Abstract
α-Boryl carbanions are valuable organoboron intermediates, but controlling their stereoselective reactions remains challenging. Here, we examine the diastereoselective generation and trapping of α-boryl lithium species formed by ring opening of substituted iodomethylcyclopropanes. After lithium–iodine exchange and selective C–C bond cleavage, these intermediates react with a range of electrophiles to give boronic esters bearing vicinal tri- and tetrasubstituted stereocenters in good yields and high diastereoselectivities. Notably, substrates bearing alkyl substituents display the opposite sense of diastereoselectivity to previously studied aryl-substituted analogues. The results suggest that the stereochemical outcome is independent of the initial configuration at C2 and is instead dictated by conformational preferences of the α-boryl lithium intermediate. A simplified steric model is proposed in which lithium coordination induces a pseudo-chelated structure that controls facial selectivity during electrophilic trapping. These findings expand the synthetic utility of α-boryl lithium intermediates and provide insight into the origins of their diastereoselectivity.

Graphical Abstract
Introduction
α-Boryl carbanions 1 (Scheme 1), also referred to as boron alkylidenes or borata alkenes, represent a fascinating class of organoboron intermediates characterized by the delocalization of electron density from the negatively charged carbon center toward the electron-deficient boron atom. This delocalization leads to the formation of a partial π C–B bond (Scheme 1) [1,2], which provides a unique stabilization to the carbanionic center and affects its reactivity towards electrophiles. In recent years, α-boryl carbanions have attracted significant attention as highly versatile intermediates capable of engaging in diverse bond-forming processes, including C–C, C–O and C–N bond formations [3-24]. Despite their growing utility, controlling the stereoselectivity in substitution reactions of α-boryl carbanions remains a formidable task [25-27]. The electronic character of these intermediates, especially for boronic ester derivatives that allow coordination between oxygen atoms and the metal counterion, complicates predictable stereochemical outcome. Developing strategies to harness the exceptional nature of these carbanions in a stereodefined manner would achieve the synthesis of polysubstituted organoboron compounds with adjacent stereocenters. The latter are highly valuable building blocks, given the pivotal role of organoboron compounds in contemporary chemistry as stable, yet reactive, intermediates that enable diverse catalytic and stereoselective transformations [28-33].
![[1860-5397-22-68-i1]](/bjoc/content/inline/1860-5397-22-68-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis and reactivity of α-boryl lithium species.
Scheme 1: Synthesis and reactivity of α-boryl lithium species.
In this context, we have recently demonstrated that a broad range of arylated (iodomethyl)cyclopropylboronic ester 2 [34] undergo ring-opening reactions, followed by electrophilic trapping of the resulting α-boryl carbanion derivative 1 to afford product 4 as single diastereomers. Specifically, iodide 2 was subjected to tert-butyllithium (t-BuLi) in diethyl ether at −95 °C, efficiently generating cyclopropylmethyllithium 3 via lithium–iodine exchange. Subsequent selective C–C bond cleavage of 3, proceeding exclusively through cleavage of the C1–C3 bond rather than the C1–C2 bond, provided direct access to α-boryl carbanion intermediate 1. Reaction of 1 with a range of electrophiles furnished tertiary pinacol boranes with a well-defined adjacent stereocenter in excellent diastereoselectivity (Scheme 1).
Theoretical studies revealed that the electrophile preferentially approaches from the face opposite the coordinating lithium cation, with the lowest-energy transition state stabilized by a lithium cation–vinyl interaction (see A in Scheme 1) [34].
Results and Discussion
The intricate interplay of the coordination and steric effects suggests that the adjacent stereocenter at C2 might play a key role in the conformational stability and the stereochemical outcome. We therefore set out to extend the scope of the transformation by probing how substitution at the C2 position of precursor 2 affects the diastereoselectivity of the reaction. We were particularly interested in examining cases with alkyl (5) instead of aryl substituents in 2 on C2 (Scheme 2).
