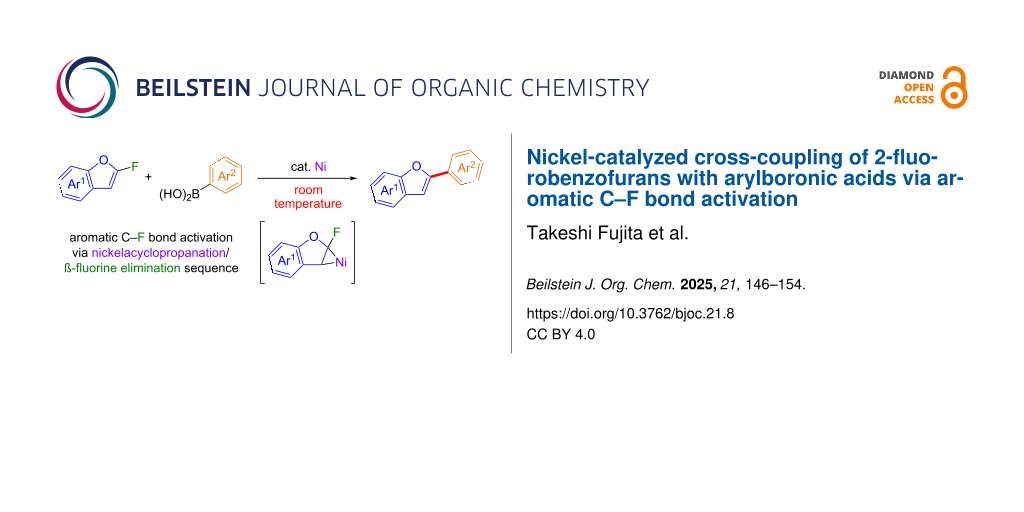
Abstract
2-Fluorobenzofurans underwent efficient nickel-catalyzed coupling with arylboronic acids through the activation of aromatic C–F bonds. This method allowed us to successfully synthesize a range of 2-arylbenzofurans with various substituents. The reaction, which proceeded under mild conditions, involved β-fluorine elimination from nickelacyclopropanes formed by the interaction of 2-fluorobenzofurans with zero-valent nickel species. This protocol facilitates orthogonal coupling reactions of aromatic C–F and C–Br bonds with arylboronic acids.

Graphical Abstract
Introduction
The metal-catalyzed activation of aromatic carbon–fluorine (C–F) bonds is widely recognized as a challenging task in synthetic organic chemistry owing to their high bond dissociation energy compared to other aromatic C–X (X = Cl, Br, I) bonds [1-7]. This activation is essential for the late-stage functionalization of stable C–F bonds in complex molecules with reactive functional groups, providing an orthogonal approach to complex molecule synthesis. Despite considerable efforts to develop various catalytic systems, the activation of aromatic C–F bonds often requires high temperatures [1-7]. Therefore, methods for activating aromatic C–F bonds at ambient temperature remain underdeveloped.
We have developed efficient metal-mediated methods for activating (i) vinylic [8-13] and (ii) allylic C–F bonds [14-18] using β-fluorine elimination under mild conditions. In these studies, (i) we discovered zirconium-mediated β-fluorine elimination from zirconacyclopropanes A, which are generated by treating 1,1-difluoroethylenes with a zirconocene equivalent (ZrCp2, Scheme 1a) [8]. The resulting 1-fluorovinylzirconocenes B then undergo palladium-catalyzed coupling with aryl iodides to produce arylated fluoroethylenes. Additionally, (ii) we observed that electron-deficient 2-(trifluoromethyl)-1-alkenes strongly interact with electron-rich zero-valent nickel species to form nickelacyclopropanes C [15-17]. These intermediates enable C–F bond activation through the formation of nickelacyclopentenes D with alkynes, followed by β-fluorine elimination, leading to defluorinative coupling between these components (Scheme 1b).
![[1860-5397-21-8-i1]](/bjoc/content/inline/1860-5397-21-8-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: C–F bond activation through β-fluorine elimination via metalacyclopropanes.
Scheme 1: C–F bond activation through β-fluorine elimination via metalacyclopropanes.
Among aromatic fluorides, we have targeted 2-fluorobenzofurans 1 for C–F bond activation [19]. These compounds, which we prepared efficiently via 5-endo-trig cyclization of β,β-difluoro-o-hydroxystyrenes [20,21], possess a C–C double bond with an electron-deficient carbon atom owing to the nearby fluorine and oxygen atoms. We expected that 2-fluorobenzofurans 1 could form nickelacyclopropanes E upon treatment with zero-valent nickel species. Subsequent β-fluorine elimination from these intermediates E would facilitate the activation of aromatic C–F bonds (Scheme 1c). In this study, we demonstrate nickel-catalyzed defluorinative cross-coupling [22-37] of 2-fluorobenzofurans 1 with arylboronic acids 2 at ambient temperature, with nickelacyclopropanes E serving as crucial intermediates for the activation of aromatic C–F bonds.
