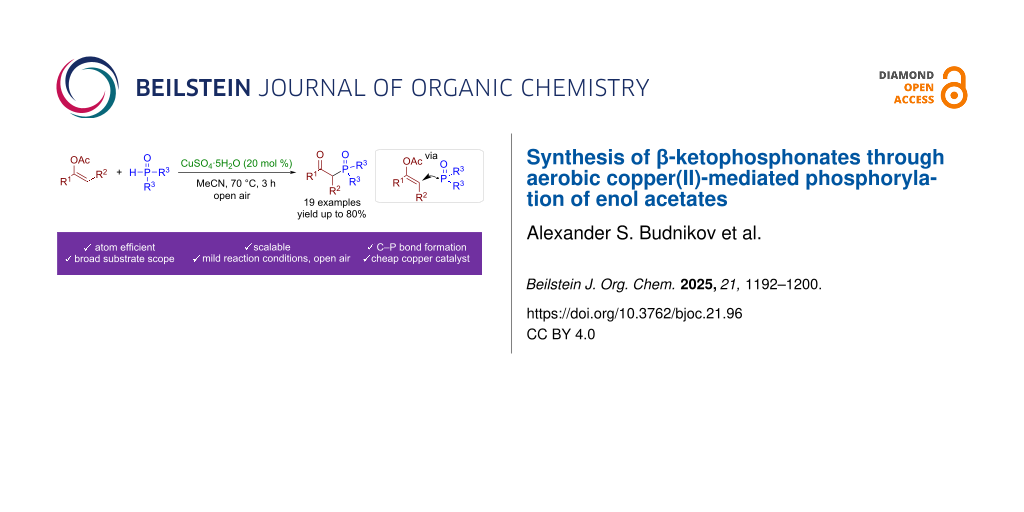
Abstract
Aerobic copper(II)-mediated phosphorylation of enol acetates with H-phosphonates leading to the formation of β-ketophosphonates was discovered. The proposed method is applicable to a wide range of H-phosphonates or phosphine oxides as PH-reagents and enol acetates. Unlike previous reports, which generally employed stoichiometric amounts of oxidants or more expensive transition metal catalysts, the present protocol employs only cheap copper sulfate pentahydrate as a catalyst under mild reaction conditions. The achieved phosphorylation proceeds via the formation of P-centered radicals produced by the oxidation of PH-reagents by copper(II)-containing species. Employing anhydrous CuSO4 instead of the pentahydrate led to a dramatic phosphorylation yield drop from 70 to <5%. It seems that the ligand environment of copper is very important for the effective reaction: other Cu(II) and Cu(I) salts, including halides, nitrate, tetrafluoroborate, or perchlorate, were much less effective or completely inert.

Graphical Abstract
Introduction
The construction of C–P bonds is a highly important task in key areas of modern chemistry [1-5] due to the numerous applications of phosphorus-containing compounds in pharmaceuticals, biology, agrochemistry, organic synthesis, and materials science [6-13].
Among various organophosphorus compounds, β-ketophosphonates have received particular attention for various synthetically useful transformations, including alkene synthesis via Horner–Wadsworth–Emmons reaction [14,15], heterocycle construction [16,17], and the synthesis of chiral β-amino and β-hydroxy phosphonic acids [18,19]. Furthermore, they exhibit metal-complexing abilities [20], and anti-inflammatory [21,22] as well as enzyme inhibition activities [23-26]. Traditionally, β-ketophosphonates were prepared via Arbuzov reaction [27], acylation of alkylphosphonates [28], and hydration of alkynylphosphonates [29-31]. However, these methods have several drawbacks, including low atom efficiency, strong basic or acidic conditions, and excess of organohalides as starting materials. Recent years have witnessed the upsurge of free-radical oxidative phosphorylation transformations that became a reliable strategy for the construction of C–P bonds in organophosphorus chemistry [2,32-39]. The primary benefit of these reactions is introducing phosphorus fragments under mild reaction conditions into a diverse array of compounds that are inaccessible for functionalization employing other traditional approaches. After the pioneering work of Ji and Wei on aerobic oxyphosphorylation of styrenes [40], this strategy was further extended [41] to phenylacetylenes [42-44], cinnamic [45-48] and α,β-alkynyl carboxylic acids [42], and vinyl azides [49,50] (Scheme 1a). As a rule, transition metal catalysts (Fe [42,44,51,52], Cu [42,44,46,51,53], Ag [54], Mn [55], etc.) and strong oxidants (K2S2O8 [46,54], Mn(OAc)3 [56,57], organic peroxides [51,58,59], etc.) are employed in these approaches. Modern photocatalytic [47,50,60,61] and electrochemical [48,62] methods were also recently reported.
![[1860-5397-21-96-i1]](/bjoc/content/inline/1860-5397-21-96-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Recent approaches for the synthesis of β-ketophosphonates by the oxyphosphorylation of unsaturated compounds.
Scheme 1: Recent approaches for the synthesis of β-ketophosphonates by the oxyphosphorylation of unsaturated ...
Although several successful oxyphosphorylation reactions leading to β-ketophosphonates have been reported [41-50,61], challenges in this area still exist primely in the search for new available synthetic equivalents of alkynes and alkenes for effective radical C–P bond formation. Enol acetates are potentially versatile precursors of α-substituted carbonyl compounds that have been recently applied as coupling partners for radical functionalization with the formation of C–C [63-66] and C–Het [67-70] bonds. The ready availability of enol acetates from the corresponding carbonyl compounds in just one synthetic step distinguishes them from the less accessible substituted vinylarenes, alkynes, cinnamic and α,β-alkynyl carboxylic acids, as well as vinyl azides. In addition, compared to similar silyl and alkyl enol ethers, enol acetates are more resistant to solvolysis, which prevents unwanted side reactions leading to the corresponding unfunctionalized carbonyl compounds. To date, only the Xu group reported oxidative phosphorylation of enol acetates with dialkyl H-phosphonates and Mn(acac)3 as an oxidant (Scheme 1b) [71]. However, the reported approach is limited by an excess of manganese salt, long reaction times, and scope limitations.
In the present work, the selective copper(II)-mediated phosphorylation of enol acetates with the formation of substituted β-ketophosphonates employing cheap copper sulfate as a catalyst and atmospheric oxygen as a terminal oxidant was carried out (Scheme 1c).
Results and Discussion
We commenced our study with 1-phenylvinyl acetate (1a) and diisopropyl H-phosphonate (2a) as the model substrates by optimizing the reaction conditions (Table 1).
Table 1: Optimization of reaction conditions for the synthesis of β-ketophosphonate 3a from enol acetate 1a and diisopropyl H-phosphonate (2a)a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-96-i6.svg?max-width=637&scale=1.0)
|
||
| Entry | Variations from standard reaction conditions | Yield 3a, %b |
| 1 | none | 70 (68) |
| 2 | without CuSO4·5H2O | n.d. |
| 3 | anhydrous CuSO4 (20 mol %) | <5 |
| 4 | 1/5/10 mol % of CuSO4·5H2O | 24/56/64 |
| 5 | rt, 24 h | 9 |
| 6 | DMF/DMSO/AcOH/toluene/EtOH instead of MeCN | <5 |
| 7 | Cu(NO3)2·2.5H2O (20 mol %) | 25 |
| 8 | Cu(ClO4)2·6H2O (20 mol %) | 23 |
| 9 | Cu(BF4)2·xH2O (20 mol %) | 39 |
| 10 | CuBr2 (20 mol %) | n.d. |
| 11 | CuBr (20 mol %) | 13 |
| 12 | CuCl (20 mol %) | n.d. |
| 13 | CuCl2 (20 mol %) | n.d. |
| 14 | Cu(OAc)2 (20 mol %) | <5 |
| 15 | Cu(acac)2 (20 mol %) | n.d. |
| 16 | Mn(acac)3 (20 mol %) | 7 |
| 17 | FeCl3 (20 mol %) | n.d. |
| 18 | CoSO4 (20 mol %) | n.d. |
| 19 | CoCO3 (20 mol %) | n.d. |
| 20 | AgNO3 (20 mol %) | 11 |
aStandard reaction conditions: enol acetate 1a (0.5 mmol, 81 mg), diisopropyl phosphite (2a, 1 mmol, 166 mg), catalyst (0–20 mol %, 0–37 mg), and a solvent (5 mL) were sequentially added to a round-bottom flask. The reaction mixture was stirred for 3 hours at 70 °C under air (air condenser); bYields of 3a were determined by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane as an internal standard. The yield of isolated product is given in parenthesis.
