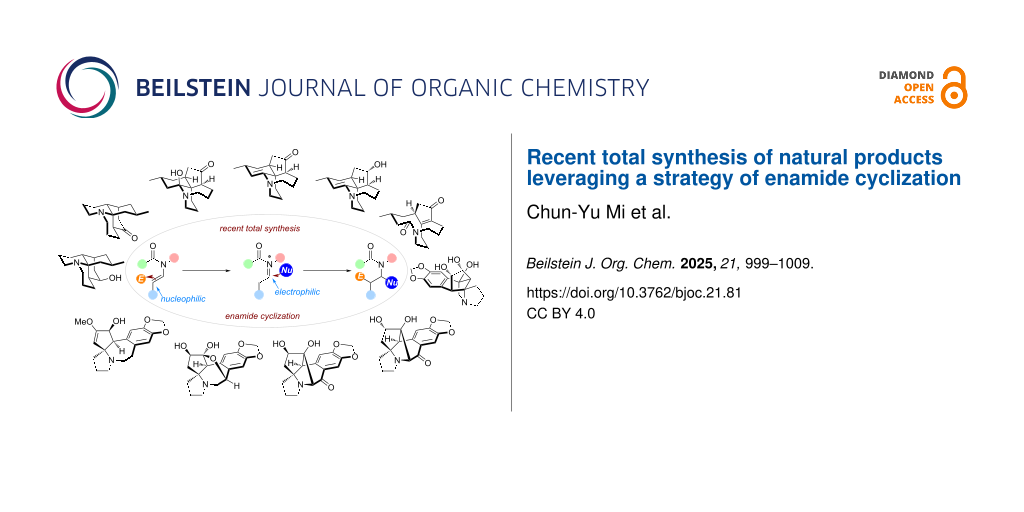
Abstract
Enamides are distinctive amphiphilic synthons that can be strategically incorporated into cyclization reactions. The iminium species generated from enamides via nucleophilic addition or substitution are capable of engaging in further electrophilic additions or isomerization processes. Exploiting the multiple reactivities of enamides facilitates the development of diverse cyclization modes that provide entries to various N-heterocycles, some of which serve as key structural motifs in natural alkaloids. This review highlights recent advancements in enamide-based cyclization reactions, including enamide–alkyne cycloisomerization, [3 + 2] annulation, and polycyclization, with a particular emphasis on their pivotal role as a strategy in the total synthesis of natural products.

Graphical Abstract
Introduction
The use of enamines as surrogates for enols in nucleophilic reactions has been well-documented for decades since their first report by Stork in the 1950s [1-3]. Compared with enols, enamines benefit from the lone pair of electrons on the nitrogen atom, which enhances the nucleophilicity of the alkene, enabling it to react with a broad range of electrophiles. This activation mode of carbonyl compounds has been so well-established that it is featured in nearly every organic chemistry textbook. However, despite their versatility, enamines themselves are not easily handling compounds in experimental settings. Their sensitivity to hydrolysis complicates their isolation and identification, and following the nucleophilic addition or substitution, the resulting iminium ions often undergo direct hydrolysis, preventing further use in a cascade nucleophilic addition. As a result, enamines are not ideal partners in tandem reactions for the synthesis of nitrogen-containing products. As analogues to enamines, the enamides contain an N-acyl group in place of the original alkyl group. The electron-withdrawing effect of the amide group delocalizes the nitrogen lone pair, thereby reducing the electron density and nucleophilicity of the enamide double bond. These features significantly diminish the reactivity of enamides as nucleophiles, rendering them more stable than enamines. This stability is reflected in their frequent occurrence in natural products [4]. As a result, research on the synthetic applications of enamides has historically lagged behind that of enamines [5,6]. Beyond their use in hydrogenation reactions [7,8], the exploration of enamides’ nucleophilic reactivity has only gained momentum in recent years. Inspired by pioneering work from various research groups [9-15], the potential of enamides in nucleophilic reactions has become recognized. Among them, enamide cyclizations have attracted considerable attention due to their promise in the total synthesis of alkaloids [16]. Notably, these valuable compounds can be employed as efficient synthons in enamide–alkyne cycloisomerization, [n + m] cycloadditions, pericyclic reactions, and radical cyclizations. A comprehensive review of these advancements up until 2015 has already been documented [16]. In this review, recent breakthroughs of these enamide cyclizations will be surveyed from the viewpoint of natural product synthesis. Leveraging the enamide–alkyne cycloisomerization cyclizations, Lycopodium alkaloids (−)-dihydrolycopodine, (−)-lycopodine, (+)-lycoposerramine Q, (+)-fawcettidine, (+)-fawcettimine, and (−)-phlegmariurine have been synthesized in a concise and efficient manner, while employment of the [2 + 3] cycloadditions or a polycyclization enables the elegant total synthesis of Cephalotaxus alkaloids cephalotaxine, cephalezomine H, (−)-cephalotaxine, (−)-cephalotine B, (−)-fortuneicyclidin A, (−)-fortuneicyclidin B, and (−)-cephalocyclidin A.