Results and Discussion
First, we explored optimal conditions for nickel-catalyzed defluorinative coupling using 2-fluoronaphtho[2,1-b]furan (1b) and m-tolylboronic acid (2b) as model substrates (Table 1). When 1b was reacted with 2b at 80 °C using Ni(cod)2 (10 mol %) as a catalyst, PCy3 (20 mol %) as a ligand, and K2CO3 (2.0 equiv) as a base, the desired arylated naphthofuran 3bb was obtained in 75% yield (Table 1, entry 1). Reducing the reaction temperature improved the yield of 3bb, reaching a quantitative yield when the reaction was performed at room temperature (Table 1, entry 3). Reducing the catalyst loading to 5 mol % slightly affected the yield of 3bb, which was 90% (Table 1, entry 4). Next, we evaluated various additives with 5 mol % of Ni(cod)2 to stabilize regenerated zero-valent nickel species (Table 1, entries 5–8). While phosphine ligands such as triphenyl phosphite were ineffective (Table 1, entry 5), the inclusion of chelating dienes improved the yield of 3bb (Table 1, entries 6–8). Among these, 5 mol % of 1,5-cyclooctadiene (cod) proved to be the most effective additive, affording 3bb in 95% yield (Table 1, entry 8). Additionally, by reducing the equivalents of 2b to 1.0 equiv and K2CO3 to 1.2 equiv, we achieved the highest yield of 98% for 3bb (Table 1, entry 9).
Table 1: Screening of conditions for coupling of 1b with 2b.
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-8-i7.svg?max-width=637&scale=1.0)
|
||||||
| Entry | X | Additive | Y | Temp. | Time (h) | 3bb (%) |
| 1 | 10 | – | – | 80 °C | 24 | 75a |
| 2 | 10 | – | – | 40 °C | 72 | 91a |
| 3 | 10 | – | – | rt | 72 | quant.a |
| 4 | 5 | – | – | rt | 28 | 90b |
| 5 | 5 | P(OPh)3 | 5 | rt | 58 | 12b |
| 6 | 5 | nbdc | 5 | rt | 58 | 93b |
| 7 | 5 | chdd | 5 | rt | 58 | 93b |
| 8 | 5 | code | 5 | rt | 52 | 95b |
| 9f | 5 | code | 5 | rt | 14 | 98b |
aYield was determined by 1H NMR spectroscopy using CH2Br2 as an internal standard. bIsolated yield. cnbd = 2,5-norbornadiene. dchd = 1,4-cyclohexadiene. ecod = 1,5-cyclooctadiene. f2b (1.0 equiv) and K2CO3 (1.2 equiv).
Under the optimized conditions, we investigated the substrate scope using 2-fluorobenzofurans 1 and arylboronic acids 2 (Scheme 2). The coupling reaction was efficient with 2-fluorobenzofuran (1a) when reacted with phenylboronic acid (2a) as well as arylboronic acids containing electron-donating groups, such as a methyl group at the 3-position (2b), two methyl groups at the 2- and 5-positions (2c), and a tert-butyl group at the 4-position (2d). The reaction with 3,5-dimethoxyphenylboronic acid (2e), which has electron-withdrawing groups on the aromatic ring, also yielded a satisfactory result of 73%. Additionally, using 2-fluoronaphtho[2,1-b]furan (1b), the reaction with phenylboronic acid (2a) and arylboronic acids with a methyl group at the 3-position (2b) or a tert-butyl group at the 4-position (2d) also produced high yields (94–98%). For arylboronic acid 2f, which has a methoxy group at the 4-position, the use of potassium phosphate as a base resulted in a 94% yield of 3bf. For arylboronic acid 2g, which features a strongly electron-withdrawing trifluoromethyl group, we optimized the coupling reaction using potassium phosphate as a base and increasing the nickel catalyst loading to 20 mol %, achieving a yield of 78% for the desired product 3bg. When 2-naphthylboronic acid (2i) was employed, its solubility was enhanced using a mixed solvent system of toluene, methanol, and water, which effectively promoted the reaction and resulted in a 70% yield of 3bi. Furthermore, when methoxy- and ethoxy-substituted benzofurans 1c and 1d were used, the corresponding coupling products 3ca and 3da were obtained with yields of 67% and 65%, respectively.
![[1860-5397-21-8-i2]](/bjoc/content/inline/1860-5397-21-8-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 2-arylbenzofurans 3 via the coupling of 1 with 2. Isolated yields are given. aNi(cod)2 (10 mol %), PCy3 (20 mol %), and cod (10 mol %). bNi(cod)2 (20 mol %), PCy3 (40 mol %), and cod (20 mol %). cK3PO4 (1.2 equiv) was used as a base. dToluene–MeOH–H2O (5:1:1) was used as a solvent.
Scheme 2: Synthesis of 2-arylbenzofurans 3 via the coupling of 1 with 2. Isolated yields are given. aNi(cod)2...
Additionally, in the coupling reaction of 2-fluorobenzothiophene (4) with 2a, increasing the amount of Ni(cod)2 to 20 mol % without adding extra cod yielded 48% of the desired product 5 (Scheme 3). This result indicates that the reaction is applicable to benzothiophenes as well as benzofurans.
![[1860-5397-21-8-i3]](/bjoc/content/inline/1860-5397-21-8-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 2-phenylbenzothiophene (5).
Scheme 3: Synthesis of 2-phenylbenzothiophene (5).