After screening reaction conditions (see Table S1 in Supporting Information File 1 for more details) it was found that the best results were obtained employing 20 mol % of copper(II) sulfate pentahydrate and 2-fold excess of 2a in MeCN at 70 °C under air atmosphere (Table 1, entry 1). In the absence of copper sulfate, no reaction product was detected (Table 1, entry 2). Notably, employing anhydrous copper sulfate afforded the desired product only in trace amounts (Table 1, entry 3). The optimal loading of the catalyst was examined (Table 1, entry 4); the best results were obtained with 20 mol % of copper sulfate, slightly lower yield (64%) was observed with 10 mol % loading of the catalyst. Increased temperature was crucial for the reaction output (Table 1, entry 5). The solvent screening revealed that MeCN is the best choice for the discovered transformation (Table 1, entry 6). Employing other copper salts as catalysts (Table 1, entries 7–15) resulted in lower yields compared to copper sulfate. In the case of copper(II) chloride and bromide (Table 1, entries 10 and 13) the formation of acetophenone as the main reaction product was observed. Presumably, in the presence of these salts, which can act as Lewis acids, the hydrolysis of the enol acetate 1a proceeds faster than the generation of the corresponding P-centered radical from diisopropyl H-phosphonate (2a). In addition, the anion affects the redox properties of the copper ion, and thus the optimal choice is important to achieve both sufficient oxidative properties and catalyst recycling by reoxidation with O2. The employment of Mn(acac)3 and FeCl3, which had previously proven themselves effective in the oxyphosphorylation of styrenes [42,44,51,52] and enol acetates [71], turned out to be unsuitable in our case: the yield of 3a did not exceed 7% (Table 1, entries 16 and 17). In the case of FeCl3 the formation of acetophenone as the main reaction product was observed. Other transition metal salts were also ineffective in the discovered transformation (Table 1, entries 18–20).
With optimal conditions in hand, the scope of the developed phosphorylation protocol was evaluated (Scheme 2). The discovered phosphorylation is applicable to various substituted enol acetates 1a–n, possessing both electron-donating and electron-withdrawing groups, leading to the desired phosphorylation products 3a–n in good yields. Notably, sterically hindered terminal substituted enol acetates also furnished the desired products 3c–f, albeit in lower yields. The substitution pattern in the aryl ring does not have a significant effect on the yield of 3 for both acceptor and donor derivatives, which shows the versatility of the developed protocol. However, among halogen-containing derivatives, higher yields were observed for bromine-substituted ones (products 3i and 3l, 80% and 79% yield). A furan moiety was also tolerated, giving the corresponding phosphorylated product 3n in 19% yield.
![[1860-5397-21-96-i2]](/bjoc/content/inline/1860-5397-21-96-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The scope of the discovered copper(II)-mediated phosphorylation of enol acetates.
Scheme 2: The scope of the discovered copper(II)-mediated phosphorylation of enol acetates.
Next, we evaluated the scope of the phosphorous component (products 3p–r). The best result was obtained with di-n-butyl H-phosphonate (product 3q). To our delight, diphenylphosphine oxide gave the corresponding product 3s in 58% yield, although earlier 3s wasn’t observed in an analogous reaction when Mn(acac)3 was used as an oxidant [71].
The synthetic utility of the developed protocol was demonstrated by performing the model reaction on a 6 mmol scale (Scheme 3), product 3a was isolated in 77% yield (1.3 g, 4.61 mmol).
![[1860-5397-21-96-i3]](/bjoc/content/inline/1860-5397-21-96-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
In order to propose the reaction mechanism, control experiments were conducted (Scheme 4).
![[1860-5397-21-96-i4]](/bjoc/content/inline/1860-5397-21-96-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The reaction is completely inhibited by the addition of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) or BHT (Scheme 4, reaction 1). A BHT-adduct derived from a P-centered radical was detected by HRMS, which supported a radical mechanism. Next, the influence of the atmosphere and the role of copper catalyst was evaluated (Scheme 4, reaction 2). Neither under argon nor under an oxygen atmosphere, the yield of 3a did not exceed 22%. However, employing 200 mol % of copper(II) sulfate under an argon atmosphere afforded 3a in 33% yield. These results indicate, that copper(II) at high loading is capable of oxidizing H-phosphonate even in inert conditions [72]; however, a balanced concentration of oxygen in the reaction mixture is required for recycling of copper catalyst and effective generation of phosphorous radicals. Under a pure O2 atmosphere, the interception of formed P-centered radicals by excess oxygen can presumably inhibit the target process by the formation of unreactive phosphoric acid from the corresponding H-phosphonate [73,74]. The formation of phosphoric acid from diisopropyl H-phosphonate (2a) was confirmed by HRMS analysis of the crude reaction mixture. Finally, vinyl azide 4a and silyl enol ether 4b were introduced into standard reaction conditions (Scheme 4, reaction 3). However, no phosphorylation product 3a was observed.
On the basis of the obtained results and previous reports on copper(II) mediated oxyphosphorylation reactions [42,44,46,51,53], a plausible reaction mechanism is proposed (Scheme 5).
![[1860-5397-21-96-i5]](/bjoc/content/inline/1860-5397-21-96-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed mechanism for copper(II) mediated phosphorylation of enol acetates.
Scheme 5: Proposed mechanism for copper(II) mediated phosphorylation of enol acetates.
The discovered transformation is unlikely to proceed via only a single route (see control experiments in Scheme 4), and the generation of phosphorus radicals and product 3 can be described by several pathways. The direct oxidation of 2 by Cu(II) A leads to the formation of P-centered radical E and Cu(I) B. Under air atmosphere, the formed Cu(I) species B can react with molecular oxygen resulting in the formation of peroxycopper intermediate C [75-79]. The latter abstracts hydrogen atom from H-phosphonate or phosphine oxide 2, thus generating P-centered radical E and hydroperoxide complex D. Subsequently, the addition of E to the double bond of enol acetate 1 leads to the formation of benzylic radical G. The latter undergoes hydroperoxide transfer from copper complex D with the formation of intermediate H and, therefore, regenerating Cu(I) B. Alternatively, the latter can be formed by oxidation of benzylic radical G with the formation of carbocation I. However, given that in the absence of oxygen the β-ketophosphonate was formed only in 33% yield (Scheme 4, reaction 2d), the direct oxidation of G to I is unlikely to be the main reaction pathway. Finally, the reduction of hydroperoxide H by initial phosphorous precursor 2 [80-82] or by Cu(I) B delivers β-ketophosphonate 3 and P-centered radical E or regenerated Cu(II) A. Alternatively, 3 can be formed by hydrolysis of benzylic cation I [71].
Conclusion
In summary, we have disclosed an aerobic copper(II)-mediated phosphorylation of enol acetates with H-phosphonates leading to substituted β-ketophosphonates. The suggested method is versatile and can also be applied to phosphine oxides as PH-reagents. The developed protocol utilizes a simple copper catalyst under mild reaction conditions and synthetically available enol acetates (compared to traditionally employed alkenes, alkynes, cinnamic, and α,β-alkynyl carboxylic acids). The present approach showed good functional group tolerance and can be applied for the gram-scale synthesis of target compounds. Conducted mechanistic studies revealed that the discovered transformation proceeds via the formation of P-centered radicals produced by the oxidation of the corresponding phosphorous precursors by copper(II)-containing species.