Unlike enamines, tertiary enamides can participate in cyclization reactions initial as nucleophiles, and upon protonation, alkenylation, or alkylation, the resultant iminium intermediates can serve as electrophiles. Due to the presence of an amide, the resulting iminiums from the enamides can be stabilized to take part in the second nucleophilic addition, though direct isomerization of the iminiums to the enamides is also possible (Figure 1). Guided by these principles, tandem reactions or annulations can be designed to efficiently access N-heterocycles. As the enamides are also easily accessible via condensations, applications of these nitrogen-containing building blocks in the synthesis of N-heterocycles are synthetically straightforward. When applied properly, these methods offer promising strategies for the total synthesis of complex natural products.
![[1860-5397-21-81-1]](/bjoc/content/figures/1860-5397-21-81-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Reactivity of enamides and enamide cyclizations.
Figure 1: Reactivity of enamides and enamide cyclizations.
Review
Aza-Prins cyclization – total synthesis of (−)-dihydrolycopodine and (−)-lycopodine
Cyclizations of enamides can proceed via several distinct pathways. If protonation of the enamide occurs first, the resulting iminium ion can be readily captured by a wide variety of nucleophiles, including alkenes and alkynes. These aza-Prins cyclizations have potential applications in the synthesis of natural alkaloids, as exemplified by She’s total synthesis of (−)-dihydrolycopodine and (−)-lycopodine [17]. These Lycopodium alkaloids have long been valued in traditional Chinese medicine for their therapeutic effects on skin disorders and as analgesics [18]. Preliminary biological evaluations also suggest their antipyretic and anticholinesterase activities [19]. As a prominent member of the lycopodine-type alkaloids, lycopodine features a characteristic tetracyclic structure with a bridged cyclohexanone. To address the challenges associated with constructing the complex ring systems of this structure, She and co-workers devised an intramolecular aza-Prins cyclization strategy to form both the bridge ring and the N-hetero quaternary center in a single step. As depicted in Scheme 1, key enamide 1 was prepared from (R)-pulegone in 6 steps. In the presence of the weak acid H3PO4, protonation of 1 generates a stabilized iminium ion 2, which then undergoes a 6-exo-trig cyclization to deliver 4 after hydration of cation 3. Notably, the terminal alkene remains intact during this process, and the initial protonation proceeds with full stereocontrol, rendering this transformation both highly chemo- and diastereoselective. From the cyclization result, it is presumed that the higher nucleophilicity of the alkyne functionality over the terminal alkene and the conformational strain of forming a bridge[3.2.1]bicycle might be responsible for a selective 6-exo-trig cyclization.
![[1860-5397-21-81-i1]](/bjoc/content/inline/1860-5397-21-81-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Total synthesis of (−)-dihydrolycopodine and (−)-lycopodine.
Scheme 1: Total synthesis of (−)-dihydrolycopodine and (−)-lycopodine.
From tricyclic compound 4, anti-Markovnikov oxidation catalyzed by palladium led to the formation of aldehyde 5. When treated with p-TsOH, the intramolecular aldol condensation of 5 provided the tetracyclic α,β-unsaturated enone 6 in 57% yield. Subsequent catalytic hydrogenation using Pd/C conditions delivered the hydrogen to the alkene from the less hindered face, producing ketone 7 with high diastereoselectivity. Final reduction of both the amide and ketone groups completed the total synthesis of (−)-dihydrolycopodine, which could then be further oxidized to (−)-lycopodine. The entire synthetic route hinges upon the development of a sterically congested aza-Prins cyclization, enabled by the presence of the enamide and its neighboring alkyne. Building on this strategy, the authors also accomplished the total synthesis of (−)-lycospidine A in only 10 steps [20], another Lycopodium alkaloid with a truncated tetracyclic skeleton and distinct oxidation levels, further highlighting the versatility and efficiency of the enamide aza-Prins approach.
Cyclization/isomerization – collective total synthesis of fawcettimine-type alkaloids
The bicyclic decahydroquinoline enamide motif can serve as a versatile precursor to access different types of tricyclic N-heterocycles. As demonstrated in the above work from She’s group, the aza-Prins cyclization renders the α-position of enamide to be an active cyclization site, with the alkyne tether acting as the nucleophile. Since it is well-established that alkynes, when activated by transition metals such as gold or platinum, can also function as electrophiles, modulating the reactivity of the decahydroquinoline enamide motif to enable an enamide–alkyne cycloisomerization is also feasible. In this case, the initial nucleophilic cyclization of the enamide is followed by isomerization, shifting the cyclization site from the α- to the β-position of the enamide, resulting in the formation of a fused triangular ring system rather than a bridged tricycle. Building on this strategy, the same research group developed a divergent synthetic route that culminated in the concise and collective total synthesis of a series of fawcettimine-type Lycopodium alkaloids (Scheme 2) [21], which are well-known for their potent acetylcholinesterase (AChE) inhibitory activities [18].