Moreover, we successfully introduced two distinct aryl groups onto a benzofuran ring through orthogonal coupling reactions, exploiting the reactivity difference between C–F and C–Br bonds (Scheme 4). Using a palladium catalyst, 5-bromo-2-fluorobenzofuran (1e) was coupled with [4-(trifluoromethyl)phenyl]boronic acid (2g). In this reaction, only the C–Br bond was transformed while the C–F bond remained intact, yielding 2-fluoro-5-[4-(trifluoromethyl)phenyl]benzofuran (1f) in 95% yield. Subsequently, nickel-catalyzed defluorinative arylation of 1f with phenylboronic acid (2a) efficiently produced 2-phenyl-5-[4-(trifluoromethyl)phenyl]benzofuran (3fa) in 81% yield.
![[1860-5397-21-8-i4]](/bjoc/content/inline/1860-5397-21-8-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Orthogonal approach to 2,5-diarylbenzofuran 3fa.
Scheme 4: Orthogonal approach to 2,5-diarylbenzofuran 3fa.
Next, we explored the mechanism of the coupling reactions between 2-fluorobenzofurans 1 and arylboronic acids 2. Because these reactions proceed under mild conditions despite involving aromatic C–F bond activation [19], direct oxidative addition of C–F bonds is unlikely (Scheme 5, path a). Instead, the reactions are thought to proceed through a formal oxidative addition involving nickelacyclopropane intermediates E [15-17,38,39], which are generated from 2-fluorobenzofurans 1 and zero-valent nickel species (Scheme 5). Following β-fluorine elimination, this results in a formal oxidative addition to form benzofuranylnickel(II) fluorides F, which then undergo transmetallation with arylboronic acids 2 to produce intermediates G (Scheme 5, path b). Alternatively, a direct transition from E to G via transition state H is also possible (Scheme 5, path c). Ultimately, reductive elimination from G yields the coupling products 3.
![[1860-5397-21-8-i5]](/bjoc/content/inline/1860-5397-21-8-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
The following experiments were performed to elucidate the mechanism. Under the same conditions as the coupling reaction, stoichiometric amounts of Ni(cod)2, PCy3, and cod were treated with fluoronaphthofuran 1b at room temperature for 13 h, excluding boronic acid 2a (Scheme 6). The reaction was monitored using 19F and 31P NMR spectroscopy. The 19F NMR analysis showed that 79% of 1b remained and revealed a new broad double doublet peak at 55.0 ppm (JFP = 53, 42 Hz) relative to internal C6F6 (δ = 0.0 ppm). The 31P NMR spectrum depicted broad singlet peaks at 32.0–33.4 ppm and 38.6–40.5 ppm, appearing in a 1:1 ratio. These new peaks were attributed to nickelacyclopropane Eb, which was formed in 19% yield. No peaks corresponding to benzofuranylnickel(II) fluoride Fb, which would arise from the oxidative addition of 1b to nickel(0), were detected [40]. High-resolution mass spectrometry (HRMS) analysis of the reaction mixture also supported the formation of Eb (calcd, 804.4474; found, 804.4449). Additionally, 79% of 1b remained, while the catalytic reaction between 1b and 2a was completed in 13 h, yielding 3ba in 96% (Scheme 2). These findings suggest that nickelacyclopropanes E and 2-fluorobenzofurans 1 are in equilibrium (see Scheme 5). Consequently, in the absence of arylboronic acids 2, the consumption of 1 was suppressed. Upon adding phenylboronic acid (2a, 1.0 equiv) to the above reaction mixture, the coupling proceeded, producing 3ba in 70% yield, with neither complex Eb nor Fb observed (Scheme 6). These results suggest that nickelacyclopropanes E are initially formed and facilitate a formal oxidative addition. Notably, the absence of F in the reaction mixture indicates that fluorine elimination and transmetallation occur simultaneously between E and the arylboronic acids 2, leading to the formation of G (Scheme 5, path c). The intermediates G then undergo reductive elimination to yield 3.
![[1860-5397-21-8-i6]](/bjoc/content/inline/1860-5397-21-8-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Formation of nickelacyclopropane Eb in a stoichiometric reaction.
Scheme 6: Formation of nickelacyclopropane Eb in a stoichiometric reaction.
To assess the impact of halogen substituents, we also examined reactions of 2-halogenated benzofurans 1a-X (1a-Cl: X = Cl; 1a-Br: X = Br; 1a-I: X = I) with (3-methylphenyl)boronic acid (2b) (Table 2). Both 2-chlorobenzofuran (1a-Cl) and 2-bromobenzofuran (1a-Br) hardly yielded 3ab under the optimized conditions for 1a (Table 2, entries 2 and 3), while the reaction of 2-iodobenzofuran (1a-I) resulted in a much lower yield (32%) of 2-arylbenzofuran 3ab (Table 2, entry 4) compared to that of 1a (X = F, quant.). The strong interaction between fluorine and boron in H likely facilitates β-fluorine elimination and transmetallation. Thus, the considerably different result observed with 1a is attributed to the distinct mechanistic aspects of the metalacyclopropanation/β-fluorine elimination sequence influenced by the fluorine substituent.