![[1860-5397-22-68-i2]](/bjoc/content/inline/1860-5397-22-68-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
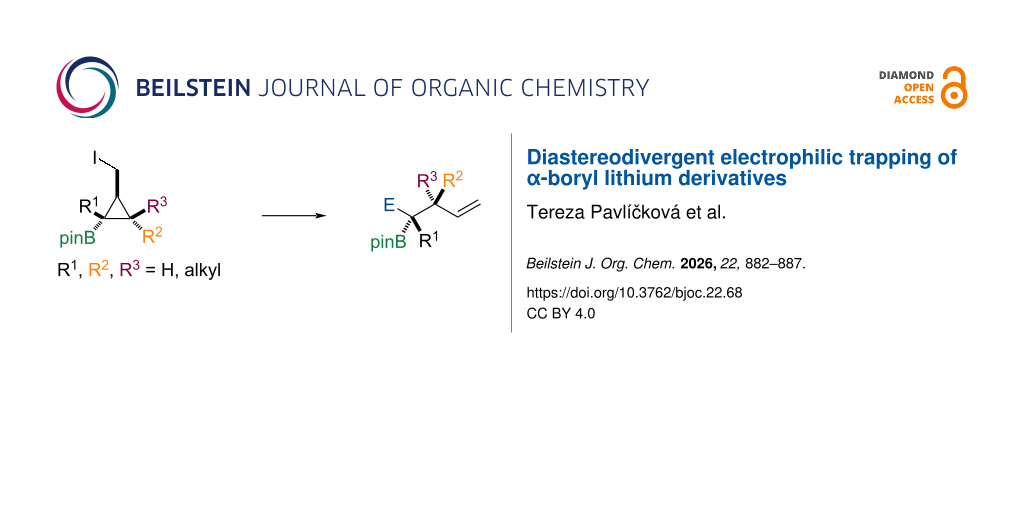
Scheme 2: Diastereodivergent electrophilic trapping of α-boryl lithium derivatives.
Scheme 2: Diastereodivergent electrophilic trapping of α-boryl lithium derivatives.
We initiated the investigation into the selectivity of the transformation with the synthesis of a library of cyclopropylmethyl iodides 5a–i, using our previously reported methodology [35-37] (Figure 1).
![[1860-5397-22-68-1]](/bjoc/content/figures/1860-5397-22-68-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Cyclopropylmethyl iodide starting materials.
Figure 1: Cyclopropylmethyl iodide starting materials.
When the substrate 5a (R1 = R3 = Me, R2 = H) was subjected to the standard reaction conditions, the ring-opening process and trapping with Me3SiCl occurred selectively, affording 6a in an excellent diastereomeric ratio (Scheme 3). Replacing Me3SiCl with the bulkier electrophile PhMe2SiCl did not alter the selectivity of the transformation, yielding 6b as a single diastereomer (Scheme 3). The relative configuration of 6b was determined by X-ray analysis of the osmate ester 7b, derived from osmium-catalyzed dihydroxylation (Scheme 3) [38], and the other products were assigned by analogy. Interestingly, the relative configuration is reversed compared to the cases with aryl group as R3 (Scheme 2) [34].
![[1860-5397-22-68-i3]](/bjoc/content/inline/1860-5397-22-68-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactivity of α-boryl lithium intermediates 5 possessing alkyl substituents at the allylic position.
Scheme 3: Reactivity of α-boryl lithium intermediates 5 possessing alkyl substituents at the allylic position....
The presence of an aliphatic substituent as R2 instead of R3 (R3 = H, R2 = allyl) has no influence on the diastereoselectivity, as 6c was obtained with high selectivity. For both 6a/6b and 6c, the alkyl substituent, whether R3 (6a, 6b) or R2 (6c), has a syn-relationship with the boronic ester group. Furthermore, performing the reaction with diastereomers 5c (R1 = R3 = Pr, R2 = H) and 5d (R1 = R2 = Pr, R3 = H), produced product 6d with the same relative configuration (described as the two enantiomers to maintain an identical configuration of C2), albeit with moderate yields and diastereoselectivities.