![[1860-5397-21-81-i2]](/bjoc/content/inline/1860-5397-21-81-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Collective total synthesis of fawcettimine-type alkaloids.
Scheme 2: Collective total synthesis of fawcettimine-type alkaloids.
In the presence of a catalytic amount of PPh3AuCl and AgSbF6, the enamide–alkyne cycloisomerization of bromo-substituted alkyne 8 proceeded via a 5-endo-dig cyclization to afford tricyclic compound 10 through the formation of iminium intermediate 9. The azepane ring was then constructed via an intramolecular reductive Heck reaction from vinyl bromide 10 with exclusive regioselectivity. Considering the strain of forming the 7-membered ring, this highly efficient 7-endo-trig (vs 6-exo-trig) transannular Heck cyclization reaction was remarkable to be realized in a regioselective manner. From tetracyclic compound 11, a one-pot facial and regioselective hydroboration/amide reduction followed by oxidation produced (+)-lycoposerramine Q, which was then converted to (+)-fawcettidine by Ley oxidation. Alternatively, hydroboration of 11 in mild conditions without the reduction of amide-generated ketone 12 after a subsequent Dess–Martin oxidation. Upon treatment of 12 with Co(acac)2 and PhSiH3 in iPrOH at 80 °C, the Mukaiyama hydration of enamide delivered hemiaminal 13. Despite the incorrect configuration of the newly formed hydroxy group, it is considered inconsequential due to the reversibility of hemiaminal. Consequently, further reduction of the amide could complete the total synthesis of (+)-fawcettimine with in situ adjustment of the hemiaminal configuration.
The incorrect configuration observed in the Mukaiyama hydration also inspired the authors to develop a fragmentation process for the total synthesis of (−)-phlegmariurine B. A one-pot epoxidation/nucleophilic epoxide opening introduced both a hydroxy group and a chloride across the cyclopentene, producing 14 in 57% yield. After oxidation of alcohol 14 to ketone 15, the Mukaiyama hydration then triggered a Grob fragmentation process of hemiaminal 16 and afforded the imide compound 17. Final regioselective reduction of one of the two carbonyls on the imide completed the synthesis of (−)-phlegmariurine B.
Annulation
Total syntheses of cephalotaxine and cephalezomine H
The [2 + 3] annulation of enamides is a relatively underexplored reaction, particularly in the context of total synthesis. Its synthetic potential remains to be fully excavated, as it offers a modular approach for disassembling molecules into segments of comparable sizes. Recently, Fan's group reported the development of this annulation and applied it in the divergent total synthesis of Cephalotaxus alkaloids (Scheme 3) [22], including cephalotaxine whose ester, homoharringtonine, has been listed as an approved FDA drug for the treatment of chronic myeloid leukemia [23]. In their elegant study, an Au-catalyzed [2 + 3] annulation was utilized to transform enamine 18 and propargyl ester 19 into 1-azaspiro[4.4]nonane 20 with high diastereoselectivity. Notably, the combination of an N-heterocyclic carbene gold catalyst and a silver salt AgSbF6 was found to be essential in guaranteeing the reactivity of the alkyne partner, probably due to the formation of a more acidic cationic gold complex. Following this annulation, reduction of the amide in 20, catalytic hydrogenation of the alkene and the N-benzyl group, and subsequent nitrogen acylation yielded chloride 21 in a 42% total yield, setting the stage for the Witkop photocyclization. This transformation was carried out using a high-pressure mercury vapor lamp to afford benzazepine 22, completing the construction of the pentacyclic framework of the natural product. Subsequent functional group manipulations, including the Chugaev elimination of the hydroxy group on the cyclopentane ring, dihydroxylation, and oxidation of the diol to a diketone, produced intermediate 25 in its enol form. From this common intermediate, regioselective etherification at the less hindered position formed an enol ether. Final reduction of both the amide and the ketone using alane completed the total synthesis of cephalotaxine. Similarly, diastereoselective reduction of 25 with KBH4 followed by alane reduction provided another alkaloid cephalezomine H.
![[1860-5397-21-81-i3]](/bjoc/content/inline/1860-5397-21-81-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Total syntheses of cephalotaxine and cephalezomine H.
Scheme 3: Total syntheses of cephalotaxine and cephalezomine H.