![[Graphic 2]](/bjoc/content/inline/1860-5397-21-8-i8.svg?max-width=637&scale=1.0)
Conclusion
In summary, we have presented a nickel-catalyzed method for synthesizing 2-arylbenzofurans through aromatic C–F bond activation, with the formation of metallacyclopropanes as an essential step. This protocol allows for the late-stage transformation of C–F bonds, as demonstrated by the orthogonal activation of both aromatic C–F and C–Br bonds, thereby facilitating the synthesis of complex 2-arylbenzofurans. Given that natural and synthetic 2-arylbenzofurans often exhibit considerable biological activities and are important in pharmaceuticals and agrochemicals [41-47], we expect that this method will provide a novel and efficient approach for producing these valuable compounds.
Experimental
General: 1H NMR, 13C NMR, 19F NMR, and 31P NMR were recorded on a Bruker Avance 500 or a JEOL ECS-400 spectrometer. Chemical shift values are given in ppm relative to internal Me4Si (for 1H NMR: δ = 0.00 ppm), CDCl3 (for 13C NMR: δ = 77.0 ppm), C6F6 (for 19F NMR: δ = 0.0 ppm), and H3PO4 (for 31P NMR: δ = 0.0 ppm). IR spectra were recorded on a Horiba FT-730 spectrometer. Mass spectra were measured on a JEOL JMS-T100GCV or a JEOL JMS-T200GC spectrometer. All the reactions were conducted under argon or nitrogen.
Materials: Column chromatography was conducted on silica gel (Silica Gel 60 N, Kanto Chemical Co., Inc.). Toluene and N,N-dimethylformamide (DMF) were purified by a solvent-purification system (GlassContour) equipped with columns of activated alumina and supported-copper catalyst (Q-5) before use. 1,4-Dioxane and methanol were distilled from sodium, and stored over 4 Å molecular sieves. Unless otherwise noted, materials were obtained from commercial sources and used directly without further purifications.
Typical procedure for coupling of 2-fluorobenzofurans 1 with arylboronic acids 2: To the mixture of 2-fluoronaphtho[2,1-b]furan (1b, 56 mg, 0.30 mmol), (3-methylphenyl)boronic acid (2b, 41 mg, 0.30 mmol), Ni(cod)2 (4.2 mg, 0.015 mmol), PCy3 (8.2 mg, 0.029 mmol), 1,5-cyclooctadiene (1.8 μL, 0.015 mmol), and K2CO3 (50 mg, 0.36 mmol) were added toluene (3.0 mL) and H2O (0.6 mL). After stirring at room temperature for 13 h, the reaction mixture was diluted with H2O. Organic materials were extracted with diethyl ether three times. The combined extracts were washed with brine and dried over Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by silica gel column chromatography (hexane/EtOAc = 10:1) to give 3bb (76 mg, 98%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.15 (d, J = 8.2 Hz, 1H), 7.93 (d, J = 8.2 Hz, 1H), 7.75–7.67 (m, 4H), 7.58 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 7.49–7.46 (m, 2H), 7.35 (dd, J = 7.7, 7.6 Hz, 1H), 7.16 (d, J = 7.6 Hz, 1H), 2.44 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 155.6, 152.3, 138.5, 130.5, 130.4, 129.1, 128.8, 128.7, 127.6, 126.2, 125.3, 125.1, 124.6, 124.5, 123.4, 121.9, 112.3, 100.3, 21.5; IR (KBr): 3051, 1606, 1487, 1387, 1280, 1255, 1163, 1053, 991, 935, 789, 690 cm–1; HREIMS m/z: [M]+ calcd for C19H14O, 258.1045; found, 258.1035.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures and spectral data. | ||
| Format: PDF | Size: 5.5 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Amii, H.; Uneyama, K. Chem. Rev. 2009, 109, 2119–2183. doi:10.1021/cr800388c
Return to citation in text: [1] [2] -
Sun, A. D.; Love, J. A. Dalton Trans. 2010, 39, 10362–10374. doi:10.1039/c0dt00540a
Return to citation in text: [1] [2] -