This observation supports the hypothesis that the reaction outcome is independent of the initial relative configuration at C2 of the starting material. Increasing the length and branching of the R1 substituent (from Me, Et to Bu, and Cy) resulted in a slight decrease in diastereomeric ratios, providing 6e–g in moderate yields and selectivities. To our delight, the reaction proceeded even when three alkyl substituents were present (5h, R1 = R3 = Me, R2 = allyl), providing compound 6h bearing two adjacent fully substituted stereocenters (Scheme 3). Although the yield was relatively low (34%), the diastereomeric remained high (dr 90:10). The relative configuration of the major diastereomers of 6h was assigned by analogy to 6c, but it could not be definitively confirmed due to the inability to obtain suitable crystals for X-ray analysis. In addition to the nucleophilic substitution of the boryl lithium species with R3SiCl, a small range of alternative electrophiles was examined (Scheme 3). Incorporation of a tin-based electrophile furnished stannane 6i in moderate yields with good diastereoselectivity. Reaction with cyclohexanone led to partial elimination, which proceeded to completion upon warming the reaction mixture to room temperature, affording the bora-Wittig product 6j in moderate yield [39]. Furthermore, an intramolecular reaction was investigated: cyclopropane 5i possessing a chloride as a leaving group underwent highly selective cyclization to deliver the stereodefined borylated cyclopentane 6k in high 84% yield as a single diastereomer. The relative configuration of 6k was unambiguously established by X-ray crystallographic analysis of an osmylated derivative (see Supporting Information File 1).
As noted above, the diastereoselectivity observed for substrates bearing alkyl substituents at R2/R3 is opposite to that previously reported for aryl-substituted analogues (Scheme 4).
![[1860-5397-22-68-i4]](/bjoc/content/inline/1860-5397-22-68-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Effect of the nature of the substituent R3 on the diastereoselectivity.
Scheme 4: Effect of the nature of the substituent R3 on the diastereoselectivity.
The solvent-coordinated lithium alkylidene model used to rationalize the aryl series [34] does not account for the selectivity trend in the alkyl case, indicating that different stereochemical factors dominate in the absence of an aryl substituent. We therefore propose a simplified, steric-based model to account for the observed diastereoselectivity. Based on previous theoretical calculations, this model assumes initial coordination of both the vinyl group and the Bpin moiety to the lithium counterion. Within this constrained geometry, when R2 = H, R3 = alkyl, the diastereocontrol is then governed by the relative steric interactions present in these two possible conformers B and C. This conformational constraint rigidifies the system through formation of a pseudo-ring, leading to potential gauche interactions between R1 and R3 in conformer B (Scheme 5).
![[1860-5397-22-68-i5]](/bjoc/content/inline/1860-5397-22-68-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mechanistic hypothesis to rationalize the diastereoselectivity.
Scheme 5: Mechanistic hypothesis to rationalize the diastereoselectivity.
In conformer C, however, substituents R1 and R3 are further apart, which expose a free diastereotopic face above the plane of the molecule and allows the electrophile to react anti to the pseudo-chelated metallacyle. When R2 = alkyl and R3 = H, the corresponding enantiomeric conformer leads to the same diastereomer, as expected from the enantiomeric relationship (see 6d from 5c and 5d, Scheme 3).
Conclusion
In conclusion, this work establishes α-boryl lithium intermediates as valuable stereogenic platforms for conducting highly diastereoselective nucleophilic transformations. The combination of the anion-stabilizing capability of boron with strain-release-driven ring fragmentation provides exceptional chemo- and regioselectivity. The use of various electrophiles enabled the synthesis of acyclic boronic esters featuring vicinal tri- and tetrasubstituted stereocenters.