Collective total syntheses of Cephalotaxus alkaloids
The cyclopentane ring in most Cephalotaxus alkaloids is characterized by the highest oxidation state within the pentacyclic framework. Installation of this cycle with suitable functional handles usually forms the key strategy of numerous total syntheses of these alkaloids [24-26]. Building upon earlier work, the same research group further advanced this approach by developing a Rh-catalyzed asymmetric [2 + 3] annulation of tertiary enamides with enoldiazoacetates, enabling highly efficient total syntheses of Cephalotaxus alkaloids (Scheme 4) [27]. In their recent study, the homopiperonyl alcohol 26 was transformed into tricyclic enamide 28 in five steps in a decagram scale. As no column chromatography was required during this process, the synthetic route is highly practical. The enantioselective annulation of tertiary enamide 28 with enoldiazoacetate 29 was then explored under the catalysis of a chiral dirhodium catalyst. While Doyle and co-workers had previously reported an elegant [2 + 3] cycloaddition of secondary enecarbamates [28], the extension of this reaction to enamides lacking an N–H group is a notable advancement. After extensive optimization, the chiral dirhodium catalyst cat. 1 was found to be most capable in terms of both stereocontrol and efficiency. The use of 0.4% amount of cat. 1 provided adduct 30 in 72% yield with 92% enantioselectivity, and the reaction could be scaled up to decagrams. Subsequent decarboxylation and recrystallization of the resulting ketone 31 yielded an enantiopure product (99% ee), which serves as a versatile intermediate for the divergent total synthesis of several Cephalotaxus alkaloids.
![[1860-5397-21-81-i4]](/bjoc/content/inline/1860-5397-21-81-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Collective total syntheses of Cephalotaxus alkaloids.
Scheme 4: Collective total syntheses of Cephalotaxus alkaloids.
The α-hydroxylation of cyclopentanone, followed by amide reduction and methanol elimination in one-pot, produced (−)-cephalotaxine in 9 steps. Alternatively, Riley SeO2 oxidation of 31, benzylic bromination/hydrolyzation, facial selective ketone reduction, and epoxidation delivered compound 33 with the required oxidation level of the cyclopentane ring. In the final stages, Meinwald rearrangement/hemiketalization in a step-wise procedure, followed by amide reduction, completed the total synthesis of (−)-cephalotine B. Alternatively, after benzylic oxidation, the Meinwald rearrangement/aldol reaction gave rise to the bridge cyclic intermediate 35, which can finally be converted into both (−)-fortuneicyclidin A and (−)-fortuneicyclidin B.
Polycyclization
Cyclization/Pictet–Spengler reaction
The hexahydropyrrolo[2,1-a]isoquinoline or tetrahydropyrrolo[2,1-a]isoquinolin-3(2H)-one framework is a pivotal core structure among various pyrrolo[2,1-a]isoquinoline alkaloids, exemplified by (+)-crispine, annosqualine, and erysotramidine, among others (Scheme 5) [29]. These bioactive alkaloids exhibit a broad spectrum of biological activities, including antitumor, antibacterial, antiviral, and antioxidizing properties. Previous synthetic strategies for these molecules typically rely on multi-step procedures to assemble the tricyclic core. However, the direct catalytic enantioselective formation of this scaffold from a linear precursor remains underexplored, despite the potential for such a tandem reaction to provide a more efficient route to these complex structures. In 2016, Wang and co-workers indigenously designed and developed a cyclization/Pictet–Spengler reaction cascade, leveraging the nucleophilicity of the tertiary enamides and the electrophilicity of the resulting acyliminium [30]. Unlike the monocyclization, which involves deprotonation of the acyliminium ion, the success of this polycyclization relies on the interception of the acyliminium ion by an aryl nucleophile, resulting in the formation of N-heterocyclic fused[6,6,5]tricycles. Optimization studies identified the tetraphenyl-substituted PyBox ligand L1 as particularly effective in controlling the stereochemistry of the polycyclization, yielding high enantioselectivity for most substrates. As illustrated in Scheme 5, tertiary enamides with a tethered electron-rich arene could undergo cyclization to form products in high yields and excellent enantioselectivities. Notably, only a single diastereomer was produced in each case. The single-crystal X-ray crystallography revealed a cis-configuration for both the alkene and ketone substituents on the enamide, indicating that the intramolecular attack of the electron-rich arene on the acyliminium ion occurs from the Si-face. This stereochemical outcome is attributed to the steric discrepancy of the phenyl or tert-butyl group and the hydroxy group. The resulting tricyclic products could be further elaborated by elimination or amide reduction to yield hexahydropyrrolo[2,1-a]isoquinoline or tetrahydropyrrolo[2,1-a]isoquinolin-3(2H)-one frameworks, characteristic of alkaloids such as crispine analogs and erysotramidine.
![[1860-5397-21-81-i5]](/bjoc/content/inline/1860-5397-21-81-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Asymmetric tandem cyclization/Pictet–Spengler reaction of tertiary enamides.
Scheme 5: Asymmetric tandem cyclization/Pictet–Spengler reaction of tertiary enamides.