Ahrens, T.; Kohlmann, J.; Ahrens, M.; Braun, T. Chem. Rev. 2015, 115, 931–972. doi:10.1021/cr500257c
Return to citation in text: [1] [2] -
Chen, W.; Bakewell, C.; Crimmin, M. R. Synthesis 2017, 49, 810–821. doi:10.1055/s-0036-1588663
Return to citation in text: [1] [2] -
Wang, M.; Shi, Z. Chem. Rev. 2020, 120, 7348–7398. doi:10.1021/acs.chemrev.9b00384
Return to citation in text: [1] [2] -
Zhao, B.; Rogge, T.; Ackermann, L.; Shi, Z. Chem. Soc. Rev. 2021, 50, 8903–8953. doi:10.1039/c9cs00571d
Return to citation in text: [1] [2] -
Zhang, J.; Geng, S.; Feng, Z. Chem. Commun. 2021, 57, 11922–11934. doi:10.1039/d1cc04729a
Return to citation in text: [1] [2] -
Fujiwara, M.; Ichikawa, J.; Okauchi, T.; Minami, T. Tetrahedron Lett. 1999, 40, 7261–7265. doi:10.1016/s0040-4039(99)01491-4
Return to citation in text: [1] [2] -
Sakoda, K.; Mihara, J.; Ichikawa, J. Chem. Commun. 2005, 4684–4686. doi:10.1039/b510039a
Return to citation in text: [1] -
Fuchibe, K.; Morikawa, T.; Shigeno, K.; Fujita, T.; Ichikawa, J. Org. Lett. 2015, 17, 1126–1129. doi:10.1021/ol503759d
Return to citation in text: [1] -
Fujita, T.; Watabe, Y.; Yamashita, S.; Tanabe, H.; Nojima, T.; Ichikawa, J. Chem. Lett. 2016, 45, 964–966. doi:10.1246/cl.160427
Return to citation in text: [1] -
Watabe, Y.; Kanazawa, K.; Fujita, T.; Ichikawa, J. Synthesis 2017, 49, 3569–3575. doi:10.1055/s-0036-1588842
Return to citation in text: [1] -
Fujita, T.; Takeishi, M.; Ichikawa, J. Org. Lett. 2020, 22, 9253–9257. doi:10.1021/acs.orglett.0c03476
Return to citation in text: [1] -
Ichikawa, J.; Nadano, R.; Ito, N. Chem. Commun. 2006, 4425–4427. doi:10.1039/b610690k
Return to citation in text: [1] -
Ichitsuka, T.; Fujita, T.; Arita, T.; Ichikawa, J. Angew. Chem., Int. Ed. 2014, 53, 7564–7568. doi:10.1002/anie.201402695
Return to citation in text: [1] [2] [3] -
Ichitsuka, T.; Fujita, T.; Ichikawa, J. ACS Catal. 2015, 5, 5947–5950. doi:10.1021/acscatal.5b01463
Return to citation in text: [1] [2] [3] -
Fujita, T.; Arita, T.; Ichitsuka, T.; Ichikawa, J. Dalton Trans. 2015, 44, 19460–19463. doi:10.1039/c5dt02160j
Return to citation in text: [1] [2] [3] -
Fujita, T.; Kobayashi, Y.; Takahashi, I.; Morioka, R.; Ichitsuka, T.; Ichikawa, J. Chem. – Eur. J. 2022, 28, e202103643. doi:10.1002/chem.202103643
Return to citation in text: [1] -
Li, Y. Monatsh. Chem. 2022, 153, 193–199. doi:10.1007/s00706-021-02881-w
Return to citation in text: [1] [2] -
Ichikawa, J.; Wada, Y.; Okauchi, T.; Minami, T. Chem. Commun. 1997, 1537–1538. doi:10.1039/a703110f
Return to citation in text: [1] -
Morioka, R.; Fujita, T.; Ichikawa, J. Helv. Chim. Acta 2020, 103, e2000159. doi:10.1002/hlca.202000159
Return to citation in text: [1] -
Tamao, K.; Sumitani, K.; Kiso, Y.; Zembayashi, M.; Fujioka, A.; Kodama, S.-i.; Nakajima, I.; Minato, A.; Kumada, M. Bull. Chem. Soc. Jpn. 1976, 49, 1958–1969. doi:10.1246/bcsj.49.1958
Return to citation in text: [1] -
Mongin, F.; Mojovic, L.; Guillamet, B.; Trécourt, F.; Quéguiner, G. J. Org. Chem. 2002, 67, 8991–8994. doi:10.1021/jo026136s
Return to citation in text: [1] -
Böhm, V. P. W.; Gstöttmayr, C. W. K.; Weskamp, T.; Herrmann, W. A. Angew. Chem., Int. Ed. 2001, 40, 3387–3389. doi:10.1002/1521-3773(20010917)40:18<3387::aid-anie3387>3.0.co;2-6
Return to citation in text: [1] -
Dankwardt, J. W. J. Organomet. Chem. 2005, 690, 932–938. doi:10.1016/j.jorganchem.2004.10.037
Return to citation in text: [1] -
Yoshikai, N.; Mashima, H.; Nakamura, E. J. Am. Chem. Soc. 2005, 127, 17978–17979. doi:10.1021/ja056327n
Return to citation in text: [1] -
Lu, Y.; Plocher, E.; Hu, Q.-S. Adv. Synth. Catal. 2006, 348, 841–845. doi:10.1002/adsc.200606002
Return to citation in text: [1] -
Schaub, T.; Backes, M.; Radius, U. J. Am. Chem. Soc. 2006, 128, 15964–15965. doi:10.1021/ja064068b
Return to citation in text: [1] -
Ackermann, L.; Wechsler, C.; Kapdi, A. R.; Althammer, A. Synlett 2010, 294–298. doi:10.1055/s-0029-1219166
Return to citation in text: [1] -
Xie, L.-G.; Wang, Z.-X. Chem. – Eur. J. 2010, 16, 10332–10336. doi:10.1002/chem.201001022
Return to citation in text: [1] -
Sun, A. D.; Love, J. A. Org. Lett. 2011, 13, 2750–2753. doi:10.1021/ol200860t
Return to citation in text: [1] -
Tobisu, M.; Xu, T.; Shimasaki, T.; Chatani, N. J. Am. Chem. Soc. 2011, 133, 19505–19511. doi:10.1021/ja207759e
Return to citation in text: [1] -
Zhang, J.; Xu, J.; Xu, Y.; Sun, H.; Shen, Q.; Zhang, Y. Organometallics 2015, 34, 5792–5800. doi:10.1021/acs.organomet.5b00874
Return to citation in text: [1] -
Malineni, J.; Jezorek, R. L.; Zhang, N.; Percec, V. Synthesis 2016, 48, 2795–2807. doi:10.1055/s-0035-1562342
Return to citation in text: [1] -
Ogawa, H.; Yang, Z.-K.; Minami, H.; Kojima, K.; Saito, T.; Wang, C.; Uchiyama, M. ACS Catal. 2017, 7, 3988–3994. doi:10.1021/acscatal.7b01058
Return to citation in text: [1] -
Kurisu, N.; Asano, E.; Hatayama, Y.; Kurihara, Y.; Hashimoto, T.; Funatsu, K.; Ueda, K.; Yamaguchi, Y. Eur. J. Inorg. Chem. 2019, 126–133. doi:10.1002/ejic.201801179
Return to citation in text: [1] -
Jacobs, E.; Keaveney, S. T. ChemCatChem 2021, 13, 637–645. doi:10.1002/cctc.202001462
Return to citation in text: [1] -
Zhang, X.; Huang, X.; Chen, Y.; Chen, B.; Ma, Y. Org. Lett. 2023, 25, 1748–1753. doi:10.1021/acs.orglett.3c00444
Return to citation in text: [1] -
Yang, P.; Yu, H.; Zhai, R.; Zhou, J. S.; Tang, B. Chem. Commun. 2024, 60, 6548–6551. doi:10.1039/d4cc00918e
Return to citation in text: [1] -
Johnson, S. A.; Huff, C. W.; Mustafa, F.; Saliba, M. J. Am. Chem. Soc. 2008, 130, 17278–17280. doi:10.1021/ja8081395
Return to citation in text: [1] -
Shi, Y.-Q.; Fukai, T.; Sakagami, H.; Chang, W.-J.; Yang, P.-Q.; Wang, F.-P.; Nomura, T. J. Nat. Prod. 2001, 64, 181–188. doi:10.1021/np000317c
Return to citation in text: [1] -
Ni, G.; Zhang, Q.-J.; Zheng, Z.-F.; Chen, R.-Y.; Yu, D.-Q. J. Nat. Prod. 2009, 72, 966–968. doi:10.1021/np800789y
Return to citation in text: [1] -
Tan, Y.-X.; Yang, Y.; Zhang, T.; Chen, R.-Y.; Yu, D.-Q. Fitoterapia 2010, 81, 742–746. doi:10.1016/j.fitote.2010.03.017
Return to citation in text: [1] -
Artini, M.; Papa, R.; Barbato, G.; Scoarughi, G. L.; Cellini, A.; Morazzoni, P.; Bombardelli, E.; Selan, L. Bioorg. Med. Chem. 2012, 20, 920–926. doi:10.1016/j.bmc.2011.11.052
Return to citation in text: [1] -
Morelli, L.; Bernardi, A.; Sattin, S. Carbohydr. Res. 2014, 390, 33–41. doi:10.1016/j.carres.2014.03.006
Return to citation in text: [1] -
Tjahjandarie, T. S.; Tanjung, M.; Saputri, R. D.; Rahayu, D. O.; Gunawan, A. N. I.; Aldin, M. F. Nat. Prod. Res. 2021, 35, 5637–5642. doi:10.1080/14786419.2020.1821016
Return to citation in text: [1] -
Heravi, M. M.; Zadsirjan, V.; Hamidi, H.; Tabar Amiri, P. H. RSC Adv. 2017, 7, 24470–24521. doi:10.1039/c7ra03551a
And references cited therein.
Return to citation in text: [1]
| 1. | Amii, H.; Uneyama, K. Chem. Rev. 2009, 109, 2119–2183. doi:10.1021/cr800388c |
| 2. | Sun, A. D.; Love, J. A. Dalton Trans. 2010, 39, 10362–10374. doi:10.1039/c0dt00540a |
| 3. | Ahrens, T.; Kohlmann, J.; Ahrens, M.; Braun, T. Chem. Rev. 2015, 115, 931–972. doi:10.1021/cr500257c |
| 4. | Chen, W.; Bakewell, C.; Crimmin, M. R. Synthesis 2017, 49, 810–821. doi:10.1055/s-0036-1588663 |
| 5. | Wang, M.; Shi, Z. Chem. Rev. 2020, 120, 7348–7398. doi:10.1021/acs.chemrev.9b00384 |
| 6. | Zhao, B.; Rogge, T.; Ackermann, L.; Shi, Z. Chem. Soc. Rev. 2021, 50, 8903–8953. doi:10.1039/c9cs00571d |
| 7. | Zhang, J.; Geng, S.; Feng, Z. Chem. Commun. 2021, 57, 11922–11934. doi:10.1039/d1cc04729a |
| 8. | Fujiwara, M.; Ichikawa, J.; Okauchi, T.; Minami, T. Tetrahedron Lett. 1999, 40, 7261–7265. doi:10.1016/s0040-4039(99)01491-4 |
| 14. | Ichikawa, J.; Nadano, R.; Ito, N. Chem. Commun. 2006, 4425–4427. doi:10.1039/b610690k |
| 15. | Ichitsuka, T.; Fujita, T.; Arita, T.; Ichikawa, J. Angew. Chem., Int. Ed. 2014, 53, 7564–7568. doi:10.1002/anie.201402695 |
| 16. | Ichitsuka, T.; Fujita, T.; Ichikawa, J. ACS Catal. 2015, 5, 5947–5950. doi:10.1021/acscatal.5b01463 |
| 17. | Fujita, T.; Arita, T.; Ichitsuka, T.; Ichikawa, J. Dalton Trans. 2015, 44, 19460–19463. doi:10.1039/c5dt02160j |
| 18. | Fujita, T.; Kobayashi, Y.; Takahashi, I.; Morioka, R.; Ichitsuka, T.; Ichikawa, J. Chem. – Eur. J. 2022, 28, e202103643. doi:10.1002/chem.202103643 |
| 8. | Fujiwara, M.; Ichikawa, J.; Okauchi, T.; Minami, T. Tetrahedron Lett. 1999, 40, 7261–7265. doi:10.1016/s0040-4039(99)01491-4 |
| 9. | Sakoda, K.; Mihara, J.; Ichikawa, J. Chem. Commun. 2005, 4684–4686. doi:10.1039/b510039a |
| 10. | Fuchibe, K.; Morikawa, T.; Shigeno, K.; Fujita, T.; Ichikawa, J. Org. Lett. 2015, 17, 1126–1129. doi:10.1021/ol503759d |
| 11. | Fujita, T.; Watabe, Y.; Yamashita, S.; Tanabe, H.; Nojima, T.; Ichikawa, J. Chem. Lett. 2016, 45, 964–966. doi:10.1246/cl.160427 |
| 12. | Watabe, Y.; Kanazawa, K.; Fujita, T.; Ichikawa, J. Synthesis 2017, 49, 3569–3575. doi:10.1055/s-0036-1588842 |
| 13. | Fujita, T.; Takeishi, M.; Ichikawa, J. Org. Lett. 2020, 22, 9253–9257. doi:10.1021/acs.orglett.0c03476 |
| 41. | Shi, Y.-Q.; Fukai, T.; Sakagami, H.; Chang, W.-J.; Yang, P.-Q.; Wang, F.-P.; Nomura, T. J. Nat. Prod. 2001, 64, 181–188. doi:10.1021/np000317c |
| 42. | Ni, G.; Zhang, Q.-J.; Zheng, Z.-F.; Chen, R.-Y.; Yu, D.-Q. J. Nat. Prod. 2009, 72, 966–968. doi:10.1021/np800789y |
| 43. | Tan, Y.-X.; Yang, Y.; Zhang, T.; Chen, R.-Y.; Yu, D.-Q. Fitoterapia 2010, 81, 742–746. doi:10.1016/j.fitote.2010.03.017 |
| 44. | Artini, M.; Papa, R.; Barbato, G.; Scoarughi, G. L.; Cellini, A.; Morazzoni, P.; Bombardelli, E.; Selan, L. Bioorg. Med. Chem. 2012, 20, 920–926. doi:10.1016/j.bmc.2011.11.052 |
| 45. | Morelli, L.; Bernardi, A.; Sattin, S. Carbohydr. Res. 2014, 390, 33–41. doi:10.1016/j.carres.2014.03.006 |
| 46. | Tjahjandarie, T. S.; Tanjung, M.; Saputri, R. D.; Rahayu, D. O.; Gunawan, A. N. I.; Aldin, M. F. Nat. Prod. Res. 2021, 35, 5637–5642. doi:10.1080/14786419.2020.1821016 |
| 47. |
Heravi, M. M.; Zadsirjan, V.; Hamidi, H.; Tabar Amiri, P. H. RSC Adv. 2017, 7, 24470–24521. doi:10.1039/c7ra03551a
And references cited therein. |
| 1. | Amii, H.; Uneyama, K. Chem. Rev. 2009, 109, 2119–2183. doi:10.1021/cr800388c |
| 2. | Sun, A. D.; Love, J. A. Dalton Trans. 2010, 39, 10362–10374. doi:10.1039/c0dt00540a |
| 3. | Ahrens, T.; Kohlmann, J.; Ahrens, M.; Braun, T. Chem. Rev. 2015, 115, 931–972. doi:10.1021/cr500257c |
| 4. | Chen, W.; Bakewell, C.; Crimmin, M. R. Synthesis 2017, 49, 810–821. doi:10.1055/s-0036-1588663 |
| 5. | Wang, M.; Shi, Z. Chem. Rev. 2020, 120, 7348–7398. doi:10.1021/acs.chemrev.9b00384 |
| 6. | Zhao, B.; Rogge, T.; Ackermann, L.; Shi, Z. Chem. Soc. Rev. 2021, 50, 8903–8953. doi:10.1039/c9cs00571d |