Experimental
General: In all experiments rt stands for 22–25 °C. 1H and 13C NMR spectra were recorded on Bruker AVANCE II 300, Bruker Fourier 300HD (300.13 MHz for 1H, 75.47 MHz for 13C and 121.49 MHz for 31P, respectively), and Bruker AVANCE II 600 (600.13 MHz for 1H, 150.92 MHz for 13C, 242.93 MHz for 31P) spectrometers in CDCl3. Chemical shifts were reported in parts per million (ppm), and the residual solvent peak was used as an internal reference: 1H (СDCl3 δ = 7.26 ppm), 13C (СDCl3 δ = 77.16 ppm). Multiplicity was indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). Coupling constants are reported in Hertz (Hz).
FTIR spectra were recorded on a Bruker Alpha instrument. High-resolution mass spectra (HRMS) were measured on a Bruker maXis instrument using electrospray ionization (ESI). The measurements were performed in a positive ion mode (interface capillary voltage – 4500 V); mass range from m/z 50 to m/z 3000 Da; external calibration with Electrospray Calibrant Solution (Fluka). A syringe injection was used for all acetonitrile solutions (flow rate 3 μL/min). Nitrogen was applied as a dry gas; interface temperature was set at 180 °C.
General reaction conditions for copper(II)-mediated phosphorylation of enolacetates (experimental data for Scheme 2)
Enol acetate 1 (0.5 mmol, 81–121 mg), H-phosphonate 2 (1.0 mmol, 110–234 mg), CuSO4·5H2O (0.1 mmol, 25 mg), and MeCN (5 mL) were sequentially added to a round-bottom flask. The reaction mixture (suspension) was stirred for 3 hours at 70 °C under air (air condenser) and then cooled to room temperature, and rotary-evaporated under reduced pressure. An additional evaporation step using a rotary vane pump (0.5 mmHg) at 80 °C was made for the evaporation of phosphite excess. The residue was isolated by column chromatography on silica gel.
Supporting Information
| Supporting Information File 1: Experimental details, compound characterization data, and NMR spectra. | ||
| Format: PDF | Size: 4.4 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Tang, J.; Ni, J.; Chen, Q. Tetrahedron Lett. 2024, 149, 155266. doi:10.1016/j.tetlet.2024.155266
Return to citation in text: [1] -
Hore, S.; Singh, R. P. Org. Biomol. Chem. 2022, 20, 498–537. doi:10.1039/d1ob02003j
Return to citation in text: [1] [2] -
Chen, T.; Zhang, J.-S.; Han, L.-B. Dalton Trans. 2016, 45, 1843–1849. doi:10.1039/c5dt01896j
Return to citation in text: [1] -
Sbei, N.; Martins, G. M.; Shirinfar, B.; Ahmed, N. Chem. Rec. 2020, 20, 1530–1552. doi:10.1002/tcr.202000096
Return to citation in text: [1] -
Muhammad, M. H.; Chen, X.-L.; Yu, B.; Qu, L.-B.; Zhao, Y.-F. Pure Appl. Chem. 2019, 91, 33–41. doi:10.1515/pac-2018-0906
Return to citation in text: [1] -
Hu, Z.-W.; Ma, M.-R.; Chen, Y.-X.; Zhao, Y.-F.; Qiang, W.; Li, Y.-M. J. Biol. Chem. 2017, 292, 8846. doi:10.1074/jbc.a116.757179
Return to citation in text: [1] -
Baumgartner, T.; Réau, R. Chem. Rev. 2006, 106, 4681–4727. doi:10.1021/cr040179m
Return to citation in text: [1] -
Chen, Y.-X.; Du, J.-T.; Zhou, L.-X.; Liu, X.-H.; Zhao, Y.-F.; Nakanishi, H.; Li, Y.-M. Chem. Biol. 2006, 13, 937–944. doi:10.1016/j.chembiol.2006.06.017
Return to citation in text: [1] -
Li, W.-H.; Wu, J.-J.; Wu, L.; Zhang, B.-D.; Hu, H.-G.; Zhao, L.; Li, Z.-B.; Yu, X.-F.; Li, Y.-M. Biomaterials 2021, 273, 120788. doi:10.1016/j.biomaterials.2021.120788
Return to citation in text: [1] -
Monge, S.; Canniccioni, B.; Graillot, A.; Robin, J.-J. Biomacromolecules 2011, 12, 1973–1982. doi:10.1021/bm2004803
Return to citation in text: [1] -
Kitamura, M.; Tokunaga, M.; Noyori, R. J. Am. Chem. Soc. 1995, 117, 2931–2932. doi:10.1021/ja00115a030
Return to citation in text: [1] -
Feng, W.; Teo, X.-Y.; Novera, W.; Ramanujulu, P. M.; Liang, D.; Huang, D.; Moore, P. K.; Deng, L.-W.; Dymock, B. W. J. Med. Chem. 2015, 58, 6456–6480. doi:10.1021/acs.jmedchem.5b00848
Return to citation in text: [1] -
Weinschenk, L.; Schols, D.; Balzarini, J.; Meier, C. J. Med. Chem. 2015, 58, 6114–6130. doi:10.1021/acs.jmedchem.5b00737
Return to citation in text: [1] -
Boutagy, J.; Thomas, R. Chem. Rev. 1974, 74, 87–99. doi:10.1021/cr60287a005
Return to citation in text: [1] -
Bisceglia, A. J.; Orelli, R. L. Curr. Org. Chem. 2015, 19, 744–775. doi:10.2174/1385272819666150311231006
Return to citation in text: [1] -
Khalladi, K.; Touil, S. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 711–718. doi:10.1080/10426507.2012.700354
Return to citation in text: [1] -
González‐Calderón, D.; Fuentes‐Benítes, A.; Díaz‐Torres, E.; González‐González, C. A.; González‐Romero, C. Eur. J. Org. Chem. 2016, 668–672. doi:10.1002/ejoc.201501465
Return to citation in text: [1] -
Ryglowski, A.; Kafarski, P. Tetrahedron 1996, 52, 10685–10692. doi:10.1016/0040-4020(96)00590-x
Return to citation in text: [1] -
Tao, X.; Li, W.; Ma, X.; Li, X.; Fan, W.; Zhu, L.; Xie, X.; Zhang, Z. J. Org. Chem. 2012, 77, 8401–8409. doi:10.1021/jo301532t
Return to citation in text: [1] -
Mitchell, L. A.; Holliday, B. J. ACS Macro Lett. 2016, 5, 1100–1103. doi:10.1021/acsmacrolett.6b00596
Return to citation in text: [1] -