Building on their previous work on cyclization/Pictet–Spengler reaction, the same group further designed cyclopentanone derived tertiary enamides as cyclization precursors (Scheme 6). The analogous polycyclization generated a tetracyclic N-heterocycle with three continuous stereogenic centers, one of them being an aza-quaternary carbon [31]. The resulting fused ring-system structurally resembles the nucleus of erysotramidine alkaloids, though it features a truncated cyclopentane rather than the characteristic cyclohexane or cyclohexene. In their optimization studies, the authors found the sequential catalysis of a chiral binol–Ti complex and BF3·Et2O to be the most efficient, providing products 39 in high yields with excellent diastereo- and enantioselectivities. The substituent on the enamide could be varied from aryl to tert-butyl groups, though the terminating aryl group still necessitates an electron-rich arene. As was found in their previous work, the steric hindrance of the phenyl or tert-butyl group was supposed to be responsible for the excellent diastereoselectivity observed in the second cyclization process. In their later studies, the authors also found cyclohexanone-derived tertiary enamides to be viable substrates for the polycyclization [32], affording erythrinane core skeletons in high yields. However, in these cases, the use of a chiral Cr(III)(salen)Cl complex in combination with InCl3 was necessary to maintain a high level of stereocontrol.
![[1860-5397-21-81-i6]](/bjoc/content/inline/1860-5397-21-81-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Tandem cyclization/Pictet–Spengler reaction for the synthesis of chiral tetracyclic compounds.
Scheme 6: Tandem cyclization/Pictet–Spengler reaction for the synthesis of chiral tetracyclic compounds.
Total synthesis of (−)-cephalocyclidin A
The bicyclic and tricyclic N-heterocycles without a fused arene are essential core structures in a wide array of alkaloids. A notable example is (−)-cephalocyclidin A, a cytotoxic pentacyclic cephalotaxus alkaloid [33,34]. Although the molecular structure contains a benzo-bridge ring system, disconnection of this bridge reveals a critical tricyclic N-heterocycle. To efficiently synthesize this tricycle, polycyclization of tertiary enamides is employed. Inspired by Wang’s earlier work, Zhang and Tu’s group developed a tandem cyclization/Mannich reaction to construct this architecture [35]. However, unlike the electron-rich arenes, the use of silyl enol ethers to terminate the second cyclization of the acyliminium intermediate would meet challenges associated with the instability of enolate derivatives. In their recent study, they successfully developed such a polycyclization taking advantage of a novel spiropyrroline-derived oxazole (SPDO) ligand (L3). As shown in Scheme 7, one-pot condensation of primary amine 40, β-silyl substituted cyclopentanone 41, and acyl chloride 42 produced enamide 43. The polycyclization then took place under the catalysis of Cu(OTf)2/L3 and In(OTf)3, delivering tricyclic product 44 in high yield with excellent enantioselectivity. Despite formation of multiple diastereomers due to the presence of silyl and aldehyde groups on the tricycle, it is inconsequential as these groups are either removed or oxidized in subsequent steps. After adjustment of the oxidation levels, the cyclopentenone 45 obtained was subjected to an intramolecular Giese reaction, producing 46 with establishment of the bridge cycle [36,37]. The excellent diastereoselectivity in this radical cyclization was further rationalized by DFT calculations, which suggests an energy discrepancy of the hydrogen atom transfer process from different faces of the resulting α-hydroxyl radical. Final reduction of the ketone and amide followed by deprotection completed the total synthesis, giving rise to (−)-cephalocyclidin A in 10 steps from known compounds.
![[1860-5397-21-81-i7]](/bjoc/content/inline/1860-5397-21-81-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Total synthesis of (−)-cephalocyclidin A.
Scheme 7: Total synthesis of (−)-cephalocyclidin A.
Conclusion
In summary, the perception of enamides as stable chemical entities with limited utilities in organic synthesis has evolved, and these compounds are now widely used in various cyclization reactions that play a pivotal role in the total synthesis of natural alkaloids. The nucleophilicity of enamides and the electrophilicity of the resulting acyliminium intermediates can be strategically manipulated in numerous ways to design cyclization and annulation reactions. Notably, these reactions – particularly tandem processes – are highly effective in constructing both fused and bridged ring systems, offering valuable new tools for chemical synthesis. Future advancements in the field could involve further applications of enamide cyclizations with other nucleophiles or in combination with other reaction patterns, potentially opening new avenues for the total synthesis of natural products.
Data Availability Statement
Data sharing is not applicable as no new data was generated or analyzed in this study.