| 7. | Zhang, J.; Geng, S.; Feng, Z. Chem. Commun. 2021, 57, 11922–11934. doi:10.1039/d1cc04729a |
| 22. | Tamao, K.; Sumitani, K.; Kiso, Y.; Zembayashi, M.; Fujioka, A.; Kodama, S.-i.; Nakajima, I.; Minato, A.; Kumada, M. Bull. Chem. Soc. Jpn. 1976, 49, 1958–1969. doi:10.1246/bcsj.49.1958 |
| 23. | Mongin, F.; Mojovic, L.; Guillamet, B.; Trécourt, F.; Quéguiner, G. J. Org. Chem. 2002, 67, 8991–8994. doi:10.1021/jo026136s |
| 24. | Böhm, V. P. W.; Gstöttmayr, C. W. K.; Weskamp, T.; Herrmann, W. A. Angew. Chem., Int. Ed. 2001, 40, 3387–3389. doi:10.1002/1521-3773(20010917)40:18<3387::aid-anie3387>3.0.co;2-6 |
| 25. | Dankwardt, J. W. J. Organomet. Chem. 2005, 690, 932–938. doi:10.1016/j.jorganchem.2004.10.037 |
| 26. | Yoshikai, N.; Mashima, H.; Nakamura, E. J. Am. Chem. Soc. 2005, 127, 17978–17979. doi:10.1021/ja056327n |
| 27. | Lu, Y.; Plocher, E.; Hu, Q.-S. Adv. Synth. Catal. 2006, 348, 841–845. doi:10.1002/adsc.200606002 |
| 28. | Schaub, T.; Backes, M.; Radius, U. J. Am. Chem. Soc. 2006, 128, 15964–15965. doi:10.1021/ja064068b |
| 29. | Ackermann, L.; Wechsler, C.; Kapdi, A. R.; Althammer, A. Synlett 2010, 294–298. doi:10.1055/s-0029-1219166 |
| 30. | Xie, L.-G.; Wang, Z.-X. Chem. – Eur. J. 2010, 16, 10332–10336. doi:10.1002/chem.201001022 |
| 31. | Sun, A. D.; Love, J. A. Org. Lett. 2011, 13, 2750–2753. doi:10.1021/ol200860t |
| 32. | Tobisu, M.; Xu, T.; Shimasaki, T.; Chatani, N. J. Am. Chem. Soc. 2011, 133, 19505–19511. doi:10.1021/ja207759e |
| 33. | Zhang, J.; Xu, J.; Xu, Y.; Sun, H.; Shen, Q.; Zhang, Y. Organometallics 2015, 34, 5792–5800. doi:10.1021/acs.organomet.5b00874 |
| 34. | Malineni, J.; Jezorek, R. L.; Zhang, N.; Percec, V. Synthesis 2016, 48, 2795–2807. doi:10.1055/s-0035-1562342 |
| 35. | Ogawa, H.; Yang, Z.-K.; Minami, H.; Kojima, K.; Saito, T.; Wang, C.; Uchiyama, M. ACS Catal. 2017, 7, 3988–3994. doi:10.1021/acscatal.7b01058 |
| 36. | Kurisu, N.; Asano, E.; Hatayama, Y.; Kurihara, Y.; Hashimoto, T.; Funatsu, K.; Ueda, K.; Yamaguchi, Y. Eur. J. Inorg. Chem. 2019, 126–133. doi:10.1002/ejic.201801179 |
| 37. | Jacobs, E.; Keaveney, S. T. ChemCatChem 2021, 13, 637–645. doi:10.1002/cctc.202001462 |
| 15. | Ichitsuka, T.; Fujita, T.; Arita, T.; Ichikawa, J. Angew. Chem., Int. Ed. 2014, 53, 7564–7568. doi:10.1002/anie.201402695 |
| 16. | Ichitsuka, T.; Fujita, T.; Ichikawa, J. ACS Catal. 2015, 5, 5947–5950. doi:10.1021/acscatal.5b01463 |
| 17. | Fujita, T.; Arita, T.; Ichitsuka, T.; Ichikawa, J. Dalton Trans. 2015, 44, 19460–19463. doi:10.1039/c5dt02160j |
| 38. | Zhang, X.; Huang, X.; Chen, Y.; Chen, B.; Ma, Y. Org. Lett. 2023, 25, 1748–1753. doi:10.1021/acs.orglett.3c00444 |
| 39. | Yang, P.; Yu, H.; Zhai, R.; Zhou, J. S.; Tang, B. Chem. Commun. 2024, 60, 6548–6551. doi:10.1039/d4cc00918e |
| 20. | Ichikawa, J.; Wada, Y.; Okauchi, T.; Minami, T. Chem. Commun. 1997, 1537–1538. doi:10.1039/a703110f |
| 21. | Morioka, R.; Fujita, T.; Ichikawa, J. Helv. Chim. Acta 2020, 103, e2000159. doi:10.1002/hlca.202000159 |
| 40. | Johnson, S. A.; Huff, C. W.; Mustafa, F.; Saliba, M. J. Am. Chem. Soc. 2008, 130, 17278–17280. doi:10.1021/ja8081395 |
| 15. | Ichitsuka, T.; Fujita, T.; Arita, T.; Ichikawa, J. Angew. Chem., Int. Ed. 2014, 53, 7564–7568. doi:10.1002/anie.201402695 |
| 16. | Ichitsuka, T.; Fujita, T.; Ichikawa, J. ACS Catal. 2015, 5, 5947–5950. doi:10.1021/acscatal.5b01463 |
| 17. | Fujita, T.; Arita, T.; Ichitsuka, T.; Ichikawa, J. Dalton Trans. 2015, 44, 19460–19463. doi:10.1039/c5dt02160j |
© 2025 Fujita et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.