Supporting Information
| Supporting Information File 1: Experimental section, characterization data and copies of spectra. | ||
| Format: PDF | Size: 3.3 MB | Download |
Funding
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Program, under Grant Agreement No. 101198411, from the Israel Science Foundation administered by the Israel Academy of Sciences and Humanities (Grant No. 1625/22) and by the Council for Higher Education from VATAT.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article. The crystal data have been deposited in the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/) deposition numbers CCDC 2537818 and 2537817.
References
-
Maza, R. J.; Fernández, E.; Carbó, J. J. Chem. – Eur. J. 2021, 27, 12352–12361. doi:10.1002/chem.202101464
Return to citation in text: [1] -
Maza, R. J.; Carbó, J. J.; Fernández, E. Adv. Synth. Catal. 2021, 363, 2274–2289. doi:10.1002/adsc.202100192
Return to citation in text: [1] -
Zhang, C.; Hu, W.; Morken, J. P. ACS Catal. 2021, 11, 10660–10680. doi:10.1021/acscatal.1c02496
Return to citation in text: [1] -
Lee, Y.; Han, S.; Cho, S. H. Acc. Chem. Res. 2021, 54, 3917–3929. doi:10.1021/acs.accounts.1c00455
Return to citation in text: [1] -
Hong, K.; Liu, X.; Morken, J. P. J. Am. Chem. Soc. 2014, 136, 10581–10584. doi:10.1021/ja505455z
Return to citation in text: [1] -
McDonald, T. R.; Rousseaux, S. A. L. Chem. Sci. 2023, 14, 963–969. doi:10.1039/d2sc06088d
Return to citation in text: [1] -
McDonald, T. R.; Turner, J. A.; Gabbey, A. L.; Balasubramanian, P.; Rousseaux, S. A. L. Org. Lett. 2024, 26, 3822–3827. doi:10.1021/acs.orglett.4c00987
Return to citation in text: [1] -
Chen, X.-Y.; Gao, F.-C.; Ning, P.-F.; Wei, Y.; Hong, K. Angew. Chem., Int. Ed. 2023, 62, e202302638. doi:10.1002/anie.202302638
Return to citation in text: [1] -
Gu, H.-Y.; Chen, X.-Y.; Yang, Y.-F.; Hong, K. Org. Lett. 2025, 27, 4208–4213. doi:10.1021/acs.orglett.5c00886
Return to citation in text: [1] -
Gava, R.; Fernández, E. Chem. – Eur. J. 2019, 25, 8013–8017. doi:10.1002/chem.201901406
Return to citation in text: [1] -
Sakai, M.; Saito, S.; Kanai, G.; Suzuki, A.; Miyaura, N. Tetrahedron 1996, 52, 915–924. doi:10.1016/0040-4020(95)00950-7
Return to citation in text: [1] -
Joannou, M. V.; Moyer, B. S.; Goldfogel, M. J.; Meek, S. J. Angew. Chem., Int. Ed. 2015, 54, 14141–14145. doi:10.1002/anie.201507171
Return to citation in text: [1] -
Nallagonda, R.; Karimov, R. R. ACS Catal. 2021, 11, 248–254. doi:10.1021/acscatal.0c04474
Return to citation in text: [1] -
Park, J.; Lee, Y.; Kim, J.; Cho, S. H. Org. Lett. 2016, 18, 1210–1213. doi:10.1021/acs.orglett.6b00376
Return to citation in text: [1] -
Pereira, S.; Srebnik, M. Tetrahedron Lett. 1995, 36, 1805–1808. doi:10.1016/0040-4039(95)00148-6
Return to citation in text: [1] -
Knochel, P. J. Am. Chem. Soc. 1990, 112, 7431–7433. doi:10.1021/ja00176a074
Return to citation in text: [1] -
Schwenzer, M.; Studer, A. Adv. Synth. Catal. 2025, 367, e70117. doi:10.1002/adsc.70117
Return to citation in text: [1] -
Murray, S. A.; Liang, M. Z.; