Abstract
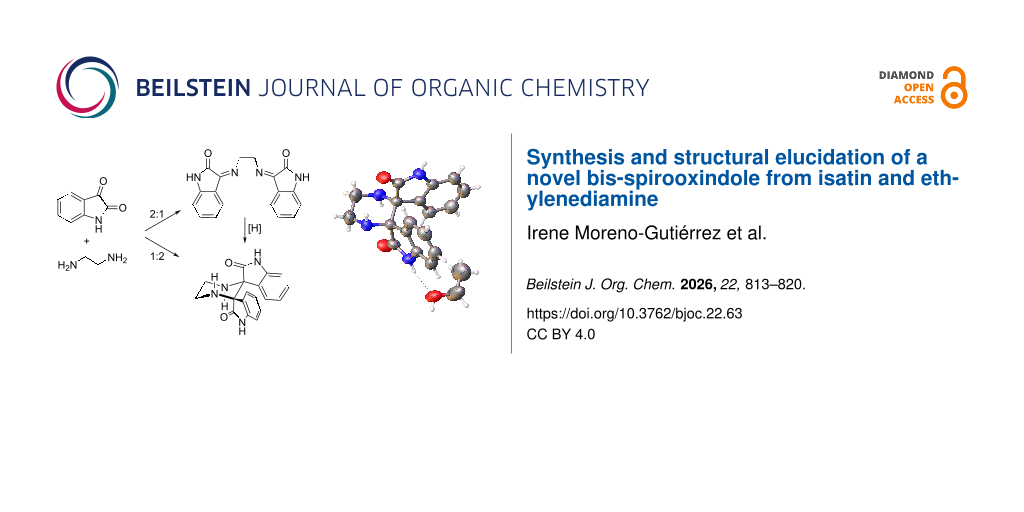
The reactivity of isatin toward ethylenediamine displays an unexpected stoichiometric divergence, affording either the anticipated diiminoisatin or a previously unreported pentacyclic bis-spirooxindole. A 2:1 isatin-to-diamine ratio provides the diiminoisatin, whereas a 1:2 ratio leads to the formation of a highly symmetric bis-spirooxindole scaffold. The new spirocyclic product was fully characterized by HRMS, IR and extensive 1D/2D NMR analysis, and its structure was unequivocally established by single-crystal X-ray diffraction. In addition, the isolated diiminoisatin can be independently reduced to the same bis-spirooxindole. These results broaden the scope of isatin–diamine condensations and demonstrate their potential to generate structurally complex spirooxindole architectures under simple conditions.

Graphical Abstract
Introduction
Isatin (1) is a highly versatile platform for heterocycle construction, particularly through skeletal editing, ring expansion, and related transformations that enable rapid access to complex molecular architectures [1]. In parallel, the design and synthesis of spiro-heterocycles – especially spirooxindoles – has continued to expand due to their broad pharmacological relevance and the wide array of modern synthetic strategies now available for their construction, including metal-free, organocatalytic, and transition-metal-mediated approaches [2-5]. In this context, comprehensive medicinal chemistry studies consistently highlight the prominence of isatin-derived frameworks among bioactive and anticancer agents [6].
The remarkable reactivity of the isatin scaffold arises from its dual functionality, combining an electrophilic C-3 carbonyl group with a γ-lactam system, which enables participation in a wide range of nucleophilic additions, condensations, and cyclizations [7]. As a result, isatin has proven to be a particularly powerful synthon for the construction of 2-oxindole derivatives, including a large variety of spiro-fused architectures. In this context, spirooxindoles have attracted considerable attention due to the conformational rigidity imparted by the spiro junction and the associated three-dimensionality, features that are often correlated with enhanced biological activity. Representative examples of bioactive spirooxindoles include the progesterone receptor modulator 2 [8], the potent MDM2–p53 inhibitor 3 [9], the CRTH2 (DP2) antagonist 4 [10] or the antimalarial agents 5 and 6 [11,12] (Figure 1). Beyond these well-known cases, several additional biologically active spirooxindoles – such as vasopressin-2 receptor antagonists, HIV-1 NNRTIs, and modulators of actin-dependent growth arrest – further exemplify the pharmacological breadth of this structural class [13-15].
![[1860-5397-22-63-1]](/bjoc/content/figures/1860-5397-22-63-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Isatin and representative bioactive spiro-fused 2-oxindoles.
Figure 1: Isatin and representative bioactive spiro-fused 2-oxindoles.
The propensity of isatin to engage in condensations with bifunctional nucleophiles has, however, also revealed a tendency toward structural complexity and, in some cases, unexpected reaction outcomes. In reactions involving amino acids or diamines, the coexistence of multiple nucleophilic and electrophilic sites can give rise to competing pathways and polycyclic architectures. A well-documented example is the condensation of isatin (1) with ʟ-proline (7), which leads to the formation of the dispirocyclic product 9 whose structure was ultimately established by X-ray diffraction after initial ambiguity based on spectroscopic data alone (Scheme 1) [16,17]. This case illustrates how condensations of isatin with bifunctional nitrogen nucleophiles may proceed beyond simple imine formation and generate highly condensed frameworks.
![[1860-5397-22-63-i1]](/bjoc/content/inline/1860-5397-22-63-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction of isatin (1) with ʟ-proline (7) [16,17].
Scheme 1: Reaction of isatin (1) with ʟ-proline (7) [16,17].
A related behavior has been observed in reactions of isatin with diamines. Whereas condensation with propan-1,3-diamine (10) under controlled stoichiometry affords the corresponding diiminoisatin 11 (Scheme 2) [18], reactions involving N-methylated ethylenediamines like 12 or 13 lead to spirocyclic products (14 and 15) [19]. These outcomes were rationalized through the involvement of imine and dipolarophilic intermediates (16 and 17), highlighting the sensitivity of the system to substitution at nitrogen.
![[1860-5397-22-63-i2]](/bjoc/content/inline/1860-5397-22-63-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Condensations of isatin with primary [18] and N-methylated diamines [19].
Scheme 2: Condensations of isatin with primary [18] and N-methylated diamines [19].
Furthermore, multicomponent systems combining isatin derivatives with diamines have been reported to furnish bis-spirocyclic architectures like 21 without detectable formation of the corresponding diimines 22 [20] (Scheme 3).
![[1860-5397-22-63-i3]](/bjoc/content/inline/1860-5397-22-63-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Catalyzed synthesis of the bis[spiro(quinazoline-oxindole)] derivative 21 [20].
Scheme 3: Catalyzed synthesis of the bis[spiro(quinazoline-oxindole)] derivative 21 [20].
Collectively, these precedents demonstrate that condensations of isatin with bifunctional nitrogen nucleophiles are highly sensitive to subtle variations in the nucleophile structure and reaction conditions, and may lead to markedly divergent outcomes. While simple diimines are often formed under controlled stoichiometry, alternative pathways can give rise to unexpectedly condensed spirocyclic architectures. In this context, we turned our attention to the reaction of isatin with unsubstituted ethylenediamine. Although analogy with related diamines would suggest preferential diiminoisatin formation, our results reveal a pronounced divergence in reactivity under different conditions, prompting a detailed structural investigation of the products obtained.
Results and Discussion
To investigate the divergent behavior of the reaction between isatin (1) and ethylenediamine (23), the system was examined under controlled stoichiometric conditions. Two distinct products were obtained depending on the isatin-to-diamine ratio (Scheme 4a,b). When isatin was reacted with ethylenediamine in a 2:1 molar ratio in methanol, the expected diiminoisatin 24 was obtained in 66% yield. The ¹H NMR spectrum displayed a diagnostic singlet at δ 4.45 ppm (4H) corresponding to the two =NCH₂ groups, together with the characteristic aromatic pattern of the isatin core. The IR spectrum showed a strong imine C=N band at 1610 cm−1, and the absence of the C-3 carbonyl stretching band confirmed complete condensation. The 13C NMR spectrum revealed multiple sets of closely related signals, consistent with the presence of three C=N geometrical isomers (E/E, E/Z, Z/Z). These spectroscopic data are in agreement with those of previously described diiminoisatin derivatives [21].
![[1860-5397-22-63-i4]](/bjoc/content/inline/1860-5397-22-63-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: (a) Ethylenediamine (0.5 equiv), MeOH, reflux, 5 h, 66%; (b) ethylenediamine (2 equiv), EtOH, reflux, 6 h, 67%; (c) 2 equiv NaBH4 (58%) or NaBH3CN (76%), reflux, MeOH.
Scheme 4: (a) Ethylenediamine (0.5 equiv), MeOH, reflux, 5 h, 66%; (b) ethylenediamine (2 equiv), EtOH, reflu...
When the reaction was carried out with an excess of ethylenediamine, a completely different product was obtained (Scheme 4b). Refluxing an ethanol solution of isatin and ethylenediamine in a 1:2 ratio afforded compound 25, which gradually precipitated from the reaction mixture. Such precipitation may contribute to driving the transformation toward product formation, as performing the reaction in methanol resulted instead in a complex mixture rather than 25 as the major product. The spectroscopic features of 25 differ markedly from those of 24 and are consistent with a highly symmetric pentacyclic bis-spirooxindole framework. The HRMS spectrum displayed an [M + H]+ ion at m/z 321.1352, in agreement with the molecular formula C18H17N4O2.
The 1H NMR spectrum reflected the symmetry of 25. The aliphatic region consisted of two doublets at δ 4.01 (2H) and 2.84 ppm (2H) with J = 9.3 Hz, corresponding to the axial and equatorial protons of a piperazine ring (H1, H2). The simple multiplicity is consistent with the presence of a C2 symmetry axis. The aromatic region retained the four-signal pattern of isatin (H4', H5', H6', H7'), slightly shifted by the new structural environment. In the 13C NMR spectrum, the amide carbonyl appeared at δ 176.5 ppm, while a key quaternary carbon at δ 61.0 ppm (C3‘) – formerly the C-3 isatin carbonyl carbon – confirmed the formation of nitrogen-bearing spiro centers. The piperazine methylenes resonated at δ 38.1 ppm, consistent with the proposed structure. The remaining signals have been assigned with the help of the two-dimensional COSY, HMBC and HSQC spectra. Altogether, these data strongly supported a bis-spirooxindole architecture.
Single-crystal X-ray diffraction analysis confirmed the structure of 25 unambiguously. The ORTEP representation (Figure 2) shows two oxindole units arranged orthogonally and connected by a central six-membered piperazine ring, with both isatin C-3 atoms converted into nitrogen-bearing spiro centers. The molecule displays C2 symmetry, consistent with the simplified NMR spectra, and the solid-state metrics support the proposed pentacyclic structure.
![[1860-5397-22-63-2]](/bjoc/content/figures/1860-5397-22-63-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP representation of bis-spirooxindole 25 (ellipsoids at 50% probability), showing the C2-symmetric arrangement of the two oxindole units and the central piperazine ring.
Figure 2: ORTEP representation of bis-spirooxindole 25 (ellipsoids at 50% probability), showing the C2-symmet...
Compound 25 (C18H17N4O2 u.d. = 13) has one degree less of unsaturation than the diimine 24 (C18H14N4O2 u.d. = 14) suggesting that a reduction is required for its formation. Indeed, when 24 was treated with either sodium borohydride or sodium cyanoborohydride in methanol, compound 25 was obtained as the sole isolable product (Scheme 4c). These experiments demonstrate that 25 can arise from 24 through an initial imine reduction followed by intramolecular C–C bond formation and cyclization.
Scheme 5 depicts a hypothetical rationalization of the process. Reaction of isatin (1) with ethylenediamine (23) should initially form the imine 26. Its reaction with a second molecule of isatine should lead to the diimine 24. Under the reaction conditions both imines 26 and 24 may coexist in equilibrium. The diimine 24 could also be in equilibrium with its tautomeric form 27. If the enol present in 27 attacks intramolecularly the other imine, it would produce a non-stabilised imine 28, which would be electrophilic and hence susceptible to reduction either by the NaBH4 or by the ethylenediamine acting as a hydride donor. In addition, if the formation of 28 by addition of the enol of 27 to the imine were reversible (and the reduction step irreversible), then the more stable, dipole-opposed C2-symmetric structure for 28 should be dominant, thus explaining the observed relative stereochemistry in 25. However, alternative pathways, cannot be excluded.
![[1860-5397-22-63-i5]](/bjoc/content/inline/1860-5397-22-63-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed mechanism for the formation of 25.
Scheme 5: Proposed mechanism for the formation of 25.
To address the generality of this transformation, a small set of substituted isatins was examined under the conditions used for the formation of 25. Reactions with 5-bromoisatin and 5-nitroisatin led to complex mixtures from which no defined bis-spirooxindole product could be isolated. In contrast, 5-methylisatin (29) afforded the corresponding methyl-substituted bis-spirooxindole 30, indicating that the transformation is not restricted to unsubstituted isatin, although it appears to be sensitive to the electronic nature of the substrate. This preliminary exploration therefore suggests that electron-neutral or weakly electron-donating substrates may be more suitable for this process, whereas strongly electron-withdrawing or halogenated derivatives require further optimization (Scheme 6).
![[1860-5397-22-63-i6]](/bjoc/content/inline/1860-5397-22-63-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of 30 from 5-methylisatin (29).
Scheme 6: Synthesis of 30 from 5-methylisatin (29).
Taken together, these results show that the reaction of isatin with ethylenediamine can be directed toward different structural outcomes depending on stoichiometry. Simple control of stoichiometry governs whether the system evolves through a conventional diimine pathway or diverges toward the assembly of a highly condensed bis-spirooxindole framework.
Conclusion
The condensation of isatin with ethylenediamine is strongly governed by stoichiometry, allowing selective access either to a diiminoisatin 24 or to the unexpected pentacyclic bis-spirooxindole 25 (Scheme 4) which was unambiguously identified by comprehensive spectroscopic analysis and single-crystal X-ray diffraction. The same bis-spirooxindole 25 can be obtained by reduction of the diiminoisatin 24. The reaction with methyl-substituted isatin 29 follows the same pattern, affording 30. Overall, we have shown that simple isatin–diamine systems can deliver structurally complex spirocyclic architectures under mild and operationally straightforward conditions, although the substrate scope appears to be limited.
Experimental
General remarks: NMR spectra were recorded on a Bruker Nanobay Avance III HD 600 MHz spectrometer. Proton-decoupled. When required, COSY, HMQC and HMBC experiments were used for signal assignment. Chemical shifts (δ) are expressed in ppm and coupling constants (J) in hertz (Hz). Chemical shifts are reported using CD3OD or DMSO-d6 as internal reference. IR spectra were recorded with a Bruker Alpha spectrometer using a single reflection ATR-platinum module. Mass spectra were recorded in a Waters Xevo LC-QTof-MS with electrospray ionization. The AQ:AcN mixture (50:50, 0.1% formic acid) was used as eluent. X-ray diffraction data were collected on a Bruker AXS SMART APEX diffractometer. Structure solution and refinement were carried out using Olex2 [22]. The structure was solved by intrinsic phasing with SHELXT [23] and refined by full-matrix least-squares methods on F2 using SHELXL [24]. Commercially available chemicals were obtained from Scharlau, TCI and Acros and used as received.
Synthesis of 25: Isatin (1, 2.98 g, 19.85 mmol, 1 equiv) was dissolved in ethanol (60 mL) and a solution of ethylenediamine (2.25 mL, 39.70 mmol, 2.0 equiv) in ethanol (60 mL) was added dropwise. The mixture was heated at reflux for 6 h and then allowed to crystallize at 4 °C for 72 h. The solid was collected by filtration and washed with cold ethanol to give 25 as yellow crystals (1.06 g, 6.62 mmol, 67%). Mp = 169.5–170.4 °C; HRMS m/z: [M + H]+ calcd. for [C18H17N4O2 + H]+, 321.1352; found, 321.1358; IR (ATR) ν (cm−1): 3276, 1710, 1619, 1472, 744; 1H NMR (600 MHz, CD3OD) δ (ppm) 7.32 (ddd, J = 7.7, 1.1, 0.6 Hz, 2H, H4’, H4’’), 7.08 (td, J = 7.7, 1.2 Hz, 2H, H6’, H6’’), 6.90 (td, J = 7.7, 1.0 Hz, 2H, H5’, H5’’), 6.79–6.77 (m, 2H, NH), 6.62 (ddd, J = 7.7, 0.9, 0.6 Hz, 2H, H7’, H7’’), 4.01 (d, J = 9.3 Hz, 2H, H1ax, H2ax), 2.84 (d, J = 9.3 Hz, 2H, H1eq, H2eq); 13CNMR (150.92 MHz, CD3OD) 176.5 (C, C2’, C2’’), 141.7 (C, C7’a, C7’’a), 129.2 (CH, C6’, C6’’), 127.7 (C, C3a’, C3’’a), 123.8 (CH, C4’, C4’’), 121.9 (CH, C5’, C5’’), 109.3 (CH, C7’, C7’’), 61.0 (C, C3’, C3’’), 38.1 (CH2, C1, C2).
X-ray analysis of 25 [22-24]. Recrystallization of 25 in EtOH afforded a yellow crystal. Crystal data for C22H26N4O4 (M = 410.47 g/mol): monoclinic, space group C2/c (no. 15), a = 16.0100(5) Å, b = 8.5258(4) Å, c = 16.6847(5) Å, β = 105.815(2)°, V = 2191.22(14) Å3, Z = 4, T = 298(2) K, μ(Cu Kα) = 0.712 mm−1, Dcalc = 1.244 g/cm3, 11324 reflections measured (11.488° ≤ 2Θ ≤ 136.964°), 2001 unique (Rint = 0.0404, Rsigma = 0.0308) which were used in all calculations. The final R1 was 0.0595 (I > 2σ(I)) and wR2 was 0.1864 (all data). CCDC 2388389 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge at https://doi.org/10.5517/ccdc.csd.cc2l59tx from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44-1223-336033; E-mail: deposit@ccdc.cam.ac.uk.
Synthesis of 24. Ethylenediamine (0.33 mL, 5 mmol, 1 equiv) was added dropwise to a solution of isatin (1, 1.47 g, 10 mmol, 2 equiv) in MeOH (50 mL). The mixture was heated at reflux for 5 h, cooled to 0 °C, and acidified with 1 M HCl (to pH ≈ 1), leading to precipitation of the product. The orange precipitate was collected by filtration, washed with cold MeOH, and dried under vacuum to give 24 (1.05 g, 3.3 mmol, 66%). HRMS m/z: [M + H]+ calcd. for [C18H14N4O2 + H]+, 319.1190; found, 319.1188; IR (ATR) ν (cm−1): 3270, 2890, 1740, 1707, 1652, 1610, 1464, 1342, 1205, 1021, 739; 1H NMR (600 MHz, DMSO-d6) δ (ppm) 10.83 (bs, 2H, NH), 7.84 (bd, J = 7.7 Hz, 2H, H4’, H4’’), 7.45 (td, J = 7.8, 1.1 Hz, 2H, H6’, H6’’), 7.08 (td, J = 7.7, 0.9 Hz, 2H, H5’,H5’’), 6.94 (bd, J = 7.8 Hz, 2H, H7’,H7’’), 4.45 (s, 4H, =NCH2); 13C NMR (125 MHz, DMSO-d6) δ 164.08 (C), 164.05 (C), 160.18(C), 155.86 (C), 155.74 (C), 154.89 (C), 146.41 (C), 146.36 (C), 145.07 (C), 134.05 (CH), 133.95 (CH), 133.49 (CH), 127.84 (CH), 127.79 (CH), 122.69 (CH), 122.64 (CH), 122.63 (CH), 122.20 (CH), 117.33 (C), 117.31 (C), 111.57 (CH), 111.51 (CH), 111.05 (CH), 55.61 (CH2), 55.47 (CH2), 53.07 (CH2).
Reduction of 24 with NaBH4. NaBH₄ (95 mg, 2.52 mmol, 2 equiv) was added in portions to a stirred solution of 24 (0.40 g, 1.26 mmol, 1 equiv) in MeOH (15 mL) at room temperature. The mixture was stirred for 2 h, poured onto ice, and the resulting yellow precipitate of 25 was collected by filtration (295 mg, 0.92 mmol, 58%).
Reduction of 24 with NaBH3CN. NaBH3CN (0.16 g, 2.52 mmol, 2 equiv) was slowly added to a solution of 24 (0.4 g, 1.26 mmol, 1 equiv) in MeOH (15 mL). The reaction mixture was stirred at reflux for 1 h 30 min, poured over ice and vacuum filtered. 25 was obtained as a yellow precipitate (305 mg, 0.95 mmol, 76%).
Synthesis of 30. 5-Methylisatin (29, 1.50 g, 9.8 mmol, 1 equiv) was dissolved in ethanol (30 mL) and a solution of ethylenediamine (1.3 mL, 19.6 mmol, 2.0 equiv) in ethanol (30 mL) was added dropwise. The mixture was heated at reflux for 6 h. The solvent was removed under reduced pressure and the mixture was purified by column chromatography (DCM/Me2CO gradient from 8:2 to 6:4) to give 30 as yellow oil (1.04 g, 2.99 mmol, 61%). HRMS m/z: [M + H]+ calcd. for [C20H21N4O2 + H]+, 349.1665; found, 349.1674; 1H NMR (300 MHz, MeOD) δ 7.19–7.17 (m, 2H, H4’, H4’’), 6.92 (ddd, J = 7.9, 1.7, 0.8 Hz, 2H, H6’, H6’’), 6.70 (bs, 1H, NH), 6.68 (bs, 1H, NH),6.54 (d, J = 7.9 Hz, 2H, H7’, H7’’), 4.01 (d, J = 9.2 Hz, 2H, H1ax, H2ax), 2.84 (d, J = 9.2 Hz, 2H, H1eq, H2eq), 2.24 (s, 6H); 13C NMR (75 MHz, MeOD) δ 176.5 (C, C2’, C2’’), 139.2 (C, C7’a, C7’’a), 131.6 (C, C5a’, C5’’a), 129.5 (CH, C6’, C6’’), 127.9 (C, C3a’, C3’’a), 124.5 (CH, C4’, C4’’), 109.2 (CH, C7’, C7’’), 61.0 (C, C3’, C3’’), 38.2 (CH2, C1, C2), 19.9 (CH3).
Supporting Information
| Supporting Information File 1: Copies of IR, NMR and MS spectra. | ||
| Format: PDF | Size: 1.6 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Zhang, T.; Feng, H. Chem. Rec. 2024, 24, e202400024. doi:10.1002/tcr.202400024
Return to citation in text: [1] -
Sohail, M.; Tanaka, F. Synlett 2023, 34, 2374–2378. doi:10.1055/a-2061-0855
Return to citation in text: [1] -
Feng, J.; Wang, Y.; Li, E.-Q.; Loh, T.-P. Chem. Rec. 2024, 24, e202400126. doi:10.1002/tcr.202400126
Return to citation in text: [1] -
D, B.; Doddamani, S. V.; S, A. C.; Siby, A.; V, S.; A, A.; Somappa, B. S. Tetrahedron 2025, 173, 134468. doi:10.1016/j.tet.2025.134468
Return to citation in text: [1] -
Buttard, F.; Guinchard, X. ACS Catal. 2023, 13, 9442–9475. doi:10.1021/acscatal.3c01417
Return to citation in text: [1] -
Banerjee, B.; Sharma, A.; Singh, A.; Kaur, M.; Priya, A. Curr. Top. Med. Chem. 2025, 25, 96–123. doi:10.2174/0115680266311332240722065652
Return to citation in text: [1] -
Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y
Return to citation in text: [1] -
Fensome, A.; Adams, W. R.; Adams, A. L.; Berrodin, T. J.; Cohen, J.; Huselton, C.; Illenberger, A.; Kern, J. C.; Hudak, V. A.; Marella, M. A.; Melenski, E. G.; McComas, C. C.; Mugford, C. A.; Slayden, O. D.; Yudt, M.; Zhang, Z.; Zhang, P.; Zhu, Y.; Winneker, R. C.; Wrobel, J. E. J. Med. Chem. 2008, 51, 1861–1873. doi:10.1021/jm701080t
Return to citation in text: [1] -
Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Tomita, Y.; Parrish, D. A.; Deschamps, J. R.; Wang, S. J. Am. Chem. Soc. 2005, 127, 10130–10131. doi:10.1021/ja051147z
Return to citation in text: [1] -
Crosignani, S.; Jorand-Lebrun, C.; Page, P.; Campbell, G.; Colovray, V.; Missotten, M.; Humbert, Y.; Cleva, C.; Arrighi, J.-F.; Gaudet, M.; Johnson, Z.; Ferro, P.; Chollet, A. ACS Med. Chem. Lett. 2011, 2, 644–649. doi:10.1021/ml2001196
Return to citation in text: [1] -
Yeung, B. K. S.; Zou, B.; Rottmann, M.; Lakshminarayana, S. B.; Ang, S. H.; Leong, S. Y.; Tan, J.; Wong, J.; Keller-Maerki, S.; Fischli, C.; Goh, A.; Schmitt, E. K.; Krastel, P.; Francotte, E.; Kuhen, K.; Plouffe, D.; Henson, K.; Wagner, T.; Winzeler, E. A.; Petersen, F.; Brun, R.; Dartois, V.; Diagana, T. T.; Keller, T. H. J. Med. Chem. 2010, 53, 5155–5164. doi:10.1021/jm100410f
Return to citation in text: [1] -
Rottmann, M.; McNamara, C.; Yeung, B. K. S.; Lee, M. C. S.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D. M.; Dharia, N. V.; Tan, J.; Cohen, S. B.; Spencer, K. R.; González-Páez, G. E.; Lakshminarayana, S. B.; Goh, A.; Suwanarusk, R.; Jegla, T.; Schmitt, E. K.; Beck, H.-P.; Brun, R.; Nosten, F.; Renia, L.; Dartois, V.; Keller, T. H.; Fidock, D. A.; Winzeler, E. A.; Diagana, T. T. Science 2010, 329, 1175–1180. doi:10.1126/science.1193225
Return to citation in text: [1] -
Farah, J.; Daifallah, S.; Zmily, H.; Ghali, J. K. Therapy 2010, 7, 409–422. doi:10.2217/thy.10.36
Return to citation in text: [1] -
Kumari, G.; Nutan; Modi, M.; Gupta, S. K.; Singh, R. K. Eur. J. Med. Chem. 2011, 46, 1181–1188. doi:10.1016/j.ejmech.2011.01.037
Return to citation in text: [1] -
Lo, M. M.-C.; Neumann, C. S.; Nagayama, S.; Perlstein, E. O.; Schreiber, S. L. J. Am. Chem. Soc. 2004, 126, 16077–16086. doi:10.1021/ja045089d
Return to citation in text: [1] -
Kumar, R. S.; Rajesh, S. M.; Perumal, S.; Banerjee, D.; Yogeeswari, P.; Sriram, D. Eur. J. Med. Chem. 2010, 45, 411–422. doi:10.1016/j.ejmech.2009.09.044
Return to citation in text: [1] [2] -
Al Mamari, K.; Ennajih, H.; Zouihri, H.; Bouhfid, R.; Ng, S. W.; Essassi, E. M. Tetrahedron Lett. 2012, 53, 2328–2331. doi:10.1016/j.tetlet.2012.02.097
Return to citation in text: [1] [2] -
McDougall, R. H.; Malik, S. H. J. Chem. Soc. C 1969, 2044–2051. doi:10.1039/j39690002044
Return to citation in text: [1] [2] -
Bergman, J.; Stålhandske, C.; Vallberg, H. Acta Chem. Scand. 1997, 51, 753–759. doi:10.3891/acta.chem.scand.51-0753
Return to citation in text: [1] [2] -
Mohammadi, A. A.; Taheri, S.; Askari, S.; Ahdenov, R. J. Heterocycl. Chem. 2015, 52, 1871–1875. doi:10.1002/jhet.2292
Return to citation in text: [1] [2] -
Rafat, F.; Siddiqi, K. S.; Siddiqi, M. Y. Pol. J. Chem. 2005, 79, 663–670.
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1] [2] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370
Return to citation in text: [1] [2] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] [2]
| 22. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 20. | Mohammadi, A. A.; Taheri, S.; Askari, S.; Ahdenov, R. J. Heterocycl. Chem. 2015, 52, 1871–1875. doi:10.1002/jhet.2292 |
| 8. | Fensome, A.; Adams, W. R.; Adams, A. L.; Berrodin, T. J.; Cohen, J.; Huselton, C.; Illenberger, A.; Kern, J. C.; Hudak, V. A.; Marella, M. A.; Melenski, E. G.; McComas, C. C.; Mugford, C. A.; Slayden, O. D.; Yudt, M.; Zhang, Z.; Zhang, P.; Zhu, Y.; Winneker, R. C.; Wrobel, J. E. J. Med. Chem. 2008, 51, 1861–1873. doi:10.1021/jm701080t |
| 19. | Bergman, J.; Stålhandske, C.; Vallberg, H. Acta Chem. Scand. 1997, 51, 753–759. doi:10.3891/acta.chem.scand.51-0753 |
| 7. | Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y |
| 20. | Mohammadi, A. A.; Taheri, S.; Askari, S.; Ahdenov, R. J. Heterocycl. Chem. 2015, 52, 1871–1875. doi:10.1002/jhet.2292 |
| 6. | Banerjee, B.; Sharma, A.; Singh, A.; Kaur, M.; Priya, A. Curr. Top. Med. Chem. 2025, 25, 96–123. doi:10.2174/0115680266311332240722065652 |
| 19. | Bergman, J.; Stålhandske, C.; Vallberg, H. Acta Chem. Scand. 1997, 51, 753–759. doi:10.3891/acta.chem.scand.51-0753 |
| 2. | Sohail, M.; Tanaka, F. Synlett 2023, 34, 2374–2378. doi:10.1055/a-2061-0855 |
| 3. | Feng, J.; Wang, Y.; Li, E.-Q.; Loh, T.-P. Chem. Rec. 2024, 24, e202400126. doi:10.1002/tcr.202400126 |
| 4. | D, B.; Doddamani, S. V.; S, A. C.; Siby, A.; V, S.; A, A.; Somappa, B. S. Tetrahedron 2025, 173, 134468. doi:10.1016/j.tet.2025.134468 |