Balg, C.; Blais, S. P.; Bernier, S.; Huot, J. L.; Couture, M.; Lapointe, J.; Chênevert, R. Bioorg. Med. Chem. 2007, 15, 295–304. doi:10.1016/j.bmc.2006.09.056
Return to citation in text: [1] -
Paterson, I.; Gardner, N. M.; Poullennec, K. G.; Wright, A. E. Bioorg. Med. Chem. Lett. 2007, 17, 2443–2447. doi:10.1016/j.bmcl.2007.02.031
Return to citation in text: [1] -
Perumal, S. K.; Adediran, S. A.; Pratt, R. F. Bioorg. Med. Chem. 2008, 16, 6987–6994. doi:10.1016/j.bmc.2008.05.045
Return to citation in text: [1] -
Bernier, S.; Akochy, P.-M.; Lapointe, J.; Chênevert, R. Bioorg. Med. Chem. 2005, 13, 69–75. doi:10.1016/j.bmc.2004.09.055
Return to citation in text: [1] -
Cavallaro, V.; Moglie, Y. F.; Murray, A. P.; Radivoy, G. E. Bioorg. Chem. 2018, 77, 420–428. doi:10.1016/j.bioorg.2018.01.030
Return to citation in text: [1] -
Auberger, N.; Frlan, R.; Al-Dabbagh, B.; Bouhss, A.; Crouvoisier, M.; Gravier-Pelletier, C.; Merrer, Y. L. Org. Biomol. Chem. 2011, 9, 8301. doi:10.1039/c1ob06124k
Return to citation in text: [1] -
Moorhoff, C. M. Synth. Commun. 2003, 33, 2069–2086. doi:10.1081/scc-120021033
Return to citation in text: [1] -
Cavalla, D.; Guéguen, C.; Nelson, A.; O'Brien, P.; Russell, M. G.; Warren, S. Tetrahedron Lett. 1996, 37, 7465–7468. doi:10.1016/0040-4039(96)01612-7
Return to citation in text: [1] -
Xiang, J.; Yi, N.; Wang, R.; Lu, L.; Zou, H.; Pan, Y.; He, W. Tetrahedron 2015, 71, 694–699. doi:10.1016/j.tet.2014.12.001
Return to citation in text: [1] -
Chen, L.-L.; Zhang, J.-W.; Yang, W.-W.; Chen, P.; Chen, D.-Y.; Wang, Y.-B. Org. Biomol. Chem. 2019, 17, 3003–3009. doi:10.1039/c9ob00251k
Return to citation in text: [1] -
Li, X.; Hu, G.; Luo, P.; Tang, G.; Gao, Y.; Xu, P.; Zhao, Y. Adv. Synth. Catal. 2012, 354, 2427–2432. doi:10.1002/adsc.201200420
Return to citation in text: [1] -
Budnikova, Y. H. Dokl. Chem. 2022, 507, 229–259. doi:10.1134/s0012500822600353
Return to citation in text: [1] -
Pan, X.-Q.; Zou, J.-P.; Yi, W.-B.; Zhang, W. Tetrahedron 2015, 71, 7481–7529. doi:10.1016/j.tet.2015.04.117
Return to citation in text: [1] -
Gao, Y.; Tang, G.; Zhao, Y. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 589–596. doi:10.1080/10426507.2017.1295965
Return to citation in text: [1] -
Liu, J.; Xiao, H.-Z.; Fu, Q.; Yu, D.-G. Chem. Synth. 2021, 1, 9. doi:10.20517/cs.2021.07
Return to citation in text: [1] -
Zeng, F.-L.; Zhang, Z.-Y.; Yin, P.-C.; Cheng, F.-K.; Chen, X.-L.; Qu, L.-B.; Cao, Z.-Y.; Yu, B. Org. Lett. 2022, 24, 7912–7917. doi:10.1021/acs.orglett.2c02930
Return to citation in text: [1] -
Liu, Y.; Chen, X.-L.; Li, X.-Y.; Zhu, S.-S.; Li, S.-J.; Song, Y.; Qu, L.-B.; Yu, B. J. Am. Chem. Soc. 2021, 143, 964–972. doi:10.1021/jacs.0c11138
Return to citation in text: [1] -
Huang, A.-X.; Fu, Y.-R.; Zhu, H.-L.; Zeng, F.-L.; Chen, X.-L.; Tang, S.; Qu, L.-B.; Yu, B. J. Org. Chem. 2022, 87, 14433–14442. doi:10.1021/acs.joc.2c01890
Return to citation in text: [1] -
Zeng, F.-L.; Chen, X.-L.; Sun, K.; Zhu, H.-L.; Yuan, X.-Y.; Liu, Y.; Qu, L.-B.; Zhao, Y.-F.; Yu, B. Org. Chem. Front. 2021, 8, 760–766. doi:10.1039/d0qo01410a
Return to citation in text: [1] -
Wei, W.; Ji, J.-X. Angew. Chem., Int. Ed. 2011, 50, 9097–9099. doi:10.1002/anie.201100219
Return to citation in text: [1] -
Bakhtiary, A.; Poor Heravi, M. R.; Hassanpour, A.; Amini, I.; Vessally, E. RSC Adv. 2021, 11, 470–483. doi:10.1039/d0ra08074h
Return to citation in text: [1] [2] -
Zhou, M.; Chen, M.; Zhou, Y.; Yang, K.; Su, J.; Du, J.; Song, Q. Org. Lett. 2015, 17, 1786–1789. doi:10.1021/acs.orglett.5b00574
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Macías-Benítez, P.; Sierra-Padilla, A.; Tenorio, M. J.; Moreno-Dorado, F. J.; Guerra, F. M. J. Org. Chem. 2021, 86, 16409–16424. doi:10.1021/acs.joc.1c01763
Return to citation in text: [1] [2] -
Yi, N.; Wang, R.; Zou, H.; He, W.; Fu, W.; He, W. J. Org. Chem. 2015, 80, 5023–5029. doi:10.1021/acs.joc.5b00408
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Chen, X.; Chen, X.; Li, X.; Qu, C.; Qu, L.; Bi, W.; Sun, K.; Zhao, Y. Tetrahedron 2017, 73, 2439–2446. doi:10.1016/j.tet.2017.03.026
Return to citation in text: [1] [2] -
Tang, L.; Wen, L.; Sun, T.; Zhang, D.; Yang, Z.; Feng, C.; Wang, Z. Asian J. Org. Chem. 2017, 6, 1683–1692. doi:10.1002/ajoc.201700434
Return to citation in text: [1] [2] [3] [4] [5] -
Qian, H.-F.; Li, C.-K.; Zhou, Z.-H.; Tao, Z.-K.; Shoberu, A.; Zou, J.-P. Org. Lett. 2018, 20, 5947–5951. doi:10.1021/acs.orglett.8b02639
Return to citation in text: [1] [2] [3] -
Yang, J.; Sun, X.; Yan, K.; Sun, H.; Sun, S.; Jia, X.; Zhang, F.; Wen, J. Adv. Synth. Catal. 2022, 364, 2735–2740. doi:10.1002/adsc.202200421
Return to citation in text: [1] [2] [3] -
Tang, P.; Zhang, C.; Chen, E.; Chen, B.; Chen, W.; Yu, Y. Tetrahedron Lett. 2017, 58, 2157–2161. doi:10.1016/j.tetlet.2017.04.069
Return to citation in text: [1] [2] -
Jung, H. I.; Kim, D. Y. Synth. Commun. 2020, 50, 380–387. doi:10.1080/00397911.2019.1696364
Return to citation in text: [1] [2] [3] -
Gu, J.; Cai, C. Org. Biomol. Chem. 2017, 15, 4226–4230. doi:10.1039/c7ob00505a
Return to citation in text: [1] [2] [3] [4] [5] -
Zhao, Y.; Xu, J.; Chen, H.; Xia, Y.; Hu, L. Adv. Synth. Catal. 2023, 365, 3290–3294. doi:10.1002/adsc.202300733
