Abstract
Borrelidin, a naturally occurring antibiotic, has attracted considerable interest due to its diverse biological activities and complex molecular architecture. Although extensive research has explored its pharmacological properties and various synthetic approaches, significant challenges remain in the efficient synthesis of borrelidin and its analogs. Existing literature largely focuses on total synthesis, bioactivity, and structural modifications, leaving a notable gap in fragment-focused synthesis, particularly for its intricate substructures. This review seeks to address this gap by offering a detailed examination of borrelidin fragment synthesis, highlighting key challenges and innovative strategies involved. By pinpointing unresolved synthetic hurdles, this work advocates for a fragment-focused approach as a crucial step toward advancing borrelidin research and expanding its potential applications.

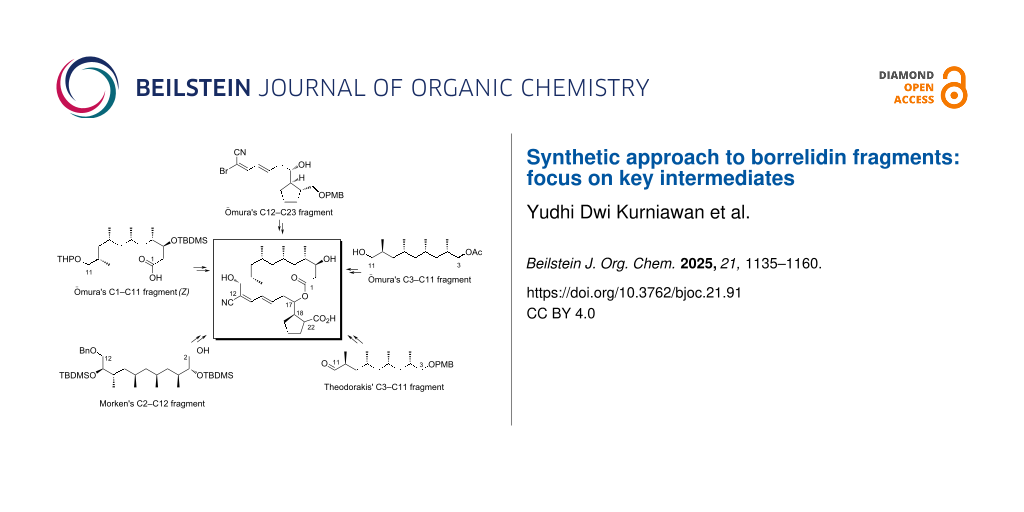
Graphical Abstract
Introduction
Borrelidin (Figure 1), a distinctive 18-membered ring macrolide, was first isolated from Streptomyces rochei by Berger et al. in 1949 [1]. This antibiotic, also known for its anti-Borrelia activity and ability to enhance penicillin’s effects, was structurally elucidated in 1967 by Keller-Schierlein [2]. Since then, borrelidin has been recognized for its potential as a cancer therapeutic [3-5], exhibiting anti-inflammatory [6], anti-angiogenic [7-9], antimicrobial [10], antifungal [11-13], and antimalarial activities [14,15]. Since then, borrelidin has been recognized for its potential as a cancer therapeutic, exhibiting anti-inflammatory, anti-angiogenic, antimicrobial, antifungal, and antimalarial activities. While Keller-Schierlein first determined its chemical structure, Anderson later confirmed its absolute configuration using X-ray crystallography [16].
![[1860-5397-21-91-1]](/bjoc/content/figures/1860-5397-21-91-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structure of borrelidin (1).
Figure 1: Chemical structure of borrelidin (1).
During the screening of a marine or hypersaline environment natural products library to identify potent drugs, a putatively novel metabolite co-produced with borrelidin was discovered, expanding the potential for new borrelidin derivatives. This led to the formation of the so-called “borrelidin family” (Table 1), with derivatives named alphabetically starting from the original borrelidin 1, designated as borrelidin A (Table 1, entry 1). A rare nitrile moiety at C12 of the macrolide ring in borrelidin A is present in most members of the borrelidin family (Table 1, entries 4, 5, 7–11, 13–16, and 18), except for borrelidin B, N-acetylborrelidin B, borrelidin CR2, borrelidin I, and borrelidin N (Table 1, entries 2, 3, 6, 12, and 17). Borrelidin B (Table 1, entry 2), a tetrahydroborrelidin derivative with an aminomethyl group instead of the nitrile in position 12 of the macrolide, was isolated from the marine-derived Streptomyces sp. RL09-241-NTF-B strain [17]. The discovery of borrelidin B, along with the novel introduction of an N-acetyl group in borrelidin B (N-acetylborrelidin B, Table 1, entry 3), expanded the borrelidin family. N-Acetylborrelidin B was obtained from Streptomyces mutabilis sp. MII [18], a marine strain from the Red Sea (Egypt), further enriching the borrelidin series.
A halophilic actinomycete strain (HYJ128) from Jeung-do Island (Shinan-gun, Jeollanamdo, Korea), belonging to the genus Nocardiopsis, inhabits a hypersaline saltern and was found to produce a series of new polyketide-derived macrolides with hydroxy groups at C20 or C7, identified as borrelidins C–E (Table 1, entries 4, 7, and 8) [19]. Borrelidins CR1 and CR2 (Table 1, entries 5 and 6), amide-containing congeners, were also isolated through bioassay-guided fractionation and purification of marine microorganisms from Costa Rica [20]. Borrelidin CR1 (Table 1, entry 5) was also discovered in Streptomyces olivaceus SCSIO LO13 from Onchidium sp. (South China Sea), alongside other borrelidin derivatives [21]. Borrelidins F–I (Table 1, entries 9–12) were obtained from Streptomyces rochei SCSIO ZJ89, a strain from mangrove-derived sediment in Yalongwan, China [22]. The C14–C15 olefin geometry of borrelidins G and H (Table 1, entries 10 and 11) exhibited a Z-configuration, as confirmed by NOESY correlations. Borrelidins J–L (Table 1, entries 13–15) were isolated from an endophytic Streptomyces sp. NA06554 from Aster tataricus in Aba County, Sichuan Province, China, along with other borrelidin derivatives, including borrelidin E (Table 1, entry 8) and 12-desnitrile-12-carboxyborrelidin (Table 1, entry 19) [23]. The latter compound, found in both endophytic bacteria and as a product of borrelidin biosynthesis, contributed to the understanding of nitrile formation [24].
Table 1: Summary of the borrelidin family: structures, sources, and related literature.
| Entry | Name | Structure | Source | Ref. |
| 1 | borrelidin A |
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-91-i25.svg?max-width=637&scale=1.0)
1 |
first encounter was isolated from Streptomyces sp. and found to be produced along with other members of the borrelidin family | [1] |
| 2 | borrelidin B |
![[Graphic 2]](/bjoc/content/inline/1860-5397-21-91-i26.svg?max-width=637&scale=1.0)
2 |
Streptomyces sp. RL09-241-NTF-B | [17] |
| 3 | N-acetylborrelidin B |
![[Graphic 3]](/bjoc/content/inline/1860-5397-21-91-i27.svg?max-width=637&scale=1.0)
3 |
Streptomyces mutabilis sp. MII | [18] |
| 4 | borrelidin C |
![[Graphic 4]](/bjoc/content/inline/1860-5397-21-91-i28.svg?max-width=637&scale=1.0)
4 |
Nocardiopsis sp. | [19] |
| 5 | borrelidin CR1 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-21-91-i29.svg?max-width=637&scale=1.0)
5 |
a terminal unfolded protein response (UPR)-inducing natural extracts (cultivated marine microorganisms)
Onchidium sp. associated Streptomyces olivaceus SCSIO LO13 |
[20,21] |
| 6 | borrelidin CR2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-21-91-i30.svg?max-width=637&scale=1.0)
6 |
a terminal unfolded protein response (UPR)-inducing natural extracts (cultivated marine microorganisms) | [20] |
| 7 | borrelidin D |
![[Graphic 7]](/bjoc/content/inline/1860-5397-21-91-i31.svg?max-width=637&scale=1.0)
7 |
Nocardiopsis sp. | [19] |
| 8 | borrelidin E |
![[Graphic 8]](/bjoc/content/inline/1860-5397-21-91-i32.svg?max-width=637&scale=1.0)
8 |
an endophytic Streptomyces sp. NA06554 from Aster tataricus
marine pulmonated mollusks Onchidium sp. associated Streptomyces olivaceus SCSIO LO13 Nocardiopsis sp. |
[19,21,23] |
| 9 | borrelidin F |
![[Graphic 9]](/bjoc/content/inline/1860-5397-21-91-i33.svg?max-width=637&scale=1.0)
9 |
Streptomyces rochei SCSIO ZJ89 | [22] |
| 10 | borrelidin G |
![[Graphic 10]](/bjoc/content/inline/1860-5397-21-91-i34.svg?max-width=637&scale=1.0)
10 |
Streptomyces rochei SCSIO ZJ89 | [22] |
| 11 | borrelidin H |
![[Graphic 11]](/bjoc/content/inline/1860-5397-21-91-i35.svg?max-width=637&scale=1.0)
11 |
Streptomyces rochei SCSIO ZJ89 | [22] |
| 12 | borrelidin I |
![[Graphic 12]](/bjoc/content/inline/1860-5397-21-91-i36.svg?max-width=637&scale=1.0)
12 |
Streptomyces rochei SCSIO ZJ89 | [22] |
| 13 | borrelidin J |
![[Graphic 13]](/bjoc/content/inline/1860-5397-21-91-i37.svg?max-width=637&scale=1.0)
13 |
an endophytic Streptomyces sp. NA06554 from Aster tataricus collected in Aba County of Sichuan Province, China. | [23] |
| 14 | borrelidin K |
![[Graphic 14]](/bjoc/content/inline/1860-5397-21-91-i38.svg?max-width=637&scale=1.0)
14 |
an endophytic Streptomyces sp. NA06554. from Aster tataricus collected in Aba County of Sichuan Province, China.
marine pulmonated mollusks Onchidium sp. associated Streptomyces olivaceus SCSIO LO13 (Daya Bay, South China Sea) |
[21,23] |
| 15 | borrelidin L |
![[Graphic 15]](/bjoc/content/inline/1860-5397-21-91-i39.svg?max-width=637&scale=1.0)
15 |
an endophytic Streptomyces sp. NA06554from Aster tataricus | [23] |
| 16 | borrelidin M |
![[Graphic 16]](/bjoc/content/inline/1860-5397-21-91-i40.svg?max-width=637&scale=1.0)
16 |
marine pulmonated mollusks Onchidium sp. associated Streptomyces olivaceus SCSIO LO13 (Daya Bay, South China Sea) | [21] |
| 17 | borrelidin N |
![[Graphic 17]](/bjoc/content/inline/1860-5397-21-91-i41.svg?max-width=637&scale=1.0)
17 |
marine pulmonated mollusks Onchidium sp. associated Streptomyces olivaceus SCSIO LO13 (Daya Bay, South China Sea) | [21] |
| 18 | borrelidin O |
![[Graphic 18]](/bjoc/content/inline/1860-5397-21-91-i42.svg?max-width=637&scale=1.0)
18 |
marine pulmonated mollusks Onchidium sp. associated Streptomyces olivaceus SCSIO LO13 (Daya Bay, South China Sea) | [21] |
| 19 |
12-desnitrile-12-
carboxyborrelidin |
![[Graphic 19]](/bjoc/content/inline/1860-5397-21-91-i43.svg?max-width=637&scale=1.0)
19 |
an endophytic Streptomyces sp. NA06554. from Aster tataricus | [23] |
There are five total syntheses of borrelidin in the literature, reported by Morken et al. (2003) [25], Hanessian et al. (2003) [26], Ōmura et al. (2004) [27], Theodorakis et al. (2004) [28], and Ōmura et al. (2007) [29]. Additionally, there are twelve synthetic studies toward borrelidin reported by Haddad et al. (1997) [30], Haddad et al. (1997) [31], Theodorakis et al. (2003) [32], Herber and Breit (2006) [33], Iqbal et al. (2006) [34], Iqbal et al. (2008) [35], Yadav et al. (2009) [36], Minnaard and Madduri (2010) [37], Laschat et al. (2011) [38], Yadav and Yadav (2013) [39], Zhou et al. (2018) [40], and Uguen and Gembus (2019) [41], with seven focusing on strategies to access key fragments of borrelidin, including Ōmura’s C3–C11 fragment [23], Theodorakis’ C3–C11 fragment [33,38], Morken’s C1–C11 fragment [25], and Ōmura’s C1–C11 fragment [36,37,39]. Moreover, several derivatives have been synthesized by Moss et al. (2006) [42], Wilkinson et al. (2006) [43], Sunazuka et al. (2013) [44], Hahn et al. (2014) [45], Laschat et al. (2016) [46], and Huang et al. (2018) [47]. This demonstrates that borrelidin, with its remarkable biological activities and complex structure, remains an attractive target for synthetic organic chemists worldwide.
Several comprehensive reviews on borrelidin have been published, with a strong focus on its synthesis. The first notable comparison of total synthesis methods was conducted by Ōmura in 2005 [48], highlighting four pioneering approaches: Morken (2003), Hanessian (2003), Ōmura (2004), and Theodorakis (2004). This review also included two synthetic studies by Haddad [30,31] and Negishi [49]. In 2011, Darna et al. expanded this scope by incorporating Omura’s 2007 method along with biosynthesis studies and investigations into fragment synthesis, providing a more holistic perspective on borrelidin’s chemical synthesis and biological relevance [50]. However, in-depth works focusing on key fragment optimizations remain limited. Hence, this review aims to address this gap.
Review
Syntheses of borrelidin fragments
Uguen’s approach for constructing Morken’s C2–C12 fragment
In 2019, Uguen and co-workers introduced a strategy to assemble Morken’s C2–C12 intermediate 20 [41]. Their approach utilized iterative base-catalyzed condensation of sulfone compounds with epoxides. As illustrated in Scheme 1, the monoalcohol 20 was prepared via PMB removal of compound 21, which, in turn, was obtained through desulfonylation of compound 22. Compound 22 originated from the condensation of epoxide 23a with sulfone 26, which was produced by desilylation of 25 followed by converting the resulting primary alcohol into a sulfone group. Intermediate 25 was prepared through TBDMS protection and desulfonylation of 24, itself derived from the condensation of epoxide 23b and sulfone 27. The precursor 27 was synthesized from Roche ester 29 via a sequence of steps, including reduction, three-carbon homologation, and enzymatic desymmetrization. An alternative route was also proposed for synthesizing 21 from compound 30, which was derived from ent-29. Notably, epoxides 23a and 23b were obtained via Sharpless epoxidation of (E)-2-butenol.
![[1860-5397-21-91-i1]](/bjoc/content/inline/1860-5397-21-91-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic strategy for Morken’s C2–C12 intermediate 20 as reported by Uguen et al. [41].
Scheme 1: Synthetic strategy for Morken’s C2–C12 intermediate 20 as reported by Uguen et al. [41].
Uguen and co-workers began their synthesis by reducing Roche esters 29 and ent-29 to their respective primary alcohols, 34 and ent-34, after recrystallization from hot hexane (100% ee by chiral phase HPLC, yield not reported). These alcohols were then treated with triphenylphosphine and iodine in the presence of imidazole to yield the iodides 35 and ent-35 (Scheme 2). The iodide intermediates were subsequently reacted with deprotonated diethyl malonate to obtain compounds 36 and ent-36 in 95 and 92% yield, respectively, over two steps from 34 and ent-34. The diols 28 (85%) and ent-28 (90%) were isolated after reduction of their parent malonates 36 and ent-36 using LiAlH4 in ether. Following several experiments with vinyl acetate as the acylating agent, Amano lipase AK (AKL) was identified as the most effective biocatalyst for achieving selective acetylation, converting diol 28 to the monoacetate 37 in 91% yield with >99.4% de (by HPLC). The diacetate byproduct 39 was formed in a small amount (9%). A similar diol desymmetrization of ent-28 was best achieved with Amano lipase PS (PSL), yielding monoacetate ent-37 (de 99.6%, 93% yield) without the formation of diacetate ent-39.
![[1860-5397-21-91-i2]](/bjoc/content/inline/1860-5397-21-91-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of monoacetates 37 and ent-38 by Uguen et al. [41].
Scheme 2: Preparation of monoacetates 37 and ent-38 by Uguen et al. [41].
Compound 37 was tosylated using tosyl chloride in pyridine with the addition of DMAP and the resulting product was treated with LiAlH4 in ether to form alcohol 40 (89%) (Scheme 3). The primary alcohol 40 was then converted to its iodide derivative 42 (89%), from which single crystals were obtained, and its structure was determined unequivocally by XRD crystallography. Iodide 42 was then reacted with sodium phenylsulfinate in DMF to afford the corresponding sulfone 41. However, Uguen found that a more efficient route involved treating alcohol 40 with tosyl chloride in pyridine and DMAP, followed by nucleophilic displacement with sodium thiophenol and oxidation of the resulting sulfide with m-CPBA, yielding sulfone 41 in 85% yield over three steps. This compound was isolated in its pure form as a white solid after recrystallization from ethanol, confirmed by HPLC and NMR. In parallel, the primary alcohol group of ent-38 was protected as a TBDMS ether, and the acetate group was converted to a tosyl ester by hydrolyzing the acetate functionality using potassium carbonate in methanol, followed by reaction with tosyl chloride in pyridine and DMAP. The product, ent-43, was obtained in 79.5% yield over three steps. This compound was reduced using LiAlH4 and treated with TBAF to remove the TBDMS group, yielding alcohol ent-40 in 94% yield. Like 40, iodination of ent-40 gave crystalline product ent-42, and its structure was confirmed by XRD crystallography. The intermediate ent-40 was then subjected to the same tosylation/thiolation/oxidation sequence used for the 40 to 41 conversion, yielding ent-41 in comparable yield (82%). Treatment of both 41 and ent-41 with acidic resin (Amberlyst-15), followed by protection of the resultant primary alcohol with TIPSOTf in the presence of 2,4,6-collidine provided the anticipated compounds 27 (97%) and ent-27 (95%).
![[1860-5397-21-91-i3]](/bjoc/content/inline/1860-5397-21-91-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of sulfones 27 and ent-27 by Uguen et al. [41].
Scheme 3: Preparation of sulfones 27 and ent-27 by Uguen et al. [41].
With 27 and ent-27 in hand, the next step was to perform the coupling of these sulfones with epoxides 23b and 23a, respectively. Following the literature procedure for a similar reaction, using n-butyllithium in the presence of BF3·Et2O at −78 °C, the coupling reaction unfortunately resulted in the decomposition of the reactants (Scheme 4). The authors hypothesized that the failure may have been due to the low reactivity of sulfones 27 and ent-27 under the reaction conditions. Additionally, treating ent-42 with excess tert-butyllithium to form the corresponding lithiated derivative and reacting it with epoxide 23a, both with or without BF3·OEt2, also led to unsatisfying results, with reactant decomposition observed. Similarly, when the lithium derivative was reacted with CuSPh to form the corresponding heterocuprate species prior to reacting with the epoxide, the same decomposition occurred, despite this strategy being successful in a related case. Finally, replacing the epoxides 23b and 23a with the non-protected variant 23c, and reacting it with sulfone 27 after pre-complexation with Ti(OiPr)4, again led only to decomposition.
![[1860-5397-21-91-i4]](/bjoc/content/inline/1860-5397-21-91-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Attempts to couple sulfones 27 and ent-27 with epoxides 23a–c reported by Uguen et al. [41].
Scheme 4: Attempts to couple sulfones 27 and ent-27 with epoxides 23a–c reported by Uguen et al. [41].
Given the unsatisfying results, Uguen and co-workers replaced the epoxides 23a–c to monoethers 46a and 46b, derived from trans-2,3-epoxy-1,4-butanediol 46c, and revised their synthetic strategy as shown in Scheme 5. The synthesis was restarted by treating the pre-cooled (−78 °C) THF mixture of sulfone ent-27 and epoxide 46a with n-butyllithium in a molar ratio of 1.5:1:2.5 (Scheme 6). After warming the mixture for 15 minutes to approximately −40 °C and leaving it at the same temperature for an additional 15 minutes, two products, 47a and 47b, were formed. After desulfonylation with sodium amalgam (Na·Hg) in methanol and column chromatographic purification, the anticipated diols 48a and 48b were obtained in 72 and 11% yield, respectively. The authors noted that using freshly prepared sodium amalgam (within two hours) was critical for achieving a good yield. Compound 48a was then tosylated using TsCl/DMAP/pyridine to give monotosylate 48c in 88% yield, with a small amount (4%) of the undesired ditosyl product 48d. The desired compound 48c was reduced with LiAlH4 to remove the OTs group, and after silyl group removal, diol 32b was obtained in 92% yield. Protection of the primary alcohol as a tosyl ester and the secondary as a TBDMS ether afforded intermediate 32c (72%). This intermediate was then treated with sodium thiophenol in ethanol, followed by oxidation with m-CPBA to provide sulfone 33, which readily was reacted with another epoxide 46b. The sequential treatments used for the condensation of ent-27 and 46a to 48a were applied to 33 and 46b, yielding the desired product 49a (77%) along with a small amount of isomeric 49b (8%). The primary alcohol of 49a was tosylated and then reduced with LiAlH4, while the secondary alcohol group was protected as a silyl ether using TBDMSOTf and collidine. The resulting product, 21, was isolated in 72.4% yield over three steps. Finally, treatment with DDQ afforded the target Morken’s C2–C12 intermediate 20 in 98% yield.
![[1860-5397-21-91-i5]](/bjoc/content/inline/1860-5397-21-91-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Modified synthetic plan for Morken’s C2–C12 intermediate by Uguen [41].
Scheme 5: Modified synthetic plan for Morken’s C2–C12 intermediate by Uguen [41].
![[1860-5397-21-91-i6]](/bjoc/content/inline/1860-5397-21-91-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Revised synthetic strategy for Morken’s C2–C12 intermediate 20 by Uguen [41].
Scheme 6: Revised synthetic strategy for Morken’s C2–C12 intermediate 20 by Uguen [41].
The authors highlighted that shifting the epoxides from 23a,b to 46a,b resulted in the additional tosylation/reduction sequence to install the C4 and C10 methyl moieties of the desired target molecule, making the overall synthesis more lengthy. Thus, 20 was obtained from ent-29 over 25 steps in 11% overall yield. On the other hand, the efficiency of the process was emphasized by the authors on the utilization of Roche esters 29 and ent-29 to prepare stereochemically pure sulfones 27 and ent-27, respectively. Moreover, the use of the trityl protecting group facilitated a simple impurity removal from the key chiral intermediates through recrystallization, from which their exact structures could be elucidated by XRD crystallography.
Zhou’s approach for constructing Omura’s C3–C11 fragment
In 2018, Zhou and co-workers developed an efficient, high-yielding iterative synthesis of polydeoxypropionate based on iridium-catalyzed asymmetric hydrogenation of α-substituted acrylic acid [40]. This method was subsequently applied to the synthesis of a promising vaccine candidate (+)-phthioceranic acid, as well as key intermediates for two natural products, ionomycin and borrelidin (C3–C11). The synthesis involved three main steps: (1) carboxymethylation using Meldrum’s acid, (2) alkenylation with Eschenmoser’s salt, and (3) asymmetric hydrogenation catalyzed by iridium complex (Ra)-50 or (Sa)-50.
The authors began their investigation by performing the hydrogenation of α-substituted acrylic acid 51 (Scheme 7). After optimization, the iridium complex (Ra)-50, in the presence of cesium carbonate, was identified as the most efficient catalyst, producing compound 52 in 97% yield with an enantiomeric excess of 97.6%. Subsequently, compound 52 was treated with Meldrum’s acid in the presence of DCC and DMAP. The reaction was allowed to proceed for 6 h at room temperature, after which the temperature was reduced to −10 °C, and sodium borohydride was added. The mixture was left to react for 12 hours, yielding compound 53 in 96%. This intermediate was then converted to acrylic acid 54 by reacting it with Eschenmoser’s salt, followed by hydrolysis with lithium hydroxide. The resulting unsaturated acid 54, isolated in 92% yield, underwent asymmetric hydrogenation using both (Ra)-50 and (Sa)-50 catalysts. This step provided the respective compounds 55 and 56 in excellent high yield and stereoselectivity. The authors emphasized that, as the alkene moiety in 54 was in a terminal position, the stereoselectivity of the products was determined solely by the chiral environment of the catalysts.
![[1860-5397-21-91-i7]](/bjoc/content/inline/1860-5397-21-91-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Iterative synthesis of polydeoxypropionates developed by Zhou et al. [40].