References
-
Stork, G.; Terrell, R.; Szmuszkovicz, J. J. Am. Chem. Soc. 1954, 76, 2029–2030. doi:10.1021/ja01636a103
Return to citation in text: [1] -
Stork, G.; Landesman, H. K. J. Am. Chem. Soc. 1956, 78, 5129–5130. doi:10.1021/ja01600a088
Return to citation in text: [1] -
Stork, G.; Brizzolara, A.; Landesman, H.; Szmuszkovicz, J.; Terrell, R. J. Am. Chem. Soc. 1963, 85, 207–222. doi:10.1021/ja00885a021
Return to citation in text: [1] -
Poulsen, T. B. Acc. Chem. Res. 2021, 54, 1830–1842. doi:10.1021/acs.accounts.0c00851
Return to citation in text: [1] -
Carbery, D. R. Org. Biomol. Chem. 2008, 6, 3455–3460. doi:10.1039/b809319a
Return to citation in text: [1] -
Beltran, F.; Miesch, L. Synthesis 2020, 52, 2497–2511. doi:10.1055/s-0040-1707403
Return to citation in text: [1] -
Gopalaiah, K.; Kagan, H. B. Chem. Rev. 2011, 111, 4599–4657. doi:10.1021/cr100031f
Return to citation in text: [1] -
Cabré, A.; Verdaguer, X.; Riera, A. Chem. Rev. 2022, 122, 269–339. doi:10.1021/acs.chemrev.1c00496
Return to citation in text: [1] -
Matsubara, R.; Kobayashi, S. Acc. Chem. Res. 2008, 41, 292–301. doi:10.1021/ar700098d
Return to citation in text: [1] -
Dake, G. R. Synlett 2012, 23, 814–824. doi:10.1055/s-0031-1290351
Return to citation in text: [1] -
Gigant, N.; Chausset‐Boissarie, L.; Gillaizeau, I. Chem. – Eur. J. 2014, 20, 7548–7564. doi:10.1002/chem.201402070
Return to citation in text: [1] -
Wang, M.-X. Chem. Commun. 2015, 51, 6039–6049. doi:10.1039/c4cc10327k
Return to citation in text: [1] -
Tong, S.; Wang, M.-X. Synlett 2021, 32, 1419–1427. doi:10.1055/a-1352-6358
Return to citation in text: [1] -
Varlet, T.; Masson, G. Chem. Commun. 2021, 57, 4089–4105. doi:10.1039/d1cc00590a
Return to citation in text: [1] -
Bouchet, D.; Varlet, T.; Masson, G. Acc. Chem. Res. 2022, 55, 3265–3283. doi:10.1021/acs.accounts.2c00540
Return to citation in text: [1] -
Courant, T.; Dagousset, G.; Masson, G. Synthesis 2015, 47, 1799–1856. doi:10.1055/s-0034-1378706
Return to citation in text: [1] [2] -
Ma, D.; Zhong, Z.; Liu, Z.; Zhang, M.; Xu, S.; Xu, D.; Song, D.; Xie, X.; She, X. Org. Lett. 2016, 18, 4328–4331. doi:10.1021/acs.orglett.6b02072
Return to citation in text: [1] -
Ma, X.; Gang, D. R. Nat. Prod. Rep. 2004, 21, 752–772. doi:10.1039/b409720n
Return to citation in text: [1] [2] -
Ortega, M. G.; Agnese, A. M.; Cabrera, J. L. Phytomedicine 2004, 11, 539–543. doi:10.1016/j.phymed.2003.07.006
Return to citation in text: [1] -
Xu, S.; Zhang, J.; Ma, D.; Xu, D.; Xie, X.; She, X. Org. Lett. 2016, 18, 4682–4685. doi:10.1021/acs.orglett.6b02322
Return to citation in text: [1] -
He, F.; Feng, S.; Zhao, Y.; Shi, H.; Duan, X.; Li, H.; Xie, X.; She, X. Angew. Chem., Int. Ed. 2022, 61, e202205439. doi:10.1002/anie.202205439
Return to citation in text: [1] -
Ma, X.-Y.; An, X.-T.; Zhao, X.-H.; Du, J.-Y.; Deng, Y.-H.; Zhang, X.-Z.; Fan, C.-A. Org. Lett. 2017, 19, 2965–2968. doi:10.1021/acs.orglett.7b01202
Return to citation in text: [1] -
Kantarjian, H. M.; O'Brien, S.; Cortes, J. Clin. Lymphoma, Myeloma Leuk. 2013, 13, 530–533. doi:10.1016/j.clml.2013.03.017
Return to citation in text: [1] -
Weinreb, S. M.; Semmelhack, M. F. Acc. Chem. Res. 1975, 8, 158–164. doi:10.1021/ar50089a003
Return to citation in text: [1] -
Chen, Y.; Li, W.-D. Z. Chin. J. Org. Chem. 2017, 37, 1885–1902. doi:10.6023/cjoc201705025
Return to citation in text: [1] -
Jeon, H. Asian J. Org. Chem. 2021, 10, 3052–3067. doi:10.1002/ajoc.202100543
Return to citation in text: [1] -
An, X.-T.; Ge, X.-M.; Liu, X.-Y.; Yang, Y.-H.; Zhao, X.-H.; Ma, X.-Y.; Peng, C.; Fan, Y.-J.; Qin, Y.; Fan, C.-A. J. Am. Chem. Soc. 2023, 145, 9233–9241. doi:10.1021/jacs.3c01572
Return to citation in text: [1] -
Deng, Y.; Yglesias, M. V.; Arman, H.; Doyle, M. P. Angew. Chem., Int. Ed. 2016, 55, 10108–10112. doi:10.1002/anie.201605438
Return to citation in text: [1] -
Yang, Y.-L.; Chang, F.-R.; Wu, Y.-C. Helv. Chim. Acta 2004, 87, 1392–1399. doi:10.1002/hlca.200490127
Return to citation in text: [1] -
Xu, X.-M.; Zhao, L.; Zhu, J.; Wang, M.-X. Angew. Chem., Int. Ed. 2016, 55, 3799–3803. doi:10.1002/anie.201600119
Return to citation in text: [1] -
Zhen, L.; Tong, S.; Zhu, J.; Wang, M.-X. Chem. – Eur. J. 2020, 26, 401–405. doi:10.1002/chem.201904596
Return to citation in text: [1] -
Zhen, L.; Tong, S.; Zhu, J.; Wang, M.-X. J. Org. Chem. 2020, 85, 13211–13219. doi:10.1021/acs.joc.0c01992
Return to citation in text: [1] -
Kobayashi, J.; Yoshinaga, M.; Yoshida, N.; Shiro, M.; Morita, H. J. Org. Chem. 2002, 67, 2283–2286. doi:10.1021/jo016327f
Return to citation in text: [1] -
Kim, J. H.; Jeon, H.; Park, C.; Park, S.; Kim, S. Angew. Chem., Int. Ed. 2021, 60, 12060–12065. doi:10.1002/anie.202101766
Return to citation in text: [1] -
Zhuang, Q.-B.; Tian, J.-R.; Lu, K.; Zhang, X.-M.; Zhang, F.-M.; Tu, Y.-Q.; Fan, R.; Li, Z.-H.; Zhang, Y.-D. J. Am. Chem. Soc. 2023, 145, 26550–26556. doi:10.1021/jacs.3c11178
Return to citation in text: [1] -
Okada, K.; Ueda, H.; Tokuyama, H. Org. Biomol. Chem. 2022, 20, 5943–5947. doi:10.1039/d2ob00274d
Return to citation in text: [1] -
Okada, K.; Ojima, K.-i.; Ueda, H.; Tokuyama, H. J. Am. Chem. Soc. 2023, 145, 16337–16343. doi:10.1021/jacs.3c05811
Return to citation in text: [1]
| 1. | Stork, G.; Terrell, R.; Szmuszkovicz, J. J. Am. Chem. Soc. 1954, 76, 2029–2030. doi:10.1021/ja01636a103 |
| 2. | Stork, G.; Landesman, H. K. J. Am. Chem. Soc. 1956, 78, 5129–5130. doi:10.1021/ja01600a088 |
| 3. | Stork, G.; Brizzolara, A.; Landesman, H.; Szmuszkovicz, J.; Terrell, R. J. Am. Chem. Soc. 1963, 85, 207–222. doi:10.1021/ja00885a021 |
| 9. | Matsubara, R.; Kobayashi, S. Acc. Chem. Res. 2008, 41, 292–301. doi:10.1021/ar700098d |
| 10. | Dake, G. R. Synlett 2012, 23, 814–824. doi:10.1055/s-0031-1290351 |
| 11. | Gigant, N.; Chausset‐Boissarie, L.; Gillaizeau, I. Chem. – Eur. J. 2014, 20, 7548–7564. doi:10.1002/chem.201402070 |
| 12. | Wang, M.-X. Chem. Commun. 2015, 51, 6039–6049. doi:10.1039/c4cc10327k |
| 13. | Tong, S.; Wang, M.-X. Synlett 2021, 32, 1419–1427. doi:10.1055/a-1352-6358 |
| 14. | Varlet, T.; Masson, G. Chem. Commun. 2021, 57, 4089–4105. doi:10.1039/d1cc00590a |
| 15. | Bouchet, D.; Varlet, T.; Masson, G. Acc. Chem. Res. 2022, 55, 3265–3283. doi:10.1021/acs.accounts.2c00540 |
| 23. | Kantarjian, H. M.; O'Brien, S.; Cortes, J. Clin. Lymphoma, Myeloma Leuk. 2013, 13, 530–533. doi:10.1016/j.clml.2013.03.017 |
| 7. | Gopalaiah, K.; Kagan, H. B. Chem. Rev. 2011, 111, 4599–4657. doi:10.1021/cr100031f |
| 8. | Cabré, A.; Verdaguer, X.; Riera, A. Chem. Rev. 2022, 122, 269–339. doi:10.1021/acs.chemrev.1c00496 |
| 24. | Weinreb, S. M.; Semmelhack, M. F. Acc. Chem. Res. 1975, 8, 158–164. doi:10.1021/ar50089a003 |
| 25. | Chen, Y.; Li, W.-D. Z. Chin. J. Org. Chem. 2017, 37, 1885–1902. doi:10.6023/cjoc201705025 |
| 26. | Jeon, H. Asian J. Org. Chem. 2021, 10, 3052–3067. doi:10.1002/ajoc.202100543 |
| 5. | Carbery, D. R. Org. Biomol. Chem. 2008, 6, 3455–3460. doi:10.1039/b809319a |
| 6. | Beltran, F.; Miesch, L. Synthesis 2020, 52, 2497–2511. doi:10.1055/s-0040-1707403 |
| 4. | Poulsen, T. B. Acc. Chem. Res. 2021, 54, 1830–1842. doi:10.1021/acs.accounts.0c00851 |
| 22. | Ma, X.-Y.; An, X.-T.; Zhao, X.-H.; Du, J.-Y.; Deng, Y.-H.; Zhang, X.-Z.; Fan, C.-A. Org. Lett. 2017, 19, 2965–2968. doi:10.1021/acs.orglett.7b01202 |
| 20. | Xu, S.; Zhang, J.; Ma, D.; Xu, D.; Xie, X.; She, X. Org. Lett. 2016, 18, 4682–4685. doi:10.1021/acs.orglett.6b02322 |
| 17. | Ma, D.; Zhong, Z.; Liu, Z.; Zhang, M.; Xu, S.; Xu, D.; Song, D.; Xie, X.; She, X. Org. Lett. 2016, 18, 4328–4331. doi:10.1021/acs.orglett.6b02072 |
| 21. | He, F.; Feng, S.; Zhao, Y.; Shi, H.; Duan, X.; Li, H.; Xie, X.; She, X. Angew. Chem., Int. Ed. 2022, 61, e202205439. doi:10.1002/anie.202205439 |
| 16. | Courant, T.; Dagousset, G.; Masson, G. Synthesis 2015, 47, 1799–1856. doi:10.1055/s-0034-1378706 |
| 16. | Courant, T.; Dagousset, G.; Masson, G. Synthesis 2015, 47, 1799–1856. doi:10.1055/s-0034-1378706 |
| 19. | Ortega, M. G.; Agnese, A. M.; Cabrera, J. L. Phytomedicine 2004, 11, 539–543. doi:10.1016/j.phymed.2003.07.006 |
| 29. | Yang, Y.-L.; Chang, F.-R.; Wu, Y.-C. Helv. Chim. Acta 2004, 87, 1392–1399. doi:10.1002/hlca.200490127 |
| 27. | An, X.-T.; Ge, X.-M.; Liu, X.-Y.; Yang, Y.-H.; Zhao, X.-H.; Ma, X.-Y.; Peng, C.; Fan, Y.-J.; Qin, Y.; Fan, C.-A. J. Am. Chem. Soc. 2023, 145, 9233–9241. doi:10.1021/jacs.3c01572 |
| 28. | Deng, Y.; Yglesias, M. V.; Arman, H.; Doyle, M. P. Angew. Chem., Int. Ed. 2016, 55, 10108–10112. doi:10.1002/anie.201605438 |
| 35. | Zhuang, Q.-B.; Tian, J.-R.; Lu, K.; Zhang, X.-M.; Zhang, F.-M.; Tu, Y.-Q.; Fan, R.; Li, Z.-H.; Zhang, Y.-D. J. Am. Chem. Soc. 2023, 145, 26550–26556. doi:10.1021/jacs.3c11178 |
| 36. | Okada, K.; Ueda, H.; Tokuyama, H. Org. Biomol. Chem. 2022, 20, 5943–5947. doi:10.1039/d2ob00274d |
| 37. | Okada, K.; Ojima, K.-i.; Ueda, H.; Tokuyama, H. J. Am. Chem. Soc. 2023, 145, 16337–16343. doi:10.1021/jacs.3c05811 |
| 32. | Zhen, L.; Tong, S.; Zhu, J.; Wang, M.-X. J. Org. Chem. 2020, 85, 13211–13219. doi:10.1021/acs.joc.0c01992 |
| 33. | Kobayashi, J.; Yoshinaga, M.; Yoshida, N.; Shiro, M.; Morita, H. J. Org. Chem. 2002, 67, 2283–2286. doi:10.1021/jo016327f |
| 34. | Kim, J. H.; Jeon, H.; Park, C.; Park, S.; Kim, S. Angew. Chem., Int. Ed. 2021, 60, 12060–12065. doi:10.1002/anie.202101766 |
| 30. | Xu, X.-M.; Zhao, L.; Zhu, J.; Wang, M.-X. Angew. Chem., Int. Ed. 2016, 55, 3799–3803. doi:10.1002/anie.201600119 |
| 31. | Zhen, L.; Tong, S.; Zhu, J.; Wang, M.-X. Chem. – Eur. J. 2020, 26, 401–405. doi:10.1002/chem.201904596 |
© 2025 Mi et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.