Return to citation in text: [1] [2] -
Fang, J.; Ren, H.; Xu, S.; Huang, C.; Jiang, Y.; Zhang, W.; You, S.; Qin, B. Org. Lett. 2024, 26, 5458–5462. doi:10.1021/acs.orglett.4c01716
Return to citation in text: [1] [2] -
Gui, Q.; Hu, L.; Chen, X.; Liu, J.; Tan, Z. Chem. Commun. 2015, 51, 13922–13924. doi:10.1039/c5cc04826e
Return to citation in text: [1] [2] -
Yamamoto, D.; Ansai, H.; Hoshino, J.; Makino, K. Chem. Pharm. Bull. 2018, 66, 873–879. doi:10.1248/cpb.c18-00381
Return to citation in text: [1] -
Zhou, P.; Hu, B.; Li, L.; Rao, K.; Yang, J.; Yu, F. J. Org. Chem. 2017, 82, 13268–13276. doi:10.1021/acs.joc.7b02391
Return to citation in text: [1] -
Zhang, G.-Y.; Li, C.-K.; Li, D.-P.; Zeng, R.-S.; Shoberu, A.; Zou, J.-P. Tetrahedron 2016, 72, 2972–2978. doi:10.1016/j.tet.2016.04.013
Return to citation in text: [1] -
Pan, J.; Feng, Y.; Lv, L.; Li, Z. Adv. Synth. Catal. 2024, 366, 3860–3867. doi:10.1002/adsc.202400606
Return to citation in text: [1] -
Moghaddam, F. M.; Daneshfar, M.; Azaryan, R.; Pirat, J.-L. Catal. Commun. 2020, 141, 106015. doi:10.1016/j.catcom.2020.106015
Return to citation in text: [1] -
Shi, Y.; Chen, R.; Guo, K.; Meng, F.; Cao, S.; Gu, C.; Zhu, Y. Tetrahedron Lett. 2018, 59, 2062–2065. doi:10.1016/j.tetlet.2018.04.040
Return to citation in text: [1] -
Bu, M.-j.; Lu, G.-p.; Cai, C. Catal. Sci. Technol. 2016, 6, 413–416. doi:10.1039/c5cy01541c
Return to citation in text: [1] [2] -
Tarasov, M. V.; Khrizanforova, V. V.; Gryaznova, T. V.; Budnikova, Y. H. Russ. J. Electrochem. 2023, 59, 896–905. doi:10.1134/s1023193523110137
Return to citation in text: [1] -
Li, Y.; Ma, Z.; Liu, X.; Li, Z.; Zhao, F.; Wu, J. Org. Chem. Front. 2023, 10, 1710–1714. doi:10.1039/d3qo00052d
Return to citation in text: [1] -
Lu, Y.; Li, Y.; Zhang, R.; Jin, K.; Duan, C. J. Fluorine Chem. 2014, 161, 128–133. doi:10.1016/j.jfluchem.2014.01.020
Return to citation in text: [1] -
Wu, F.; Liu, S.; Lv, X.; Pan, M.; Liu, X.; Zhang, J.; Rong, L. J. Org. Chem. 2023, 88, 13749–13759. doi:10.1021/acs.joc.3c01407
Return to citation in text: [1] -
Vil’, V. A.; Merkulova, V. M.; Ilovaisky, A. I.; Paveliev, S. A.; Nikishin, G. I.; Terent’ev, A. O. Org. Lett. 2021, 23, 5107–5112. doi:10.1021/acs.orglett.1c01643
Return to citation in text: [1] -
Zaikina, L. A.; Mulina, O. M.; Merkulova, V. M.; Ilovaisky, A. I.; Vil’, V. A.; Terent’ev, A. O. ChemistrySelect 2024, 9, e202403708. doi:10.1002/slct.202403708
Return to citation in text: [1] -
Zhang, P.; Ma, J.; Liu, X.; Xue, F.; Zhang, Y.; Wang, B.; Jin, W.; Xia, Y.; Liu, C. J. Org. Chem. 2023, 88, 16122–16131. doi:10.1021/acs.joc.3c01417
Return to citation in text: [1] -
de Souza, A. A. N.; Bartolomeu, A. d. A.; Brocksom, T. J.; Noël, T.; de Oliveira, K. T. J. Org. Chem. 2022, 87, 5856–5865. doi:10.1021/acs.joc.2c00147
Return to citation in text: [1] -
Li, Y.-L.; Shi, Z.; Shen, T.; Ye, K.-Y. Beilstein J. Org. Chem. 2022, 18, 1026–1031. doi:10.3762/bjoc.18.103
Return to citation in text: [1] -
Huo, B.; Guo, C.; Xu, Z. Chin. J. Org. Chem. 2023, 43, 3989. doi:10.6023/cjoc202304013
Return to citation in text: [1] [2] [3] [4] -
Zhou, Y.; Yin, S.; Gao, Y.; Zhao, Y.; Goto, M.; Han, L.-B. Angew. Chem., Int. Ed. 2010, 49, 6852–6855. doi:10.1002/anie.201003484
Return to citation in text: [1] -
Kang, D.; Kim, T.; Lee, H.; Hong, S. Org. Lett. 2018, 20, 7571–7575. doi:10.1021/acs.orglett.8b03309
Return to citation in text: [1] -
Peng, P.; Peng, L.; Wang, G.; Wang, F.; Luo, Y.; Lei, A. Org. Chem. Front. 2016, 3, 749–752. doi:10.1039/c6qo00049e
Return to citation in text: [1] -
Allen, S. E.; Walvoord, R. R.; Padilla-Salinas, R.; Kozlowski, M. C. Chem. Rev. 2013, 113, 6234–6458. doi:10.1021/cr300527g
Return to citation in text: [1] -
Punniyamurthy, T.; Rout, L. Coord. Chem. Rev. 2008, 252, 134–154. doi:10.1016/j.ccr.2007.04.003
Return to citation in text: [1] -
Mills, S. A.; Klinman, J. P. J. Am. Chem. Soc. 2000, 122, 9897–9904. doi:10.1021/ja000325f
Return to citation in text: [1] -
Toh, K. K.; Wang, Y.-F.; Ng, E. P. J.; Chiba, S. J. Am. Chem. Soc. 2011, 133, 13942–13945. doi:10.1021/ja206580j
Return to citation in text: [1] -
Maiti, D.; Fry, H. C.; Woertink, J. S.; Vance, M. A.; Solomon, E. I.; Karlin, K. D. J. Am. Chem. Soc. 2007, 129, 264–265. doi:10.1021/ja067411l
Return to citation in text: [1] -
Gutierrez, V.; Mascaró, E.; Alonso, F.; Moglie, Y.; Radivoy, G. RSC Adv. 2015, 5, 65739–65744. doi:10.1039/c5ra10223e
Return to citation in text: [1] -
Fu, Q.; Yi, D.; Zhang, Z.; Liang, W.; Chen, S.; Yang, L.; Zhang, Q.; Ji, J.; Wei, W. Org. Chem. Front. 2017, 4, 1385–1389. doi:10.1039/c7qo00202e
Return to citation in text: [1] -
Zhong, W.-W.; Zhang, Q.; Li, M.-S.; Hu, D.-Y.; Cheng, M.; Du, F.-T.; Ji, J.-X.; Wei, W. Synth. Commun. 2016, 46, 1377–1385. doi:10.1080/00397911.2016.1205196
Return to citation in text: [1]
| 41. | Bakhtiary, A.; Poor Heravi, M. R.; Hassanpour, A.; Amini, I.; Vessally, E. RSC Adv. 2021, 11, 470–483. doi:10.1039/d0ra08074h |
| 42. | Zhou, M.; Chen, M.; Zhou, Y.; Yang, K.; Su, J.; Du, J.; Song, Q. Org. Lett. 2015, 17, 1786–1789. doi:10.1021/acs.orglett.5b00574 |
| 43. | Macías-Benítez, P.; Sierra-Padilla, A.; Tenorio, M. J.; Moreno-Dorado, F. J.; Guerra, F. M. J. Org. Chem. 2021, 86, 16409–16424. doi:10.1021/acs.joc.1c01763 |
| 44. | Yi, N.; Wang, R.; Zou, H.; He, W.; Fu, W.; He, W. J. Org. Chem. 2015, 80, 5023–5029. doi:10.1021/acs.joc.5b00408 |
| 45. | Chen, X.; Chen, X.; Li, X.; Qu, C.; Qu, L.; Bi, W.; Sun, K.; Zhao, Y. Tetrahedron 2017, 73, 2439–2446. doi:10.1016/j.tet.2017.03.026 |
| 46. | Tang, L.; Wen, L.; Sun, T.; Zhang, D.; Yang, Z.; Feng, C.; Wang, Z. Asian J. Org. Chem. 2017, 6, 1683–1692. doi:10.1002/ajoc.201700434 |
| 47. | Qian, H.-F.; Li, C.-K.; Zhou, Z.-H.; Tao, Z.-K.; Shoberu, A.; Zou, J.-P. Org. Lett. 2018, 20, 5947–5951. doi:10.1021/acs.orglett.8b02639 |
| 48. | Yang, J.; Sun, X.; Yan, K.; Sun, H.; Sun, S.; Jia, X.; Zhang, F.; Wen, J. Adv. Synth. Catal. 2022, 364, 2735–2740. doi:10.1002/adsc.202200421 |
| 49. | Tang, P.; Zhang, C.; Chen, E.; Chen, B.; Chen, W.; Yu, Y. Tetrahedron Lett. 2017, 58, 2157–2161. doi:10.1016/j.tetlet.2017.04.069 |
| 50. | Jung, H. I.; Kim, D. Y. Synth. Commun. 2020, 50, 380–387. doi:10.1080/00397911.2019.1696364 |
| 61. | Bu, M.-j.; Lu, G.-p.; Cai, C. Catal. Sci. Technol. 2016, 6, 413–416. doi:10.1039/c5cy01541c |
| 63. | Li, Y.; Ma, Z.; Liu, X.; Li, Z.; Zhao, F.; Wu, J. Org. Chem. Front. 2023, 10, 1710–1714. doi:10.1039/d3qo00052d |
| 64. | Lu, Y.; Li, Y.; Zhang, R.; Jin, K.; Duan, C. J. Fluorine Chem. 2014, 161, 128–133. doi:10.1016/j.jfluchem.2014.01.020 |
| 65. | Wu, F.; Liu, S.; Lv, X.; Pan, M.; Liu, X.; Zhang, J.; Rong, L. J. Org. Chem. 2023, 88, 13749–13759. doi:10.1021/acs.joc.3c01407 |
| 66. | Vil’, V. A.; Merkulova, V. M.; Ilovaisky, A. I.; Paveliev, S. A.; Nikishin, G. I.; Terent’ev, A. O. Org. Lett. 2021, 23, 5107–5112. doi:10.1021/acs.orglett.1c01643 |
| 67. | Zaikina, L. A.; Mulina, O. M.; Merkulova, V. M.; Ilovaisky, A. I.; Vil’, V. A.; Terent’ev, A. O. ChemistrySelect 2024, 9, e202403708. doi:10.1002/slct.202403708 |
| 68. | Zhang, P.; Ma, J.; Liu, X.; Xue, F.; Zhang, Y.; Wang, B.; Jin, W.; Xia, Y.; Liu, C. J. Org. Chem. 2023, 88, 16122–16131. doi:10.1021/acs.joc.3c01417 |
| 69. | de Souza, A. A. N.; Bartolomeu, A. d. A.; Brocksom, T. J.; Noël, T.; de Oliveira, K. T. J. Org. Chem. 2022, 87, 5856–5865. doi:10.1021/acs.joc.2c00147 |
| 70. | Li, Y.-L.; Shi, Z.; Shen, T.; Ye, K.-Y. Beilstein J. Org. Chem. 2022, 18, 1026–1031. doi:10.3762/bjoc.18.103 |
| 1. | Tang, J.; Ni, J.; Chen, Q. Tetrahedron Lett. 2024, 149, 155266. doi:10.1016/j.tetlet.2024.155266 |
| 2. | Hore, S.; Singh, R. P. Org. Biomol. Chem. 2022, 20, 498–537. doi:10.1039/d1ob02003j |
| 3. | Chen, T.; Zhang, J.-S.; Han, L.-B. Dalton Trans. 2016, 45, 1843–1849. doi:10.1039/c5dt01896j |
| 4. | Sbei, N.; Martins, G. M.; Shirinfar, B.; Ahmed, N. Chem. Rec. 2020, 20, 1530–1552. doi:10.1002/tcr.202000096 |
| 5. | Muhammad, M. H.; Chen, X.-L.; Yu, B.; Qu, L.-B.; Zhao, Y.-F. Pure Appl. Chem. 2019, 91, 33–41. doi:10.1515/pac-2018-0906 |
| 18. | Ryglowski, A.; Kafarski, P. Tetrahedron 1996, 52, 10685–10692. doi:10.1016/0040-4020(96)00590-x |
| 19. | Tao, X.; Li, W.; Ma, X.; Li, X.; Fan, W.; Zhu, L.; Xie, X.; Zhang, Z. J. Org. Chem. 2012, 77, 8401–8409. doi:10.1021/jo301532t |
| 42. | Zhou, M.; Chen, M.; Zhou, Y.; Yang, K.; Su, J.; Du, J.; Song, Q. Org. Lett. 2015, 17, 1786–1789. doi:10.1021/acs.orglett.5b00574 |
| 43. | Macías-Benítez, P.; Sierra-Padilla, A.; Tenorio, M. J.; Moreno-Dorado, F. J.; Guerra, F. M. J. Org. Chem. 2021, 86, 16409–16424. doi:10.1021/acs.joc.1c01763 |
| 44. | Yi, N.; Wang, R.; Zou, H.; He, W.; Fu, W.; He, W. J. Org. Chem. 2015, 80, 5023–5029. doi:10.1021/acs.joc.5b00408 |
| 42. | Zhou, M.; Chen, M.; Zhou, Y.; Yang, K.; Su, J.; Du, J.; Song, Q. Org. Lett. 2015, 17, 1786–1789. doi:10.1021/acs.orglett.5b00574 |
| 44. | Yi, N.; Wang, R.; Zou, H.; He, W.; Fu, W.; He, W. J. Org. Chem. 2015, 80, 5023–5029. doi:10.1021/acs.joc.5b00408 |
| 46. | Tang, L.; Wen, L.; Sun, T.; Zhang, D.; Yang, Z.; Feng, C.; Wang, Z. Asian J. Org. Chem. 2017, 6, 1683–1692. doi:10.1002/ajoc.201700434 |
| 51. | Gu, J.; Cai, C. Org. Biomol. Chem. 2017, 15, 4226–4230. doi:10.1039/c7ob00505a |
| 53. | Fang, J.; Ren, H.; Xu, S.; Huang, C.; Jiang, Y.; Zhang, W.; You, S.; Qin, B. Org. Lett. 2024, 26, 5458–5462. doi:10.1021/acs.orglett.4c01716 |
| 16. | Khalladi, K.; Touil, S. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 711–718. doi:10.1080/10426507.2012.700354 |
| 17. | González‐Calderón, D.; Fuentes‐Benítes, A.; Díaz‐Torres, E.; González‐González, C. A.; González‐Romero, C. Eur. J. Org. Chem. 2016, 668–672. doi:10.1002/ejoc.201501465 |
| 45. | Chen, X.; Chen, X.; Li, X.; Qu, C.; Qu, L.; Bi, W.; Sun, K.; Zhao, Y. Tetrahedron 2017, 73, 2439–2446. doi:10.1016/j.tet.2017.03.026 |
| 46. | Tang, L.; Wen, L.; Sun, T.; Zhang, D.; Yang, Z.; Feng, C.; Wang, Z. Asian J. Org. Chem. 2017, 6, 1683–1692. doi:10.1002/ajoc.201700434 |
| 47. | Qian, H.-F.; Li, C.-K.; Zhou, Z.-H.; Tao, Z.-K.; Shoberu, A.; Zou, J.-P. Org. Lett. 2018, 20, 5947–5951. doi:10.1021/acs.orglett.8b02639 |
| 48. | Yang, J.; Sun, X.; Yan, K.; Sun, H.; Sun, S.; Jia, X.; Zhang, F.; Wen, J. Adv. Synth. Catal. 2022, 364, 2735–2740. doi:10.1002/adsc.202200421 |