Scheme 7: Iterative synthesis of polydeoxypropionates developed by Zhou et al. [40].
The application of this method to construct the C3–C11 fragment 60 of borrelidin is summarized in Scheme 8. Starting from ent-52, obtained via the asymmetric hydrogenation of 51 using the catalyst (Sa)-50, the previously developed three-steps reaction sequence was adopted and repeated three times, yielding polydeoxypropionic acid 57 in an overall yield of 54%. The iridium catalyst (Ra)-50 was chosen to ensure the correct stereochemistry of the newly formed three stereocenters. Additionally, replacing cesium carbonate with triethylamine proved crucial for achieving efficient asymmetric hydrogenation in this case. Subsequently, the carboxylic acid group of 57 was reduced with LiAlH4 in THF to produce primary alcohol 58 in 93% yield. This alcohol was then acetylated using acetic anhydride and pyridine reagent. Finally, the resulting acetate 59 was treated with DDQ, affording the target compound 60 in 99% yield, corresponding to an overall yield of 49% over 18 steps starting from 51.
![[1860-5397-21-91-i8]](/bjoc/content/inline/1860-5397-21-91-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Application of iterative synthesis of polydeoxypropionate to construct the C3–C11 fragment 60 of borrelidin 1.
Scheme 8: Application of iterative synthesis of polydeoxypropionate to construct the C3–C11 fragment 60 of bo...
Yadav’s approach for constructing Omura’s C1–C11 fragment
Yadav and Yadav, in 2013, reported their work on preparing the C1–C11 fragment 61 of borrelidin. Their approach employed an iterative sequence of oxidation, Wittig olefination, hydrogenation, and asymmetric methylation for carbon homologation, alongside Sharpless epoxidation and regioselective reduction to install the hydroxy group at the C3 position [39]. In their retrosynthetic analysis, the target molecule 61 was envisioned to be obtained from epoxide 63 through regioselective opening of the epoxide ring, oxidation of the resulting primary alcohol to a carboxylic acid, and protection of the secondary alcohol as a TBDMS ether (Scheme 9). Intermediate 63 was planned to be derived from Evans’ amide 64 by reducing the amide moiety to a primary alcohol, oxidizing it to an aldehyde, performing a Wittig olefination to install an unsaturated ester, reducing the ester to a primary alcohol, and then conducting asymmetric epoxidation of the double bond. Evan’s amide 64 would be synthesized from primary alcohol 65 through a sequence of oxidation to aldehyde, Wittig olefination to an unsaturated ester, hydrogenation of the olefin, conversion of the ester to Evans’ amide, and asymmetric methylation. Intermediate 65, in turn, would be obtained from the known five-carbon precursor 66 through the iterative sequence of oxidation, Wittig olefination, hydrogenation, asymmetric methylation, followed by reduction of the carboxyl group and protection of the resulting alcohol as a THP ether.
![[1860-5397-21-91-i9]](/bjoc/content/inline/1860-5397-21-91-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Retrosynthetic analysis of borrelidin by Yadav et al. [39].
Scheme 9: Retrosynthetic analysis of borrelidin by Yadav et al. [39].
Yadav and Yadav commenced their synthesis with the enzymatic desymmetrization of meso-diol 67 to monoacetate 66, achieving a 47% yield with an enantiomeric excess greater than 95%, using porcine pancreatic lipase (PPL) and vinyl acetate in THF (Scheme 10). The remaining primary alcohol in 66 was oxidized to its corresponding aldehyde using IBX. Subsequent the two-carbon elongation of this aldehyde yielded unsaturated ester 68 in 91% yield with an E/Z ratio of 90:10. The double bond in ester 68 was reduced using sodium borohydride in the presence of NiCl2·6H2O, affording ester 69 in 96% yield. Hydrolysis of the acetate group in 69 with potassium carbonate followed by treatment with TBDMSCl and imidazole converted it into silyl ether 70. The ester group in 70 was then hydrolyzed using lithium hydroxide, and the resulting acid was coupled with Evans’ chiral oxazolidinone in the presence of pivaloyl chloride, triethylamine, and lithium chloride to produce compound 71 in 86% yield. Diastereoselective methylation of 71 was achieved by treating it with NaHMDS at low temperature, followed by the addition of methyl iodide, resulting in a diastereomeric ratio greater than 98:2. Reduction of the product to remove the Evans auxiliary furnished primary alcohol 72 in 84% yield. This alcohol was then protected as a THP ether, and the TBDMS group was removed using a fluoride source, yielding another primary alcohol 65 in 85% yield, thus completing the synthesis of the left-hand portion of the target molecule.
![[1860-5397-21-91-i10]](/bjoc/content/inline/1860-5397-21-91-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Two-carbon homologation of precursor 66 in the synthesize C1–C11 fragment 61 of borrelidin [39].
Scheme 10: Two-carbon homologation of precursor 66 in the synthesize C1–C11 fragment 61 of borrelidin [39].
Another series of oxidation, Wittig olefination, reduction, and asymmetric methylation was applied to the right-hand side of compound 65, affording compound 76. This transformation added two carbons to the backbone and introduced a methyl branch with the correct stereochemistry (Scheme 11). A further two-carbon homologation of compound 76 through an oxidation and Wittig olefination sequence yielded unsaturated ester 77 in 92% yield. The ester group in 77 was reduced using DIBAL-H, achieving a 95% yield. Sharpless epoxidation of alcohol 78 was then performed using (−)-DET, tert-butyl hydroperoxide (TBHP), and Ti(OiPr)4, resulting in the desired epoxide 63 with a 90% yield. Regioselective reductive opening of this epoxide was successfully carried out with Red-Al®, yielding diol 79. The more reactive primary alcohol in diol 79 was selectively masked as TBDPS ether 80 (94%), followed by protection of the secondary alcohol as TBDMS ether 81 (98%). The primary alcohol was then liberated using ammonium fluoride in hot methanol (60 °C). Oxidation of this alcohol to a carboxylic acid was achieved using TEMPO and (diacetoxyiodo)benzene (BAIB), completing the synthesis of the target molecule 61. The final compound 61 was obtained in 18.4% overall yield over 27 steps starting from precursor 66.
![[1860-5397-21-91-i11]](/bjoc/content/inline/1860-5397-21-91-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthesis of the C1–C11 fragment 61 of borrelidin from monoalcohol 65 [39].
Scheme 11: Synthesis of the C1–C11 fragment 61 of borrelidin from monoalcohol 65 [39].
Laschat’s strategy for constructing Theodorakis’ C3–C11 fragment
Laschat and co-workers, in 2011, developed a chemoenzymatic strategy for synthesizing Theodorakis’ C3–C11 fragment 82 of borrelidin [38]. They highlighted a significant limitation in existing methods for constructing the deoxypropionate unit of borrelidin, which features four 1,3-alternating methyl groups with a syn,syn,anti-configuration. These methods typically required at least three synthetic steps to iteratively form each stereocenter, significantly reducing overall efficiency. To overcome this challenge, Laschat’s approach leveraged a chiral pool building block – methyl-branched preen gland wax ester – as the starting material. This ester already contained three methyl groups pre-installed with the stereochemistry necessary for borrelidin, streamlining the synthesis process.
The retrosynthetic analysis by Laschat and co-workers is described in Scheme 12. The target molecule 82 was envisioned to be derived from esters 83 or 86, depending on the chosen synthetic pathway. In route A, ester 83 was designed to originate from compound 84 through a series of sequential steps, including chemoenzymatic (ω-1)-hydroxylation, regioselective dehydration of the resulting alcohol to form a terminal alkene, ozonolysis of the alkene to yield an aldehyde, reduction of the aldehyde product to a primary alcohol, and protection of the alcohol as a PMB ether. Compound 84 could be obtained from the all-syn isomer 85 through an epimerization process. In route B, compound 86 was proposed to be synthesized from methyl ester 87. The transformation involved reduction of 87 to a primary alcohol, conversion of the alcohol into the corresponding iodide, and subsequent nucleophilic substitution with deprotonated (R,R)-91. The introduction of the OPMB functionality in compound 88 could then be achieved by following the steps employed in the transformation of 84 to 83.
![[1860-5397-21-91-i12]](/bjoc/content/inline/1860-5397-21-91-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Synthetic plan for Theodorakis’ C3–C11 fragment 82 of borrelidin by Laschat et al. [38].
Scheme 12: Synthetic plan for Theodorakis’ C3–C11 fragment 82 of borrelidin by Laschat et al. [38].
The synthesis via route A began with efforts to optimize the epimerization of compound 85 to produce 84 (Scheme 13). After numerous attempts, it was found that treating 85 with LDA at low temperature followed by the addition of various acids yielded 84 as the minor product, with the product ratio of 85/84 ranging from 2:1 to 3:1. Attempts to influence the stereochemical outcome by employing chiral proton sources, such as ᴅ- and ʟ-menthol, (+)- and (–)-camphorsulfonic acid, or pseudoephedrinamide (R,R)-91, proved unsuccessful, as they did not significantly alter the stereochemical preference. Additionally, reversing the quenching order by adding the enolates to acids also failed to impact the outcome. Subsequently, separation of 84 from 85 was explored using various lipases and an esterase. However, preliminary experiments with these enzymes did not yield satisfactory results.
![[1860-5397-21-91-i13]](/bjoc/content/inline/1860-5397-21-91-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Synthesis of Theodorakis’ C3–C11 fragment 82 from compound 88 [38].
Scheme 13: Synthesis of Theodorakis’ C3–C11 fragment 82 from compound 88 [38].
The focus then shifted to route B, utilizing methyl 2,4,6-trimethyloctanoate 88 as the starting material. Hydroxylation at the (ω-1) position was achieved using the NADH-dependent mutated enzyme variant CYP102A1 3 mDS, a p450 monooxygenase derived from Bacillus megaterium CYP102A1. After chromatographic purification, alcohol 89 was obtained in a 34% yield with a diastereomeric ratio of 82:18. This alcohol was subsequently dehydrated using Martin’s sulfurane to produce terminal alkene 90 in 97% yield. A sequence of ozonolysis, reduction with sodium borohydride, and PMB protection using camphorsulfonic acid and PMB-trichloroacetimidate reagents followed, yielding compound 87 in 62% yield. Next, the ester functionality of 87 was reduced to the corresponding primary alcohol with DIBAL-H and converted to the iodide derivative using Ph3P/I2/imidazole reagents. The resulting iodide was treated with lithiated (R,R)-91 to afford compound 86 in a 76% yield with excellent diastereoselectivity (>99:1 dr). Finally, reductive cleavage of the chiral auxiliary using a combination of LDA and BH3·NH3 provided the target molecule 82 in a 76% yield. The authors emphasized that the Theodorakis’ C3–C11 fragment 82 of borrelidin was synthesized via a concise 8-step route, achieving a 36% overall yield from the chiral pool precursor 88.
Minnaard’s strategy for constructing Ōmura’s C1–C11 and C12–C23 fragments
Minnaard and Madduri in 2010 [37] developed a novel strategy to prepare the C1–C11 and C12–C23 fragments of borrelidin, representing the upper and lower parts of this natural product as classified by Ōmura [27,29]. Their method was based on the concept of “catalytic total synthesis”, wherein all stereocenters were installed under the control of catalysts. Minnaard and Madduri proposed the synthesis of the C1–C11 fragment from unsaturated thioester 92 through iterative, previously developed asymmetric 1,4-addition reactions (key step), reduction of the thioester moiety to an aldehyde, and olefination to produce another unsaturated ester (Scheme 14). In contrast, the synthesis of the lower part, the C12–C23 fragment, was designed to proceed from ester 93 via asymmetric hydrogenation (key step), sequential protection and deprotection steps, functional group transformations, stereocontrolled allylation, cross-metathesis, and Horner–Wadsworth–Emmons (HWE) olefination. This method highlights the power of catalytic stereocontrol, achieving the complex architecture of borrelidin fragments with efficiency and precision.
![[1860-5397-21-91-i14]](/bjoc/content/inline/1860-5397-21-91-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Retrosynthesis of 61 and 62b by Minnaard and Madduri [37].
Scheme 14: Retrosynthesis of 61 and 62b by Minnaard and Madduri [37].