| 5. | Buttard, F.; Guinchard, X. ACS Catal. 2023, 13, 9442–9475. doi:10.1021/acscatal.3c01417 |
| 18. | McDougall, R. H.; Malik, S. H. J. Chem. Soc. C 1969, 2044–2051. doi:10.1039/j39690002044 |
| 13. | Farah, J.; Daifallah, S.; Zmily, H.; Ghali, J. K. Therapy 2010, 7, 409–422. doi:10.2217/thy.10.36 |
| 14. | Kumari, G.; Nutan; Modi, M.; Gupta, S. K.; Singh, R. K. Eur. J. Med. Chem. 2011, 46, 1181–1188. doi:10.1016/j.ejmech.2011.01.037 |
| 15. | Lo, M. M.-C.; Neumann, C. S.; Nagayama, S.; Perlstein, E. O.; Schreiber, S. L. J. Am. Chem. Soc. 2004, 126, 16077–16086. doi:10.1021/ja045089d |
| 16. | Kumar, R. S.; Rajesh, S. M.; Perumal, S.; Banerjee, D.; Yogeeswari, P.; Sriram, D. Eur. J. Med. Chem. 2010, 45, 411–422. doi:10.1016/j.ejmech.2009.09.044 |
| 17. | Al Mamari, K.; Ennajih, H.; Zouihri, H.; Bouhfid, R.; Ng, S. W.; Essassi, E. M. Tetrahedron Lett. 2012, 53, 2328–2331. doi:10.1016/j.tetlet.2012.02.097 |
| 22. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 23. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
| 24. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 11. | Yeung, B. K. S.; Zou, B.; Rottmann, M.; Lakshminarayana, S. B.; Ang, S. H.; Leong, S. Y.; Tan, J.; Wong, J.; Keller-Maerki, S.; Fischli, C.; Goh, A.; Schmitt, E. K.; Krastel, P.; Francotte, E.; Kuhen, K.; Plouffe, D.; Henson, K.; Wagner, T.; Winzeler, E. A.; Petersen, F.; Brun, R.; Dartois, V.; Diagana, T. T.; Keller, T. H. J. Med. Chem. 2010, 53, 5155–5164. doi:10.1021/jm100410f |
| 12. | Rottmann, M.; McNamara, C.; Yeung, B. K. S.; Lee, M. C. S.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D. M.; Dharia, N. V.; Tan, J.; Cohen, S. B.; Spencer, K. R.; González-Páez, G. E.; Lakshminarayana, S. B.; Goh, A.; Suwanarusk, R.; Jegla, T.; Schmitt, E. K.; Beck, H.-P.; Brun, R.; Nosten, F.; Renia, L.; Dartois, V.; Keller, T. H.; Fidock, D. A.; Winzeler, E. A.; Diagana, T. T. Science 2010, 329, 1175–1180. doi:10.1126/science.1193225 |
| 18. | McDougall, R. H.; Malik, S. H. J. Chem. Soc. C 1969, 2044–2051. doi:10.1039/j39690002044 |
| 10. | Crosignani, S.; Jorand-Lebrun, C.; Page, P.; Campbell, G.; Colovray, V.; Missotten, M.; Humbert, Y.; Cleva, C.; Arrighi, J.-F.; Gaudet, M.; Johnson, Z.; Ferro, P.; Chollet, A. ACS Med. Chem. Lett. 2011, 2, 644–649. doi:10.1021/ml2001196 |
| 23. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
| 9. | Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P. P.; Tomita, Y.; Parrish, D. A.; Deschamps, J. R.; Wang, S. J. Am. Chem. Soc. 2005, 127, 10130–10131. doi:10.1021/ja051147z |
| 16. | Kumar, R. S.; Rajesh, S. M.; Perumal, S.; Banerjee, D.; Yogeeswari, P.; Sriram, D. Eur. J. Med. Chem. 2010, 45, 411–422. doi:10.1016/j.ejmech.2009.09.044 |
| 17. | Al Mamari, K.; Ennajih, H.; Zouihri, H.; Bouhfid, R.; Ng, S. W.; Essassi, E. M. Tetrahedron Lett. 2012, 53, 2328–2331. doi:10.1016/j.tetlet.2012.02.097 |
| 24. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
© 2026 Moreno-Gutiérrez et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.