| 75. | Allen, S. E.; Walvoord, R. R.; Padilla-Salinas, R.; Kozlowski, M. C. Chem. Rev. 2013, 113, 6234–6458. doi:10.1021/cr300527g |
| 76. | Punniyamurthy, T.; Rout, L. Coord. Chem. Rev. 2008, 252, 134–154. doi:10.1016/j.ccr.2007.04.003 |
| 77. | Mills, S. A.; Klinman, J. P. J. Am. Chem. Soc. 2000, 122, 9897–9904. doi:10.1021/ja000325f |
| 78. | Toh, K. K.; Wang, Y.-F.; Ng, E. P. J.; Chiba, S. J. Am. Chem. Soc. 2011, 133, 13942–13945. doi:10.1021/ja206580j |
| 79. | Maiti, D.; Fry, H. C.; Woertink, J. S.; Vance, M. A.; Solomon, E. I.; Karlin, K. D. J. Am. Chem. Soc. 2007, 129, 264–265. doi:10.1021/ja067411l |
| 14. | Boutagy, J.; Thomas, R. Chem. Rev. 1974, 74, 87–99. doi:10.1021/cr60287a005 |
| 15. | Bisceglia, A. J.; Orelli, R. L. Curr. Org. Chem. 2015, 19, 744–775. doi:10.2174/1385272819666150311231006 |
| 40. | Wei, W.; Ji, J.-X. Angew. Chem., Int. Ed. 2011, 50, 9097–9099. doi:10.1002/anie.201100219 |
| 72. | Zhou, Y.; Yin, S.; Gao, Y.; Zhao, Y.; Goto, M.; Han, L.-B. Angew. Chem., Int. Ed. 2010, 49, 6852–6855. doi:10.1002/anie.201003484 |
| 6. | Hu, Z.-W.; Ma, M.-R.; Chen, Y.-X.; Zhao, Y.-F.; Qiang, W.; Li, Y.-M. J. Biol. Chem. 2017, 292, 8846. doi:10.1074/jbc.a116.757179 |
| 7. | Baumgartner, T.; Réau, R. Chem. Rev. 2006, 106, 4681–4727. doi:10.1021/cr040179m |
| 8. | Chen, Y.-X.; Du, J.-T.; Zhou, L.-X.; Liu, X.-H.; Zhao, Y.-F.; Nakanishi, H.; Li, Y.-M. Chem. Biol. 2006, 13, 937–944. doi:10.1016/j.chembiol.2006.06.017 |
| 9. | Li, W.-H.; Wu, J.-J.; Wu, L.; Zhang, B.-D.; Hu, H.-G.; Zhao, L.; Li, Z.-B.; Yu, X.-F.; Li, Y.-M. Biomaterials 2021, 273, 120788. doi:10.1016/j.biomaterials.2021.120788 |
| 10. | Monge, S.; Canniccioni, B.; Graillot, A.; Robin, J.-J. Biomacromolecules 2011, 12, 1973–1982. doi:10.1021/bm2004803 |
| 11. | Kitamura, M.; Tokunaga, M.; Noyori, R. J. Am. Chem. Soc. 1995, 117, 2931–2932. doi:10.1021/ja00115a030 |
| 12. | Feng, W.; Teo, X.-Y.; Novera, W.; Ramanujulu, P. M.; Liang, D.; Huang, D.; Moore, P. K.; Deng, L.-W.; Dymock, B. W. J. Med. Chem. 2015, 58, 6456–6480. doi:10.1021/acs.jmedchem.5b00848 |
| 13. | Weinschenk, L.; Schols, D.; Balzarini, J.; Meier, C. J. Med. Chem. 2015, 58, 6114–6130. doi:10.1021/acs.jmedchem.5b00737 |
| 41. | Bakhtiary, A.; Poor Heravi, M. R.; Hassanpour, A.; Amini, I.; Vessally, E. RSC Adv. 2021, 11, 470–483. doi:10.1039/d0ra08074h |
| 73. | Kang, D.; Kim, T.; Lee, H.; Hong, S. Org. Lett. 2018, 20, 7571–7575. doi:10.1021/acs.orglett.8b03309 |
| 74. | Peng, P.; Peng, L.; Wang, G.; Wang, F.; Luo, Y.; Lei, A. Org. Chem. Front. 2016, 3, 749–752. doi:10.1039/c6qo00049e |
| 27. | Moorhoff, C. M. Synth. Commun. 2003, 33, 2069–2086. doi:10.1081/scc-120021033 |
| 29. | Xiang, J.; Yi, N.; Wang, R.; Lu, L.; Zou, H.; Pan, Y.; He, W. Tetrahedron 2015, 71, 694–699. doi:10.1016/j.tet.2014.12.001 |
| 30. | Chen, L.-L.; Zhang, J.-W.; Yang, W.-W.; Chen, P.; Chen, D.-Y.; Wang, Y.-B. Org. Biomol. Chem. 2019, 17, 3003–3009. doi:10.1039/c9ob00251k |
| 31. | Li, X.; Hu, G.; Luo, P.; Tang, G.; Gao, Y.; Xu, P.; Zhao, Y. Adv. Synth. Catal. 2012, 354, 2427–2432. doi:10.1002/adsc.201200420 |
| 71. | Huo, B.; Guo, C.; Xu, Z. Chin. J. Org. Chem. 2023, 43, 3989. doi:10.6023/cjoc202304013 |
| 23. | Perumal, S. K.; Adediran, S. A.; Pratt, R. F. Bioorg. Med. Chem. 2008, 16, 6987–6994. doi:10.1016/j.bmc.2008.05.045 |
| 24. | Bernier, S.; Akochy, P.-M.; Lapointe, J.; Chênevert, R. Bioorg. Med. Chem. 2005, 13, 69–75. doi:10.1016/j.bmc.2004.09.055 |
| 25. | Cavallaro, V.; Moglie, Y. F.; Murray, A. P.; Radivoy, G. E. Bioorg. Chem. 2018, 77, 420–428. doi:10.1016/j.bioorg.2018.01.030 |
| 26. | Auberger, N.; Frlan, R.; Al-Dabbagh, B.; Bouhss, A.; Crouvoisier, M.; Gravier-Pelletier, C.; Merrer, Y. L. Org. Biomol. Chem. 2011, 9, 8301. doi:10.1039/c1ob06124k |
| 2. | Hore, S.; Singh, R. P. Org. Biomol. Chem. 2022, 20, 498–537. doi:10.1039/d1ob02003j |
| 32. | Budnikova, Y. H. Dokl. Chem. 2022, 507, 229–259. doi:10.1134/s0012500822600353 |
| 33. | Pan, X.-Q.; Zou, J.-P.; Yi, W.-B.; Zhang, W. Tetrahedron 2015, 71, 7481–7529. doi:10.1016/j.tet.2015.04.117 |
| 34. | Gao, Y.; Tang, G.; Zhao, Y. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 589–596. doi:10.1080/10426507.2017.1295965 |
| 35. | Liu, J.; Xiao, H.-Z.; Fu, Q.; Yu, D.-G. Chem. Synth. 2021, 1, 9. doi:10.20517/cs.2021.07 |
| 36. | Zeng, F.-L.; Zhang, Z.-Y.; Yin, P.-C.; Cheng, F.-K.; Chen, X.-L.; Qu, L.-B.; Cao, Z.-Y.; Yu, B. Org. Lett. 2022, 24, 7912–7917. doi:10.1021/acs.orglett.2c02930 |
| 37. | Liu, Y.; Chen, X.-L.; Li, X.-Y.; Zhu, S.-S.; Li, S.-J.; Song, Y.; Qu, L.-B.; Yu, B. J. Am. Chem. Soc. 2021, 143, 964–972. doi:10.1021/jacs.0c11138 |
| 38. | Huang, A.-X.; Fu, Y.-R.; Zhu, H.-L.; Zeng, F.-L.; Chen, X.-L.; Tang, S.; Qu, L.-B.; Yu, B. J. Org. Chem. 2022, 87, 14433–14442. doi:10.1021/acs.joc.2c01890 |
| 39. | Zeng, F.-L.; Chen, X.-L.; Sun, K.; Zhu, H.-L.; Yuan, X.-Y.; Liu, Y.; Qu, L.-B.; Zhao, Y.-F.; Yu, B. Org. Chem. Front. 2021, 8, 760–766. doi:10.1039/d0qo01410a |
| 71. | Huo, B.; Guo, C.; Xu, Z. Chin. J. Org. Chem. 2023, 43, 3989. doi:10.6023/cjoc202304013 |
| 21. | Balg, C.; Blais, S. P.; Bernier, S.; Huot, J. L.; Couture, M.; Lapointe, J.; Chênevert, R. Bioorg. Med. Chem. 2007, 15, 295–304. doi:10.1016/j.bmc.2006.09.056 |
| 22. | Paterson, I.; Gardner, N. M.; Poullennec, K. G.; Wright, A. E. Bioorg. Med. Chem. Lett. 2007, 17, 2443–2447. doi:10.1016/j.bmcl.2007.02.031 |
| 71. | Huo, B.; Guo, C.; Xu, Z. Chin. J. Org. Chem. 2023, 43, 3989. doi:10.6023/cjoc202304013 |
| 20. | Mitchell, L. A.; Holliday, B. J. ACS Macro Lett. 2016, 5, 1100–1103. doi:10.1021/acsmacrolett.6b00596 |
| 28. | Cavalla, D.; Guéguen, C.; Nelson, A.; O'Brien, P.; Russell, M. G.; Warren, S. Tetrahedron Lett. 1996, 37, 7465–7468. doi:10.1016/0040-4039(96)01612-7 |
| 42. | Zhou, M.; Chen, M.; Zhou, Y.; Yang, K.; Su, J.; Du, J.; Song, Q. Org. Lett. 2015, 17, 1786–1789. doi:10.1021/acs.orglett.5b00574 |
| 44. | Yi, N.; Wang, R.; Zou, H.; He, W.; Fu, W.; He, W. J. Org. Chem. 2015, 80, 5023–5029. doi:10.1021/acs.joc.5b00408 |
| 51. | Gu, J.; Cai, C. Org. Biomol. Chem. 2017, 15, 4226–4230. doi:10.1039/c7ob00505a |
| 52. | Zhao, Y.; Xu, J.; Chen, H.; Xia, Y.; Hu, L. Adv. Synth. Catal. 2023, 365, 3290–3294. doi:10.1002/adsc.202300733 |
| 42. | Zhou, M.; Chen, M.; Zhou, Y.; Yang, K.; Su, J.; Du, J.; Song, Q. Org. Lett. 2015, 17, 1786–1789. doi:10.1021/acs.orglett.5b00574 |
| 44. | Yi, N.; Wang, R.; Zou, H.; He, W.; Fu, W.; He, W. J. Org. Chem. 2015, 80, 5023–5029. doi:10.1021/acs.joc.5b00408 |
| 51. | Gu, J.; Cai, C. Org. Biomol. Chem. 2017, 15, 4226–4230. doi:10.1039/c7ob00505a |
| 52. | Zhao, Y.; Xu, J.; Chen, H.; Xia, Y.; Hu, L. Adv. Synth. Catal. 2023, 365, 3290–3294. doi:10.1002/adsc.202300733 |
| 42. | Zhou, M.; Chen, M.; Zhou, Y.; Yang, K.; Su, J.; Du, J.; Song, Q. Org. Lett. 2015, 17, 1786–1789. doi:10.1021/acs.orglett.5b00574 |
| 80. | Gutierrez, V.; Mascaró, E.; Alonso, F.; Moglie, Y.; Radivoy, G. RSC Adv. 2015, 5, 65739–65744. doi:10.1039/c5ra10223e |
| 81. | Fu, Q.; Yi, D.; Zhang, Z.; Liang, W.; Chen, S.; Yang, L.; Zhang, Q.; Ji, J.; Wei, W. Org. Chem. Front. 2017, 4, 1385–1389. doi:10.1039/c7qo00202e |
| 82. | Zhong, W.-W.; Zhang, Q.; Li, M.-S.; Hu, D.-Y.; Cheng, M.; Du, F.-T.; Ji, J.-X.; Wei, W. Synth. Commun. 2016, 46, 1377–1385. doi:10.1080/00397911.2016.1205196 |
| 49. | Tang, P.; Zhang, C.; Chen, E.; Chen, B.; Chen, W.; Yu, Y. Tetrahedron Lett. 2017, 58, 2157–2161. doi:10.1016/j.tetlet.2017.04.069 |
| 50. | Jung, H. I.; Kim, D. Y. Synth. Commun. 2020, 50, 380–387. doi:10.1080/00397911.2019.1696364 |
| 71. | Huo, B.; Guo, C.; Xu, Z. Chin. J. Org. Chem. 2023, 43, 3989. doi:10.6023/cjoc202304013 |
| 47. | Qian, H.-F.; Li, C.-K.; Zhou, Z.-H.; Tao, Z.-K.; Shoberu, A.; Zou, J.-P. Org. Lett. 2018, 20, 5947–5951. doi:10.1021/acs.orglett.8b02639 |
| 50. | Jung, H. I.; Kim, D. Y. Synth. Commun. 2020, 50, 380–387. doi:10.1080/00397911.2019.1696364 |
| 60. | Shi, Y.; Chen, R.; Guo, K.; Meng, F.; Cao, S.; Gu, C.; Zhu, Y. Tetrahedron Lett. 2018, 59, 2062–2065. doi:10.1016/j.tetlet.2018.04.040 |
| 61. | Bu, M.-j.; Lu, G.-p.; Cai, C. Catal. Sci. Technol. 2016, 6, 413–416. doi:10.1039/c5cy01541c |
| 48. | Yang, J.; Sun, X.; Yan, K.; Sun, H.; Sun, S.; Jia, X.; Zhang, F.; Wen, J. Adv. Synth. Catal. 2022, 364, 2735–2740. doi:10.1002/adsc.202200421 |
| 62. | Tarasov, M. V.; Khrizanforova, V. V.; Gryaznova, T. V.; Budnikova, Y. H. Russ. J. Electrochem. 2023, 59, 896–905. doi:10.1134/s1023193523110137 |
| 56. | Zhou, P.; Hu, B.; Li, L.; Rao, K.; Yang, J.; Yu, F. J. Org. Chem. 2017, 82, 13268–13276. doi:10.1021/acs.joc.7b02391 |
| 57. | Zhang, G.-Y.; Li, C.-K.; Li, D.-P.; Zeng, R.-S.; Shoberu, A.; Zou, J.-P. Tetrahedron 2016, 72, 2972–2978. doi:10.1016/j.tet.2016.04.013 |
| 51. | Gu, J.; Cai, C. Org. Biomol. Chem. 2017, 15, 4226–4230. doi:10.1039/c7ob00505a |
| 58. | Pan, J.; Feng, Y.; Lv, L.; Li, Z. Adv. Synth. Catal. 2024, 366, 3860–3867. doi:10.1002/adsc.202400606 |
| 59. | Moghaddam, F. M.; Daneshfar, M.; Azaryan, R.; Pirat, J.-L. Catal. Commun. 2020, 141, 106015. doi:10.1016/j.catcom.2020.106015 |
| 55. | Yamamoto, D.; Ansai, H.; Hoshino, J.; Makino, K. Chem. Pharm. Bull. 2018, 66, 873–879. doi:10.1248/cpb.c18-00381 |
| 46. | Tang, L.; Wen, L.; Sun, T.; Zhang, D.; Yang, Z.; Feng, C.; Wang, Z. Asian J. Org. Chem. 2017, 6, 1683–1692. doi:10.1002/ajoc.201700434 |
| 54. | Gui, Q.; Hu, L.; Chen, X.; Liu, J.; Tan, Z. Chem. Commun. 2015, 51, 13922–13924. doi:10.1039/c5cc04826e |
| 42. | Zhou, M.; Chen, M.; Zhou, Y.; Yang, K.; Su, J.; Du, J.; Song, Q. Org. Lett. 2015, 17, 1786–1789. doi:10.1021/acs.orglett.5b00574 |
| 44. | Yi, N.; Wang, R.; Zou, H.; He, W.; Fu, W.; He, W. J. Org. Chem. 2015, 80, 5023–5029. doi:10.1021/acs.joc.5b00408 |
| 46. | Tang, L.; Wen, L.; Sun, T.; Zhang, D.; Yang, Z.; Feng, C.; Wang, Z. Asian J. Org. Chem. 2017, 6, 1683–1692. doi:10.1002/ajoc.201700434 |
| 51. | Gu, J.; Cai, C. Org. Biomol. Chem. 2017, 15, 4226–4230. doi:10.1039/c7ob00505a |
| 53. | Fang, J.; Ren, H.; Xu, S.; Huang, C.; Jiang, Y.; Zhang, W.; You, S.; Qin, B. Org. Lett. 2024, 26, 5458–5462. doi:10.1021/acs.orglett.4c01716 |
| 54. | Gui, Q.; Hu, L.; Chen, X.; Liu, J.; Tan, Z. Chem. Commun. 2015, 51, 13922–13924. doi:10.1039/c5cc04826e |
© 2025 Budnikov et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.