The synthesis commenced with the treatment of precursor 92, obtained in three steps from ethylene glycol, with MeMgBr and catalytic 94/CuBr, yielding the 1,4-addition product 93 in 96% yield and excellent regioselectivity (ee 98%) (Scheme 15). The authors highlighted the scalability of this reaction, successfully processing up to 15 g of starting material. Compound 93 was reduced to its corresponding aldehyde using DIBAL-H, followed by Horner–Wadsworth–Emmons (HWE) olefination with (EtO)2P(O)CH2COSEt, resulting in the unsaturated thioester 95. Reapplying the 1,4-addition reaction conditions to 95 produced the syn-product 96a in 90% yield with a diastereomeric ratio of 98:2. Interestingly, substituting the catalyst with ent-94 delivered 96b in 89% yield and dr 95:5. Subsequently, 96a was subjected to a similar sequence of reduction, HWE olefination, asymmetric 1,4-addition, culminating in compound 98 in 70% overall yield across three steps, with a dr exceeding 98:2.
![[1860-5397-21-91-i15]](/bjoc/content/inline/1860-5397-21-91-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Synthesis of intermediate 98 by Minnaard and Madduri [37].
Scheme 15: Synthesis of intermediate 98 by Minnaard and Madduri [37].
The reaction continued with the reduction of thioester 98 to aldehyde 99, followed by HWE olefination with (EtO)2P(O)CH2COMe, yielding compound 100 in 92% yield (Scheme 16). The stereocontrol achieved by the catalysts was again demonstrated when compound 100 was treated with MeMgBr/CuBr and either 94 or ent-94, affording 101a and 101b, respectively, with high yield and exclusive diastereoselectivity. Compound 101a, featuring the relevant stereochemistry of borrelidin at the C4, C6, C8, and C10, underwent Baeyer–Villiger oxidation using m-CPBA. Subsequent hydrolysis of the resulting ester with K2CO3 in methanol provided alcohol 102 in 82% yield. The free primary alcohol of 102 was protected as a THP ether, and the TBDPS group was removed to expose the opposite free primary alcohol, which was oxidized to aldehyde 103 in 83% yield over three steps using TPAP/NMO reagents. Compound 103 was subjected to a SmI2-mediated Reformatsky-type reaction with 4-methoxybenzyl 2-bromoacetate, followed by oxidation of the resulting β-hydroxy intermediate with TPAP/NMO, producing keto ester 104 in 77% yield over two steps. Catalytic asymmetric hydrogenation of 104, employing (R)-[(RuCl(Tol-BINAP))2(μ-Cl)3[NH2Me2], yielded the product in 90% yield with nearly complete diastereoselectivity (de >99%). Finally, the secondary alcohol was protected as a TBS ether, and the ester group was hydrolyzed to deliver Ōmura’s C1–C11 fragment 61 in 85% yield.
![[1860-5397-21-91-i16]](/bjoc/content/inline/1860-5397-21-91-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Synthesis of Ōmura’s C1–C11 fragment 61 by Minnaard and Madduri [37].
Scheme 16: Synthesis of Ōmura’s C1–C11 fragment 61 by Minnaard and Madduri [37].
The synthesis of part 62b began with the asymmetric hydrogenation of 93 to yield β-hydroxy ester 106 (Scheme 17). Initial experiments, following the procedure of Noyori et al. [51] and using [RuCl2(p-cymene)]2 metal complex with BINAP as the chiral ligand, produced 106 in 92% yield (92% ee, 99:1 dr). Optimized conditions were achieved by employing [RuI2(p-cymene)]2 with the chiral ligand 3,5-xylyl-BINAP, resulting in 106 with an improved yield of 98% (97% ee, 99:1 dr). The secondary alcohol of 106 was protected as a THP ether, and the ester group was reduced to a primary alcohol 107 in 89% yield. This primary alcohol was then protected as a PMB ether. After deprotecting the THP group, the resulting secondary alcohol was converted to a tosyl ester, which underwent an SN2 reaction with sodium cyanide in DMSO, yielding compound 109 with stereochemical inversion. Interestingly, reduction of the cyanide group with DIBAL-H to aldehyde 110 also resulted in stereochemical inversion (85%, anti/syn >15:1). Subsequent chelation-controlled allylation of aldehyde 110, following Ōmura’s method [27,29], employed allyltrimethylsilane and MgBr2·OEt2, yielding allyl alcohol 111 in 86% yield with exclusive diastereoselectivity (20:1 dr). Direct cross-metathesis of 111 with acrolein was envisioned as an efficient method to introduce an aldehyde functionality adjacent to the alkene moiety without prior protection of the free allylic alcohol. This hypothesis was successfully realized by reacting 111 with acrolein diethyl acetal in the presence of Hoveyda–Grubbs’ second-generation catalyst, affording aldehyde 112 in 75% yield as the E-isomer after a careful acidic workup. Finally, HWE olefination of aldehyde 112 with (EtO)2P(O)CHBrCN completed the synthesis of fragment 62b of borrelidin.
![[1860-5397-21-91-i17]](/bjoc/content/inline/1860-5397-21-91-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Synthesis of fragment 62b of borrelidin as proposed by Minnaard and Madduri [37].
Scheme 17: Synthesis of fragment 62b of borrelidin as proposed by Minnaard and Madduri [37].
Minnaard and Madduri emphasized the significant role of asymmetric catalysis in their strategy, utilizing a copper-catalyzed asymmetric 1,4-addition and a ruthenium-catalyzed asymmetric ketone hydrogenation. Fragment 61 was synthesized in 15% overall yield across 19 steps, while fragment 62b was achieved in 32% yield over 11 steps.
Herber and Breit’s strategy for constructing Theodorakis’ C3–C11 fragment
In 2006, Herber and Breit utilized an iterative deoxypropionate synthesis to construct the Theodorakis’ C3–C11 fragment of borrelidin. This approach was based on a copper-mediated directed allylic substitution previously developed in their laboratory. The strategy primarily involved the reaction of a chiral organometallic reagent 115 with a chiral allyl electrophile 114, as depicted in Scheme 18. The resulting deoxypropionate 113 was obtained with the newly formed stereocenter controlled by the reagent directing group (RDG) attached to the allyl precursor 114. Iteration of this process required ozonolysis of 113, followed by its conversion to an organometallic intermediate 116, which was then reacted with allyl 114 to yield another deoxypropionate product, 117.
![[1860-5397-21-91-i18]](/bjoc/content/inline/1860-5397-21-91-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Iterative directed allylation for the synthesis of deoxypropionates by Herber and Breit [33].
Scheme 18: Iterative directed allylation for the synthesis of deoxypropionates by Herber and Breit [33].
The synthesis began with the preparation of the precursor chiral allyl ester (R)-120 via enzymatic kinetic resolution of readily available rac-118 using Novozyme 435 and vinyl acetate in pentane at 30 °C (Scheme 19). The reaction was halted at the conversion of 54%, yielding the remaining alcohol (S)-118 with >99% ee. The product (R)-119 was subsequently treated with Novozyme 435 in a pH 7 buffer to hydrolyze the acetate group. The resulting alcohol, (R)-118, was isolated in 73% yield with 96% ee. This alcohol was then reacted with o-diphenylphosphanylbenzoate (o-DPPB) in the presence of DCC, affording (R)-120 in 83% yield (>99% ee, E/Z >99:1) after recrystallization. Similarly, the enantiomer (S)-118 was esterified with o-DPPB under the same conditions, providing (S)-120 in 88% yield (>99% ee, E/Z >99:1) after recrystallization. The authors noted that both (R)- and (S)-120 could be stored in their crystalline forms for months without significant decomposition or undesired oxidation to the corresponding phosphane oxide.
![[1860-5397-21-91-i19]](/bjoc/content/inline/1860-5397-21-91-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Iterative copper-mediated directed allyl substitution for the synthesis of Theodorakis’ C3–C11 fragment 82 developed by Herber and Breit [33].
Scheme 19: Iterative copper-mediated directed allyl substitution for the synthesis of Theodorakis’ C3–C11 frag...
The work continued with the preparation of the chiral organometallic reagent 124. Starting from known bromide 122, which was readily accessible from Roche ester (R)-121 [52] or 1,3-diol 123 [53,54] through literature procedures, the Grignard reagent 124 was obtained by reaction with magnesium in anhydrous diethyl ether. Herber and Breit emphasized that activating the magnesium using a dry stirring method was crucial for smooth magnesiation. The freshly prepared Grignard reagent 124 was added to the allyl electrophile (R)-120 in the presence of CuBr·SMe2, facilitating the formation of deoxypropionate 125 in 80% yield with excellent regioselectivity (>99:1) and stereoselectivity (99:1 dr). Ozonolysis of 125 followed by reductive work up with NaBH4 afforded alcohol 126 in 92% yield. At this stage, the authors explored an alternative procedure for preparing the organometallic reagent using halogen-metal exchange, which proved feasible for small scale operations. Alcohol 126 was converted into its iodide derivative 127 in 93% yield using Ph3P/I2/imidazole reagent. Halogen-metal exchange of 127 with tert-butyllithium proceeded efficiently, and subsequent transmetallation with MgBr2·OEt2 yielded the desired magnesium species. Reaction of this reagent with (R)-120 in the presence of CuBr·SMe2 afforded product (–)-128 in 86% yield, with perfect regioselectivity (>99:1) and excellent stereochemistry (98:2 dr). Repeating the sequence of ozonolysis, reductive work up, iodination, halogen-metal exchange, and transmetallation for compound (–)-128, followed by reaction with (S)-120, provided the deoxypropionate product (+)-131 in 78% yield over three steps, with dr >95:5. Finally, another ozonolysis followed by reductive work up with triphenylphospine afforded the target molecule 82 in 89% yield.
Herber and Breit synthesized Theodorakis’ C3-C11 fragment 82 of borrelidin with a 41% overall yield over eight steps, starting from the known precursor 122.
Synthetic studies of borrelidin
Iqbal’s strategy for constructing the C3–C17 fragment of borrelidin using cross-metathesis
In 2008, Iqbal and co-workers conducted a synthetic study in which they successfully synthesized the C3–C17 fragment of borrelidin using a cross-metathesis reaction [35]. In the retrosynthesis, compound 132, representing the synthetic target, was realized through an addition reaction of a Grignard reagent derived from 133 to the aldehyde counterpart (Scheme 20). Compound 133 was prepared via a cross-metathesis reaction of 2-bromohexa-2,4-dienenitrile 134 and a selection of alkenes.
![[1860-5397-21-91-i20]](/bjoc/content/inline/1860-5397-21-91-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Retrosynthesis of the C3–C17 fragment of borrelidin by Iqbal and co-workers [35].
Scheme 20: Retrosynthesis of the C3–C17 fragment of borrelidin by Iqbal and co-workers [35].
Iqbal noted from the literature that cyano alkenes, such as acrylonitrile, predominantly yielded the Z-product during the cross-metathesis reactions. Therefore, the synthetic study was initiated by performing cross-metathesis reactions between both (E,E)- and (Z,E)-134 with various alkenes to investigate the Z- or E-selectivity of the reaction. Fortunately, Iqbal observed that all cross-metathesis reactions in the study exhibited high E-selectivity. As a result, the reaction of (Z,E)-134 with olefin 135 provided the desired product 136 in 56% yield, with a (Z,E)/(Z,Z) ratio of 4:1 (Scheme 21). Compound 136 was subsequently protected as a TBS ether, 137 [35].
![[1860-5397-21-91-i21]](/bjoc/content/inline/1860-5397-21-91-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Synthesis of key intermediates 137 and 147 for the synthesis of the C3–C17 fragment of borrelidin.
Scheme 21: Synthesis of key intermediates 137 and 147 for the synthesis of the C3–C17 fragment of borrelidin.
The aldehyde counterpart 147 for the late stage coupling with 137 was prepared from the known lactone 138 (Scheme 21). Following a literature procedure, compound 139 was obtained from lactone 138 and subsequently treated with CrO3 to yield ketone 140 in 90% yield [55]. The ketone moiety of this compound was then reacted with a Wittig reagent derived from methyltriphenylphosphonium bromide and n-BuLi as base to provide olefin 141 in 66% yield. The ester group in this compound was reduced with LiAlH4 to afford primary alcohol 142 in 94% yield. The alcohol functionality was then converted to its corresponding iodide 143 (96%) upon treatment with Ph3P/I2/imidazole reagents and reacted with lithiated pseudoephedrine propionamide. The resulting product 144 was obtained in 88% yield. Basic hydrolysis of this compound successfully removed the chiral auxiliary, yielding acid 145 in 91% yield. Sequential reduction of this carboxylic acid with LiAlH4, followed by oxidation of the resulting primary alcohol with Dess–Martin periodinane, gave the anticipated aldehyde 147 in 76% yield over two steps.
In the final stage, compound 137 was converted to organomagnesium intermediate 148 upon treatment with isopropylmagnesium bromide in THF and then reacted with aldehyde 147 (Scheme 22). After careful chromatographic purification, a pair of (Z,E)-150a,b (9%, 4%) corresponding to the C3–C17 fragment of borrelidin, as well as a pair of (E,E)-151a,b (6%, 4%), were isolated. Additionally, the debrominated product 149 was also isolated as the major product (70%).
![[1860-5397-21-91-i22]](/bjoc/content/inline/1860-5397-21-91-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Synthesis of the C3–C17 fragment 150a,b of borrelidin.
Scheme 22: Synthesis of the C3–C17 fragment 150a,b of borrelidin.
In summary, Iqbal and co-workers synthesized the C1–C17 fragment of borrelidin using cross-metathesis as the main strategy. The desired compounds, 150a and 150b, were isolated in 2.8 and 1.2% yields, respectively, after 10 linear steps from compound 139.
Iqbal’s strategy to construct the C11–C15 fragment of borrelidin and macrocyclization using a model compound based on metathesis reaction
Two years earlier, in 2006, Iqbal and co-workers developed a strategy for synthesizing the C11–C15 fragment of borrelidin [34]. Additionally, they conducted a synthetic study to achieve the macrocyclization using a model system, employing a ring-closing metathesis reaction. As shown in Scheme 23, ylide 152, derived from triphenylphosphine and chloroacetonitrile, was treated with bromine in the presence of sodium hexamethyldisilazide to afford compound 153 in 72% yield. Reaction of this intermediate with (E)-crotonaldehyde produced a mixture of (E,Z)- and (E,E)-isomers of 154 in a combined yield of 58%. Subsequent treatment of 154 with isopropylmagnesium bromide, followed by reaction with undecylenic aldehyde, provided compounds 155a and 155b (65% yield), representing the (E,Z)- and (E,E)-configurations, respectively.
![[1860-5397-21-91-i23]](/bjoc/content/inline/1860-5397-21-91-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Synthesis of the C11–C15 fragment 155a of borrelidin.
Scheme 23: Synthesis of the C11–C15 fragment 155a of borrelidin.
The macrocyclization study of borrelidin was subsequently carried out using model compounds 155a and 155b (Scheme 24). Heating compound 155a in dichloromethane for 18 hours in the presence of 5 mol % Grubbs’ second generation catalyst yielded coupling product 156a and unreacted starting material 155a. Encouragingly, the addition of an extra 5 mol % of the catalyst followed by further heating for 28 hours successfully converted the remaining 155a into 156a, which was isolated in 54% yield. A trace amount of an unwanted dimer of 155a was also detected. In contrast, different results were obtained when compound 155b was used. Addition of 5 mol % catalyst produced the expected macrocyclic product and dimer 157 in yields of 24 and 22%, respectively, while a significant amount of unreacted 155b (26%) remained under these conditions.
![[1860-5397-21-91-i24]](/bjoc/content/inline/1860-5397-21-91-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Macrocyclization of borrelidin model compounds 155a and 155b using ring-closing metathesis.
Scheme 24: Macrocyclization of borrelidin model compounds 155a and 155b using ring-closing metathesis.
Conclusion
In summary, this review has examined the various synthetic strategies employed in the construction of borrelidin fragments. By focusing on key intermediates and synthetic methods explored in recent literature, we have highlighted the feasibility and versatility of different approaches. These methods offer valuable insights into the efficient design and synthesis of borrelidin fragments, aiding the advancement of borrelidin-based drug development. Future research in this area should continue to explore novel synthetic strategies to optimize the synthesis and functionalization of borrelidin fragments, further supporting their potential applications in medicinal chemistry.
Data Availability Statement
Data sharing is not applicable as no new data was generated or analyzed in this study.
References
-
Berger, J.; Jampolsky, L. M.; Goldberg, M. W. Arch. Biochem. 1949, 22, 476–478.
Return to citation in text: [1] [2] -
Keller‐Schierlein, W. Helv. Chim. Acta 1967, 50, 731–753. doi:10.1002/hlca.19670500303
Return to citation in text: [1] -
Gafiuc, D.; Weiß, M.; Mylonas, I.; Brüning, A. J. Appl. Toxicol. 2014, 34, 1109–1113. doi:10.1002/jat.2946
Return to citation in text: [1] -
Gao, X.; Jiang, Y.; Han, L.; Chen, X.; Hu, C.; Su, H.; Mu, Y.; Guan, P.; Huang, X. RSC Adv. 2017, 7, 44401–44409. doi:10.1039/c7ra08290h
Return to citation in text: [1] -
Jeong, M.; Kim, H.; Kim, S.; Park, J.-H. Drug Delivery Transl. Res. 2018, 8, 1380–1388. doi:10.1007/s13346-018-0563-z
Return to citation in text: [1] -
Jackson, L.; Bechler, S.; Miller, J.; Brownell, A.; Garshott, D.; Callaghan, M.; Fribley, A. M. Blood 2011, 118, 4260. doi:10.1182/blood.v118.21.4260.4260
Return to citation in text: [1] -
Kawamura, T.; Liu, D.; Towle, M. J.; Kageyama, R.; Tsukahara, N.; Wakabayashi, T.; Littlefield, B. A. J. Antibiot. 2003, 56, 709–715. doi:10.7164/antibiotics.56.709
Return to citation in text: [1] -
Woolard, J.; Vousden, W.; Moss, S. J.; Krishnakumar, A.; Gammons, M. V. R.; Nowak, D. G.; Dixon, N.; Micklefield, J.; Spannhoff, A.; Bedford, M. T.; Gregory, M. A.; Martin, C. J.; Leadlay, P. F.; Zhang, M. Q.; Harper, S. J.; Bates, D. O.; Wilkinson, B. Chem. Sci. 2011, 2, 273–278. doi:10.1039/c0sc00297f
Return to citation in text: [1] -
Olano, C.; Wilkinson, B.; Sánchez, C.; Moss, S. J.; Sheridan, R.; Math, V.; Weston, A. J.; Braña, A. F.; Martin, C. J.; Oliynyk, M.; Méndez, C.; Leadlay, P. F.; Salas, J. A. Chem. Biol. 2004, 11, 87–97. doi:10.1016/j.chembiol.2003.12.018
Return to citation in text: [1] -
Sibero, M. T.; Frederick, E. H.; Wijayanti, D. P.; Haryanti, D.; Siswanto, A. P.; Igarashi, Y. J. Appl. Pharm. Sci. 2024, 14, 132–140. doi:10.7324/japs.2024.139056
Return to citation in text: [1] -
Gao, Y.-M.; Wang, X.-J.; Zhang, J.; Li, M.; Liu, C.-X.; An, J.; Jiang, L.; Xiang, W.-S. J. Agric. Food Chem. 2012, 60, 9874–9881. doi:10.1021/jf302857x
Return to citation in text: [1] -
Liu, C.-X.; Zhang, J.; Wang, X.-J.; Qian, P.-T.; Wang, J.-D.; Gao, Y.-M.; Yan, Y.-J.; Zhang, S.-Z.; Xu, P.-F.; Li, W.-B.; Xiang, W.-S. J. Agric. Food Chem. 2012, 60, 1251–1257. doi:10.1021/jf2044982
Return to citation in text: [1] -
Wang, N.; Huang, W.; Jia, Q.; Song, B.; Wang, S.; Wu, L.; Sun, M.; Wang, Y.; Zhang, L.; Wang, W. J. Appl. Microbiol. 2025, 136, lxaf073. doi:10.1093/jambio/lxaf073
Return to citation in text: [1] -
Ishiyama, A.; Iwatsuki, M.; Namatame, M.; Nishihara-Tsukashima, A.; Sunazuka, T.; Takahashi, Y.; Ōmura, S.; Otoguro, K. J. Antibiot. 2011, 64, 381–384. doi:10.1038/ja.2011.6
Return to citation in text: [1] -
Novita, R.; Suprayogi, A.; Agusta, A.; Nugraha, A. B.; Nozaki, T.; Agustini, K.; Darusman, H. S. Open Vet. J. 2024, 14, 2007. doi:10.5455/ovj.2024.v14.i8.30
Return to citation in text: [1] -
Anderson, B. F.; Herlt, A. J.; Rickards, R. W.; Robertson, G. B. Aust. J. Chem. 1989, 42, 717–730. doi:10.1071/ch9890717
Return to citation in text: [1] -
Schulze, C. J.; Bray, W. M.; Loganzo, F.; Lam, M.-H.; Szal, T.; Villalobos, A.; Koehn, F. E.; Linington, R. G. J. Nat. Prod. 2014, 77, 2570–2574. doi:10.1021/np500727g
Return to citation in text: [1] [2] -
Hamed, A.; Abdel-Razek, A. S.; Frese, M.; Wibberg, D.; El-Haddad, A. F.; Ibrahim, T. M.; Kalinowski, J.; Sewald, N.; Shaaban, M. Z. Naturforsch., C: J. Biosci. 2018, 73, 49–57. doi:10.1515/znc-2017-0140
Return to citation in text: [1] [2] -
Kim, J.; Shin, D.; Kim, S.-H.; Park, W.; Shin, Y.; Kim, W. K.; Lee, S. K.; Oh, K.-B.; Shin, J.; Oh, D.-C. Mar. Drugs 2017, 15, 166. doi:10.3390/md15060166
Return to citation in text: [1] [2] [3] [4] -
Sidhu, A.; Miller, J. R.; Tripathi, A.; Garshott, D. M.; Brownell, A. L.; Chiego, D. J.; Arevang, C.; Zeng, Q.; Jackson, L. C.; Bechler, S. A.; Callaghan, M. U.; Yoo, G. H.; Sethi, S.; Lin, H.-S.; Callaghan, J. H.; Tamayo-Castillo, G.; Sherman, D. H.; Kaufman, R. J.; Fribley, A. M. ACS Med. Chem. Lett. 2015, 6, 1122–1127. doi:10.1021/acsmedchemlett.5b00133
Return to citation in text: [1] [2] [3] -
Zhou, Z.; Wu, Q.; Xie, Q.; Ling, C.; Zhang, H.; Sun, C.; Ju, J. Chem. Biodiversity 2020, 17, e1900560. doi:10.1002/cbdv.201900560
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Sun, J.; Shao, J.; Sun, C.; Song, Y.; Li, Q.; Lu, L.; Hu, Y.; Gui, C.; Zhang, H.; Ju, J. Bioorg. Med. Chem. 2018, 26, 1488–1494. doi:10.1016/j.bmc.2018.01.010
Return to citation in text: [1] [2] [3] [4] [5] -
Zhang, L.; Shi, J.; Liu, C. L.; Xiang, L.; Ma, S. Y.; Li, W.; Jiao, R. H.; Tan, R. X.; Ge, H. M. Tetrahedron Lett. 2018, 59, 4517–4520. doi:10.1016/j.tetlet.2018.11.023
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Olano, C.; Moss, S. J.; Braña, A. F.; Sheridan, R. M.; Math, V.; Weston, A. J.; Méndez, C.; Leadlay, P. F.; Wilkinson, B.; Salas, J. A. Mol. Microbiol. 2004, 52, 1745–1756. doi:10.1111/j.1365-2958.2004.04090.x
Return to citation in text: [1] -
Duffey, M. O.; LeTiran, A.; Morken, J. P. J. Am. Chem. Soc. 2003, 125, 1458–1459. doi:10.1021/ja028941u
Return to citation in text: [1] [2] -
Hanessian, S.; Yang, Y.; Giroux, S.; Mascitti, V.; Ma, J.; Raeppel, F. J. Am. Chem. Soc. 2003, 125, 13784–13792. doi:10.1021/ja030139k
Return to citation in text: [1] -
Nagamitsu, T.; Takano, D.; Fukuda, T.; Otoguro, K.; Kuwajima, I.; Harigaya, Y.; Ōmura, S. Org. Lett. 2004, 6, 1865–1867. doi:10.1021/ol049356w
Return to citation in text: [1] [2] [3] -
Vong, B. G.; Kim, S. H.; Abraham, S.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2004, 43, 3947–3951. doi:10.1002/anie.200460203
Return to citation in text: [1] -
Nagamitsu, T.; Takano, D.; Marumoto, K.; Fukuda, T.; Furuya, K.; Otoguro, K.; Takeda, K.; Kuwajima, I.; Harigaya, Y.; Ōmura, S. J. Org. Chem. 2007, 72, 2744–2756. doi:10.1021/jo062089i
Return to citation in text: [1] [2] [3] -
Haddad, N.; Brik, A.; Grishko, M. Tetrahedron Lett. 1997, 38, 6079–6082. doi:10.1016/s0040-4039(97)01372-5
Return to citation in text: [1] [2] -
Haddad, N.; Grishko, M.; Brik, A. Tetrahedron Lett. 1997, 38, 6075–6078. doi:10.1016/s0040-4039(97)01371-3
Return to citation in text: [1] [2] -
Vong, B. G.; Abraham, S.; Xiang, A. X.; Theodorakis, E. A. Org. Lett. 2003, 5, 1617–1620. doi:10.1021/ol034243i
Return to citation in text: [1] -
Herber, C.; Breit, B. Chem. – Eur. J. 2006, 12, 6684–6691. doi:10.1002/chem.200600343
Return to citation in text: [1] [2] [3] [4] -
Krishna, C. V.; Maitra, S.; Dev, R. V.; Mukkanti, K.; Iqbal, J. Tetrahedron Lett. 2006, 47, 6103–6106. doi:10.1016/j.tetlet.2006.06.092
Return to citation in text: [1] [2] -
Vamsee Krishna, C.; Bhonde, V. R.; Devendar, A.; Maitra, S.; Mukkanti, K.; Iqbal, J. Tetrahedron Lett. 2008, 49, 2013–2017. doi:10.1016/j.tetlet.2008.01.055
Return to citation in text: [1] [2] [3] [4] -
Yadav, J. S.; Bezawada, P.; Chenna, V. Tetrahedron Lett. 2009, 50, 3772–3775. doi:10.1016/j.tetlet.2009.03.196
Return to citation in text: [1] [2] -
Madduri, A. V. R.; Minnaard, A. J. Chem. – Eur. J. 2010, 16, 11726–11731. doi:10.1002/chem.201001284
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Theurer, M.; El Baz, Y.; Koschorreck, K.; Urlacher, V. B.; Rauhut, G.; Baro, A.; Laschat, S. Eur. J. Org. Chem. 2011, 4241–4249. doi:10.1002/ejoc.201100412
Return to citation in text: [1] [2] [3] [4] [5] -
Yadav, J. S.; Yadav, N. N. RSC Adv. 2013, 3, 4024–4032. doi:10.1039/c3ra22754e
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Che, W.; Wen, D. C.; Zhu, S.-F.; Zhou, Q.-L. Org. Lett. 2018, 20, 3305–3309. doi:10.1021/acs.orglett.8b01193
Return to citation in text: [1] [2] [3] -
Gembus, V.; Karmazin, L.; Uguen, D.; Zoller, T. Bull. Chem. Soc. Jpn. 2019, 92, 359–380. doi:10.1246/bcsj.20180292
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Moss, S. J.; Carletti, I.; Olano, C.; Sheridan, R. M.; Ward, M.; Math, V.; Nur-E-Alam, M.; Braña, A. F.; Zhang, M. Q.; Leadlay, P. F.; Méndez, C.; Salas, J. A.; Wilkinson, B. Chem. Commun. 2006, 2341–2343. doi:10.1039/b602931k
Return to citation in text: [1] -
Wilkinson, B.; Gregory, M. A.; Moss, S. J.; Carletti, I.; Sheridan, R. M.; Kaja, A.; Ward, M.; Olano, C.; Mendez, C.; Salas, J. A.; Leadlay, P. F.; vanGinckel, R.; Zhang, M.-Q. Bioorg. Med. Chem. Lett. 2006, 16, 5814–5817. doi:10.1016/j.bmcl.2006.08.073
Return to citation in text: [1] -
Sugawara, A.; Tanaka, T.; Hirose, T.; Ishiyama, A.; Iwatsuki, M.; Takahashi, Y.; Otoguro, K.; Ōmura, S.; Sunazuka, T. Bioorg. Med. Chem. Lett. 2013, 23, 2302–2305. doi:10.1016/j.bmcl.2013.02.075
Return to citation in text: [1] -
Hahn, F.; Kandziora, N.; Friedrich, S.; Leadlay, P. F. Beilstein J. Org. Chem. 2014, 10, 634–640. doi:10.3762/bjoc.10.55
Return to citation in text: [1] -
Gündemir-Durmaz, T.; Schmid, F.; El Baz, Y.; Häusser, A.; Schneider, C.; Bilitewski, U.; Rauhut, G.; Garnier, D.; Baro, A.; Laschat, S. Org. Biomol. Chem. 2016, 14, 8261–8269. doi:10.1039/c6ob01358a
Return to citation in text: [1] -
Hu, C.; Su, H.; Luo, J.; Han, L.; Liu, Q.; Wu, W.; Mu, Y.; Guan, P.; Sun, T.; Huang, X. Bioorg. Med. Chem. 2018, 26, 6035–6049. doi:10.1016/j.bmc.2018.11.005
Return to citation in text: [1] -
Nagamitsu, T.; Harigaya, Y.; Omura, S. Proc. Jpn. Acad., Ser. B 2005, 81, 244–256. doi:10.2183/pjab.81.244
Return to citation in text: [1] -
Novak, T.; Tan, Z.; Liang, B.; Negishi, E.-i. J. Am. Chem. Soc. 2005, 127, 2838–2839. doi:10.1021/ja043534z
Return to citation in text: [1] -
Darna, B.; Kulkarni, R.; Uppuluru, A. K.; Gannu, P. K.; Gadiraju, R. R. J. J. Pharm. Res. (Mohali, India) 2011, 4, 1025–1030.
Return to citation in text: [1] -
Noyori, R.; Ikeda, T.; Ohkuma, T.; Widhalm, M.; Kitamura, M.; Takaya, H.; Akutagawa, S.; Sayo, N.; Saito, T.; Taketomi, T.; Kumobayashi, H. J. Am. Chem. Soc. 1989, 111, 9134–9135. doi:10.1021/ja00207a038
Return to citation in text: [1] -
Meyer, S. D.; Miwa, T.; Nakatsuka, M.; Schreiber, S. L. J. Org. Chem. 1992, 57, 5058–5060. doi:10.1021/jo00045a007
Return to citation in text: [1] -
Santaniello, E.; Ferraboschi, P.; Grisenti, P. Tetrahedron Lett. 1990, 31, 5657–5660. doi:10.1016/s0040-4039(00)97925-5
Return to citation in text: [1] -
Bertucci, C.; Petri, A.; Felix, G.; Perini, B.; Salvadori, P. Tetrahedron: Asymmetry 1999, 10, 4455–4462. doi:10.1016/s0957-4166(99)00478-4
Return to citation in text: [1] -
Mori, K.; Kuwahara, S. Tetrahedron 1986, 42, 5545–5550. doi:10.1016/s0040-4020(01)88158-8
Return to citation in text: [1]
| 41. | Gembus, V.; Karmazin, L.; Uguen, D.; Zoller, T. Bull. Chem. Soc. Jpn. 2019, 92, 359–380. doi:10.1246/bcsj.20180292 |
| 41. | Gembus, V.; Karmazin, L.; Uguen, D.; Zoller, T. Bull. Chem. Soc. Jpn. 2019, 92, 359–380. doi:10.1246/bcsj.20180292 |
| 41. | Gembus, V.; Karmazin, L.; Uguen, D.; Zoller, T. Bull. Chem. Soc. Jpn. 2019, 92, 359–380. doi:10.1246/bcsj.20180292 |
| 41. | Gembus, V.; Karmazin, L.; Uguen, D.; Zoller, T. Bull. Chem. Soc. Jpn. 2019, 92, 359–380. doi:10.1246/bcsj.20180292 |
| 41. | Gembus, V.; Karmazin, L.; Uguen, D.; Zoller, T. Bull. Chem. Soc. Jpn. 2019, 92, 359–380. doi:10.1246/bcsj.20180292 |
| 39. | Yadav, J. S.; Yadav, N. N. RSC Adv. 2013, 3, 4024–4032. doi:10.1039/c3ra22754e |
| 39. | Yadav, J. S.; Yadav, N. N. RSC Adv. 2013, 3, 4024–4032. doi:10.1039/c3ra22754e |
| 39. | Yadav, J. S.; Yadav, N. N. RSC Adv. 2013, 3, 4024–4032. doi:10.1039/c3ra22754e |
| 40. | Che, W.; Wen, D. C.; Zhu, S.-F.; Zhou, Q.-L. Org. Lett. 2018, 20, 3305–3309. doi:10.1021/acs.orglett.8b01193 |
| 40. | Che, W.; Wen, D. C.; Zhu, S.-F.; Zhou, Q.-L. Org. Lett. 2018, 20, 3305–3309. doi:10.1021/acs.orglett.8b01193 |
| 37. | Madduri, A. V. R.; Minnaard, A. J. Chem. – Eur. J. 2010, 16, 11726–11731. doi:10.1002/chem.201001284 |
| 27. | Nagamitsu, T.; Takano, D.; Fukuda, T.; Otoguro, K.; Kuwajima, I.; Harigaya, Y.; Ōmura, S. Org. Lett. 2004, 6, 1865–1867. doi:10.1021/ol049356w |
| 29. | Nagamitsu, T.; Takano, D.; Marumoto, K.; Fukuda, T.; Furuya, K.; Otoguro, K.; Takeda, K.; Kuwajima, I.; Harigaya, Y.; Ōmura, S. J. Org. Chem. 2007, 72, 2744–2756. doi:10.1021/jo062089i |
| 38. | Theurer, M.; El Baz, Y.; Koschorreck, K.; Urlacher, V. B.; Rauhut, G.; Baro, A.; Laschat, S. Eur. J. Org. Chem. 2011, 4241–4249. doi:10.1002/ejoc.201100412 |
| 38. | Theurer, M.; El Baz, Y.; Koschorreck, K.; Urlacher, V. B.; Rauhut, G.; Baro, A.; Laschat, S. Eur. J. Org. Chem. 2011, 4241–4249. doi:10.1002/ejoc.201100412 |
| 39. | Yadav, J. S.; Yadav, N. N. RSC Adv. 2013, 3, 4024–4032. doi:10.1039/c3ra22754e |
| 38. | Theurer, M.; El Baz, Y.; Koschorreck, K.; Urlacher, V. B.; Rauhut, G.; Baro, A.; Laschat, S. Eur. J. Org. Chem. 2011, 4241–4249. doi:10.1002/ejoc.201100412 |
| 34. | Krishna, C. V.; Maitra, S.; Dev, R. V.; Mukkanti, K.; Iqbal, J. Tetrahedron Lett. 2006, 47, 6103–6106. doi:10.1016/j.tetlet.2006.06.092 |
| 37. | Madduri, A. V. R.; Minnaard, A. J. Chem. – Eur. J. 2010, 16, 11726–11731. doi:10.1002/chem.201001284 |
| 51. | Noyori, R.; Ikeda, T.; Ohkuma, T.; Widhalm, M.; Kitamura, M.; Takaya, H.; Akutagawa, S.; Sayo, N.; Saito, T.; Taketomi, T.; Kumobayashi, H. J. Am. Chem. Soc. 1989, 111, 9134–9135. doi:10.1021/ja00207a038 |
| 37. | Madduri, A. V. R.; Minnaard, A. J. Chem. – Eur. J. 2010, 16, 11726–11731. doi:10.1002/chem.201001284 |
| 37. | Madduri, A. V. R.; Minnaard, A. J. Chem. – Eur. J. 2010, 16, 11726–11731. doi:10.1002/chem.201001284 |
| 39. | Yadav, J. S.; Yadav, N. N. RSC Adv. 2013, 3, 4024–4032. doi:10.1039/c3ra22754e |
| 40. | Che, W.; Wen, D. C.; Zhu, S.-F.; Zhou, Q.-L. Org. Lett. 2018, 20, 3305–3309. doi:10.1021/acs.orglett.8b01193 |
| 38. | Theurer, M.; El Baz, Y.; Koschorreck, K.; Urlacher, V. B.; Rauhut, G.; Baro, A.; Laschat, S. Eur. J. Org. Chem. 2011, 4241–4249. doi:10.1002/ejoc.201100412 |
| 1. | Berger, J.; Jampolsky, L. M.; Goldberg, M. W. Arch. Biochem. 1949, 22, 476–478. |
| 7. | Kawamura, T.; Liu, D.; Towle, M. J.; Kageyama, R.; Tsukahara, N.; Wakabayashi, T.; Littlefield, B. A. J. Antibiot. 2003, 56, 709–715. doi:10.7164/antibiotics.56.709 |
| 8. | Woolard, J.; Vousden, W.; Moss, S. J.; Krishnakumar, A.; Gammons, M. V. R.; Nowak, D. G.; Dixon, N.; Micklefield, J.; Spannhoff, A.; Bedford, M. T.; Gregory, M. A.; Martin, C. J.; Leadlay, P. F.; Zhang, M. Q.; Harper, S. J.; Bates, D. O.; Wilkinson, B. Chem. Sci. 2011, 2, 273–278. doi:10.1039/c0sc00297f |
| 9. | Olano, C.; Wilkinson, B.; Sánchez, C.; Moss, S. J.; Sheridan, R.; Math, V.; Weston, A. J.; Braña, A. F.; Martin, C. J.; Oliynyk, M.; Méndez, C.; Leadlay, P. F.; Salas, J. A. Chem. Biol. 2004, 11, 87–97. doi:10.1016/j.chembiol.2003.12.018 |
| 43. | Wilkinson, B.; Gregory, M. A.; Moss, S. J.; Carletti, I.; Sheridan, R. M.; Kaja, A.; Ward, M.; Olano, C.; Mendez, C.; Salas, J. A.; Leadlay, P. F.; vanGinckel, R.; Zhang, M.-Q. Bioorg. Med. Chem. Lett. 2006, 16, 5814–5817. doi:10.1016/j.bmcl.2006.08.073 |
| 6. | Jackson, L.; Bechler, S.; Miller, J.; Brownell, A.; Garshott, D.; Callaghan, M.; Fribley, A. M. Blood 2011, 118, 4260. doi:10.1182/blood.v118.21.4260.4260 |
| 3. | Gafiuc, D.; Weiß, M.; Mylonas, I.; Brüning, A. J. Appl. Toxicol. 2014, 34, 1109–1113. doi:10.1002/jat.2946 |
| 4. | Gao, X.; Jiang, Y.; Han, L.; Chen, X.; Hu, C.; Su, H.; Mu, Y.; Guan, P.; Huang, X. RSC Adv. 2017, 7, 44401–44409. doi:10.1039/c7ra08290h |
| 5. | Jeong, M.; Kim, H.; Kim, S.; Park, J.-H. Drug Delivery Transl. Res. 2018, 8, 1380–1388. doi:10.1007/s13346-018-0563-z |
| 36. | Yadav, J. S.; Bezawada, P.; Chenna, V. Tetrahedron Lett. 2009, 50, 3772–3775. doi:10.1016/j.tetlet.2009.03.196 |
| 37. | Madduri, A. V. R.; Minnaard, A. J. Chem. – Eur. J. 2010, 16, 11726–11731. doi:10.1002/chem.201001284 |
| 39. | Yadav, J. S.; Yadav, N. N. RSC Adv. 2013, 3, 4024–4032. doi:10.1039/c3ra22754e |
| 2. | Keller‐Schierlein, W. Helv. Chim. Acta 1967, 50, 731–753. doi:10.1002/hlca.19670500303 |
| 42. | Moss, S. J.; Carletti, I.; Olano, C.; Sheridan, R. M.; Ward, M.; Math, V.; Nur-E-Alam, M.; Braña, A. F.; Zhang, M. Q.; Leadlay, P. F.; Méndez, C.; Salas, J. A.; Wilkinson, B. Chem. Commun. 2006, 2341–2343. doi:10.1039/b602931k |
| 16. | Anderson, B. F.; Herlt, A. J.; Rickards, R. W.; Robertson, G. B. Aust. J. Chem. 1989, 42, 717–730. doi:10.1071/ch9890717 |
| 33. | Herber, C.; Breit, B. Chem. – Eur. J. 2006, 12, 6684–6691. doi:10.1002/chem.200600343 |
| 38. | Theurer, M.; El Baz, Y.; Koschorreck, K.; Urlacher, V. B.; Rauhut, G.; Baro, A.; Laschat, S. Eur. J. Org. Chem. 2011, 4241–4249. doi:10.1002/ejoc.201100412 |
| 14. | Ishiyama, A.; Iwatsuki, M.; Namatame, M.; Nishihara-Tsukashima, A.; Sunazuka, T.; Takahashi, Y.; Ōmura, S.; Otoguro, K. J. Antibiot. 2011, 64, 381–384. doi:10.1038/ja.2011.6 |
| 15. | Novita, R.; Suprayogi, A.; Agusta, A.; Nugraha, A. B.; Nozaki, T.; Agustini, K.; Darusman, H. S. Open Vet. J. 2024, 14, 2007. doi:10.5455/ovj.2024.v14.i8.30 |
| 25. | Duffey, M. O.; LeTiran, A.; Morken, J. P. J. Am. Chem. Soc. 2003, 125, 1458–1459. doi:10.1021/ja028941u |
| 11. | Gao, Y.-M.; Wang, X.-J.; Zhang, J.; Li, M.; Liu, C.-X.; An, J.; Jiang, L.; Xiang, W.-S. J. Agric. Food Chem. 2012, 60, 9874–9881. doi:10.1021/jf302857x |
| 12. | Liu, C.-X.; Zhang, J.; Wang, X.-J.; Qian, P.-T.; Wang, J.-D.; Gao, Y.-M.; Yan, Y.-J.; Zhang, S.-Z.; Xu, P.-F.; Li, W.-B.; Xiang, W.-S. J. Agric. Food Chem. 2012, 60, 1251–1257. doi:10.1021/jf2044982 |
| 13. | Wang, N.; Huang, W.; Jia, Q.; Song, B.; Wang, S.; Wu, L.; Sun, M.; Wang, Y.; Zhang, L.; Wang, W. J. Appl. Microbiol. 2025, 136, lxaf073. doi:10.1093/jambio/lxaf073 |
| 41. | Gembus, V.; Karmazin, L.; Uguen, D.; Zoller, T. Bull. Chem. Soc. Jpn. 2019, 92, 359–380. doi:10.1246/bcsj.20180292 |
| 10. | Sibero, M. T.; Frederick, E. H.; Wijayanti, D. P.; Haryanti, D.; Siswanto, A. P.; Igarashi, Y. J. Appl. Pharm. Sci. 2024, 14, 132–140. doi:10.7324/japs.2024.139056 |
| 23. | Zhang, L.; Shi, J.; Liu, C. L.; Xiang, L.; Ma, S. Y.; Li, W.; Jiao, R. H.; Tan, R. X.; Ge, H. M. Tetrahedron Lett. 2018, 59, 4517–4520. doi:10.1016/j.tetlet.2018.11.023 |
| 46. | Gündemir-Durmaz, T.; Schmid, F.; El Baz, Y.; Häusser, A.; Schneider, C.; Bilitewski, U.; Rauhut, G.; Garnier, D.; Baro, A.; Laschat, S. Org. Biomol. Chem. 2016, 14, 8261–8269. doi:10.1039/c6ob01358a |
| 47. | Hu, C.; Su, H.; Luo, J.; Han, L.; Liu, Q.; Wu, W.; Mu, Y.; Guan, P.; Sun, T.; Huang, X. Bioorg. Med. Chem. 2018, 26, 6035–6049. doi:10.1016/j.bmc.2018.11.005 |
| 44. | Sugawara, A.; Tanaka, T.; Hirose, T.; Ishiyama, A.; Iwatsuki, M.; Takahashi, Y.; Otoguro, K.; Ōmura, S.; Sunazuka, T. Bioorg. Med. Chem. Lett. 2013, 23, 2302–2305. doi:10.1016/j.bmcl.2013.02.075 |
| 45. | Hahn, F.; Kandziora, N.; Friedrich, S.; Leadlay, P. F. Beilstein J. Org. Chem. 2014, 10, 634–640. doi:10.3762/bjoc.10.55 |
| 41. | Gembus, V.; Karmazin, L.; Uguen, D.; Zoller, T. Bull. Chem. Soc. Jpn. 2019, 92, 359–380. doi:10.1246/bcsj.20180292 |
| 41. | Gembus, V.; Karmazin, L.; Uguen, D.; Zoller, T. Bull. Chem. Soc. Jpn. 2019, 92, 359–380. doi:10.1246/bcsj.20180292 |
| 49. | Novak, T.; Tan, Z.; Liang, B.; Negishi, E.-i. J. Am. Chem. Soc. 2005, 127, 2838–2839. doi:10.1021/ja043534z |
| 50. | Darna, B.; Kulkarni, R.; Uppuluru, A. K.; Gannu, P. K.; Gadiraju, R. R. J. J. Pharm. Res. (Mohali, India) 2011, 4, 1025–1030. |
| 48. | Nagamitsu, T.; Harigaya, Y.; Omura, S. Proc. Jpn. Acad., Ser. B 2005, 81, 244–256. doi:10.2183/pjab.81.244 |
| 30. | Haddad, N.; Brik, A.; Grishko, M. Tetrahedron Lett. 1997, 38, 6079–6082. doi:10.1016/s0040-4039(97)01372-5 |
| 31. | Haddad, N.; Grishko, M.; Brik, A. Tetrahedron Lett. 1997, 38, 6075–6078. doi:10.1016/s0040-4039(97)01371-3 |
| 22. | Sun, J.; Shao, J.; Sun, C.; Song, Y.; Li, Q.; Lu, L.; Hu, Y.; Gui, C.; Zhang, H.; Ju, J. Bioorg. Med. Chem. 2018, 26, 1488–1494. doi:10.1016/j.bmc.2018.01.010 |
| 22. | Sun, J.; Shao, J.; Sun, C.; Song, Y.; Li, Q.; Lu, L.; Hu, Y.; Gui, C.; Zhang, H.; Ju, J. Bioorg. Med. Chem. 2018, 26, 1488–1494. doi:10.1016/j.bmc.2018.01.010 |
| 23. | Zhang, L.; Shi, J.; Liu, C. L.; Xiang, L.; Ma, S. Y.; Li, W.; Jiao, R. H.; Tan, R. X.; Ge, H. M. Tetrahedron Lett. 2018, 59, 4517–4520. doi:10.1016/j.tetlet.2018.11.023 |
| 25. | Duffey, M. O.; LeTiran, A.; Morken, J. P. J. Am. Chem. Soc. 2003, 125, 1458–1459. doi:10.1021/ja028941u |
| 26. | Hanessian, S.; Yang, Y.; Giroux, S.; Mascitti, V.; Ma, J.; Raeppel, F. J. Am. Chem. Soc. 2003, 125, 13784–13792. doi:10.1021/ja030139k |
| 21. | Zhou, Z.; Wu, Q.; Xie, Q.; Ling, C.; Zhang, H.; Sun, C.; Ju, J. Chem. Biodiversity 2020, 17, e1900560. doi:10.1002/cbdv.201900560 |
| 23. | Zhang, L.; Shi, J.; Liu, C. L.; Xiang, L.; Ma, S. Y.; Li, W.; Jiao, R. H.; Tan, R. X.; Ge, H. M. Tetrahedron Lett. 2018, 59, 4517–4520. doi:10.1016/j.tetlet.2018.11.023 |
| 21. | Zhou, Z.; Wu, Q.; Xie, Q.; Ling, C.; Zhang, H.; Sun, C.; Ju, J. Chem. Biodiversity 2020, 17, e1900560. doi:10.1002/cbdv.201900560 |
| 21. | Zhou, Z.; Wu, Q.; Xie, Q.; Ling, C.; Zhang, H.; Sun, C.; Ju, J. Chem. Biodiversity 2020, 17, e1900560. doi:10.1002/cbdv.201900560 |
| 21. | Zhou, Z.; Wu, Q.; Xie, Q.; Ling, C.; Zhang, H.; Sun, C.; Ju, J. Chem. Biodiversity 2020, 17, e1900560. doi:10.1002/cbdv.201900560 |
| 23. | Zhang, L.; Shi, J.; Liu, C. L.; Xiang, L.; Ma, S. Y.; Li, W.; Jiao, R. H.; Tan, R. X.; Ge, H. M. Tetrahedron Lett. 2018, 59, 4517–4520. doi:10.1016/j.tetlet.2018.11.023 |
| 23. | Zhang, L.; Shi, J.; Liu, C. L.; Xiang, L.; Ma, S. Y.; Li, W.; Jiao, R. H.; Tan, R. X.; Ge, H. M. Tetrahedron Lett. 2018, 59, 4517–4520. doi:10.1016/j.tetlet.2018.11.023 |
| 27. | Nagamitsu, T.; Takano, D.; Fukuda, T.; Otoguro, K.; Kuwajima, I.; Harigaya, Y.; Ōmura, S. Org. Lett. 2004, 6, 1865–1867. doi:10.1021/ol049356w |
| 28. | Vong, B. G.; Kim, S. H.; Abraham, S.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2004, 43, 3947–3951. doi:10.1002/anie.200460203 |
| 29. | Nagamitsu, T.; Takano, D.; Marumoto, K.; Fukuda, T.; Furuya, K.; Otoguro, K.; Takeda, K.; Kuwajima, I.; Harigaya, Y.; Ōmura, S. J. Org. Chem. 2007, 72, 2744–2756. doi:10.1021/jo062089i |
| 36. | Yadav, J. S.; Bezawada, P.; Chenna, V. Tetrahedron Lett. 2009, 50, 3772–3775. doi:10.1016/j.tetlet.2009.03.196 |
| 37. | Madduri, A. V. R.; Minnaard, A. J. Chem. – Eur. J. 2010, 16, 11726–11731. doi:10.1002/chem.201001284 |
| 34. | Krishna, C. V.; Maitra, S.; Dev, R. V.; Mukkanti, K.; Iqbal, J. Tetrahedron Lett. 2006, 47, 6103–6106. doi:10.1016/j.tetlet.2006.06.092 |
| 35. | Vamsee Krishna, C.; Bhonde, V. R.; Devendar, A.; Maitra, S.; Mukkanti, K.; Iqbal, J. Tetrahedron Lett. 2008, 49, 2013–2017. doi:10.1016/j.tetlet.2008.01.055 |
| 32. | Vong, B. G.; Abraham, S.; Xiang, A. X.; Theodorakis, E. A. Org. Lett. 2003, 5, 1617–1620. doi:10.1021/ol034243i |
| 33. | Herber, C.; Breit, B. Chem. – Eur. J. 2006, 12, 6684–6691. doi:10.1002/chem.200600343 |
| 30. | Haddad, N.; Brik, A.; Grishko, M. Tetrahedron Lett. 1997, 38, 6079–6082. doi:10.1016/s0040-4039(97)01372-5 |
| 31. | Haddad, N.; Grishko, M.; Brik, A. Tetrahedron Lett. 1997, 38, 6075–6078. doi:10.1016/s0040-4039(97)01371-3 |
| 53. | Santaniello, E.; Ferraboschi, P.; Grisenti, P. Tetrahedron Lett. 1990, 31, 5657–5660. doi:10.1016/s0040-4039(00)97925-5 |
| 54. | Bertucci, C.; Petri, A.; Felix, G.; Perini, B.; Salvadori, P. Tetrahedron: Asymmetry 1999, 10, 4455–4462. doi:10.1016/s0957-4166(99)00478-4 |
| 35. | Vamsee Krishna, C.; Bhonde, V. R.; Devendar, A.; Maitra, S.; Mukkanti, K.; Iqbal, J. Tetrahedron Lett. 2008, 49, 2013–2017. doi:10.1016/j.tetlet.2008.01.055 |
| 33. | Herber, C.; Breit, B. Chem. – Eur. J. 2006, 12, 6684–6691. doi:10.1002/chem.200600343 |
| 52. | Meyer, S. D.; Miwa, T.; Nakatsuka, M.; Schreiber, S. L. J. Org. Chem. 1992, 57, 5058–5060. doi:10.1021/jo00045a007 |
| 37. | Madduri, A. V. R.; Minnaard, A. J. Chem. – Eur. J. 2010, 16, 11726–11731. doi:10.1002/chem.201001284 |
| 33. | Herber, C.; Breit, B. Chem. – Eur. J. 2006, 12, 6684–6691. doi:10.1002/chem.200600343 |
| 27. | Nagamitsu, T.; Takano, D.; Fukuda, T.; Otoguro, K.; Kuwajima, I.; Harigaya, Y.; Ōmura, S. Org. Lett. 2004, 6, 1865–1867. doi:10.1021/ol049356w |
| 29. | Nagamitsu, T.; Takano, D.; Marumoto, K.; Fukuda, T.; Furuya, K.; Otoguro, K.; Takeda, K.; Kuwajima, I.; Harigaya, Y.; Ōmura, S. J. Org. Chem. 2007, 72, 2744–2756. doi:10.1021/jo062089i |
| 22. | Sun, J.; Shao, J.; Sun, C.; Song, Y.; Li, Q.; Lu, L.; Hu, Y.; Gui, C.; Zhang, H.; Ju, J. Bioorg. Med. Chem. 2018, 26, 1488–1494. doi:10.1016/j.bmc.2018.01.010 |
| 23. | Zhang, L.; Shi, J.; Liu, C. L.; Xiang, L.; Ma, S. Y.; Li, W.; Jiao, R. H.; Tan, R. X.; Ge, H. M. Tetrahedron Lett. 2018, 59, 4517–4520. doi:10.1016/j.tetlet.2018.11.023 |
| 20. | Sidhu, A.; Miller, J. R.; Tripathi, A.; Garshott, D. M.; Brownell, A. L.; Chiego, D. J.; Arevang, C.; Zeng, Q.; Jackson, L. C.; Bechler, S. A.; Callaghan, M. U.; Yoo, G. H.; Sethi, S.; Lin, H.-S.; Callaghan, J. H.; Tamayo-Castillo, G.; Sherman, D. H.; Kaufman, R. J.; Fribley, A. M. ACS Med. Chem. Lett. 2015, 6, 1122–1127. doi:10.1021/acsmedchemlett.5b00133 |
| 21. | Zhou, Z.; Wu, Q.; Xie, Q.; Ling, C.; Zhang, H.; Sun, C.; Ju, J. Chem. Biodiversity 2020, 17, e1900560. doi:10.1002/cbdv.201900560 |
| 18. | Hamed, A.; Abdel-Razek, A. S.; Frese, M.; Wibberg, D.; El-Haddad, A. F.; Ibrahim, T. M.; Kalinowski, J.; Sewald, N.; Shaaban, M. Z. Naturforsch., C: J. Biosci. 2018, 73, 49–57. doi:10.1515/znc-2017-0140 |
| 55. | Mori, K.; Kuwahara, S. Tetrahedron 1986, 42, 5545–5550. doi:10.1016/s0040-4020(01)88158-8 |
| 19. | Kim, J.; Shin, D.; Kim, S.-H.; Park, W.; Shin, Y.; Kim, W. K.; Lee, S. K.; Oh, K.-B.; Shin, J.; Oh, D.-C. Mar. Drugs 2017, 15, 166. doi:10.3390/md15060166 |
| 35. | Vamsee Krishna, C.; Bhonde, V. R.; Devendar, A.; Maitra, S.; Mukkanti, K.; Iqbal, J. Tetrahedron Lett. 2008, 49, 2013–2017. doi:10.1016/j.tetlet.2008.01.055 |
| 17. | Schulze, C. J.; Bray, W. M.; Loganzo, F.; Lam, M.-H.; Szal, T.; Villalobos, A.; Koehn, F. E.; Linington, R. G. J. Nat. Prod. 2014, 77, 2570–2574. doi:10.1021/np500727g |
| 35. | Vamsee Krishna, C.; Bhonde, V. R.; Devendar, A.; Maitra, S.; Mukkanti, K.; Iqbal, J. Tetrahedron Lett. 2008, 49, 2013–2017. doi:10.1016/j.tetlet.2008.01.055 |
| 17. | Schulze, C. J.; Bray, W. M.; Loganzo, F.; Lam, M.-H.; Szal, T.; Villalobos, A.; Koehn, F. E.; Linington, R. G. J. Nat. Prod. 2014, 77, 2570–2574. doi:10.1021/np500727g |
| 24. | Olano, C.; Moss, S. J.; Braña, A. F.; Sheridan, R. M.; Math, V.; Weston, A. J.; Méndez, C.; Leadlay, P. F.; Wilkinson, B.; Salas, J. A. Mol. Microbiol. 2004, 52, 1745–1756. doi:10.1111/j.1365-2958.2004.04090.x |
| 1. | Berger, J.; Jampolsky, L. M.; Goldberg, M. W. Arch. Biochem. 1949, 22, 476–478. |
| 22. | Sun, J.; Shao, J.; Sun, C.; Song, Y.; Li, Q.; Lu, L.; Hu, Y.; Gui, C.; Zhang, H.; Ju, J. Bioorg. Med. Chem. 2018, 26, 1488–1494. doi:10.1016/j.bmc.2018.01.010 |
| 22. | Sun, J.; Shao, J.; Sun, C.; Song, Y.; Li, Q.; Lu, L.; Hu, Y.; Gui, C.; Zhang, H.; Ju, J. Bioorg. Med. Chem. 2018, 26, 1488–1494. doi:10.1016/j.bmc.2018.01.010 |
| 19. | Kim, J.; Shin, D.; Kim, S.-H.; Park, W.; Shin, Y.; Kim, W. K.; Lee, S. K.; Oh, K.-B.; Shin, J.; Oh, D.-C. Mar. Drugs 2017, 15, 166. doi:10.3390/md15060166 |
| 19. | Kim, J.; Shin, D.; Kim, S.-H.; Park, W.; Shin, Y.; Kim, W. K.; Lee, S. K.; Oh, K.-B.; Shin, J.; Oh, D.-C. Mar. Drugs 2017, 15, 166. doi:10.3390/md15060166 |
| 21. | Zhou, Z.; Wu, Q.; Xie, Q.; Ling, C.; Zhang, H.; Sun, C.; Ju, J. Chem. Biodiversity 2020, 17, e1900560. doi:10.1002/cbdv.201900560 |
| 23. | Zhang, L.; Shi, J.; Liu, C. L.; Xiang, L.; Ma, S. Y.; Li, W.; Jiao, R. H.; Tan, R. X.; Ge, H. M. Tetrahedron Lett. 2018, 59, 4517–4520. doi:10.1016/j.tetlet.2018.11.023 |
| 20. | Sidhu, A.; Miller, J. R.; Tripathi, A.; Garshott, D. M.; Brownell, A. L.; Chiego, D. J.; Arevang, C.; Zeng, Q.; Jackson, L. C.; Bechler, S. A.; Callaghan, M. U.; Yoo, G. H.; Sethi, S.; Lin, H.-S.; Callaghan, J. H.; Tamayo-Castillo, G.; Sherman, D. H.; Kaufman, R. J.; Fribley, A. M. ACS Med. Chem. Lett. 2015, 6, 1122–1127. doi:10.1021/acsmedchemlett.5b00133 |
| 21. | Zhou, Z.; Wu, Q.; Xie, Q.; Ling, C.; Zhang, H.; Sun, C.; Ju, J. Chem. Biodiversity 2020, 17, e1900560. doi:10.1002/cbdv.201900560 |
| 20. | Sidhu, A.; Miller, J. R.; Tripathi, A.; Garshott, D. M.; Brownell, A. L.; Chiego, D. J.; Arevang, C.; Zeng, Q.; Jackson, L. C.; Bechler, S. A.; Callaghan, M. U.; Yoo, G. H.; Sethi, S.; Lin, H.-S.; Callaghan, J. H.; Tamayo-Castillo, G.; Sherman, D. H.; Kaufman, R. J.; Fribley, A. M. ACS Med. Chem. Lett. 2015, 6, 1122–1127. doi:10.1021/acsmedchemlett.5b00133 |
| 18. | Hamed, A.; Abdel-Razek, A. S.; Frese, M.; Wibberg, D.; El-Haddad, A. F.; Ibrahim, T. M.; Kalinowski, J.; Sewald, N.; Shaaban, M. Z. Naturforsch., C: J. Biosci. 2018, 73, 49–57. doi:10.1515/znc-2017-0140 |
| 19. | Kim, J.; Shin, D.; Kim, S.-H.; Park, W.; Shin, Y.; Kim, W. K.; Lee, S. K.; Oh, K.-B.; Shin, J.; Oh, D.-C. Mar. Drugs 2017, 15, 166. doi:10.3390/md15060166 |
© 2025 Kurniawan et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.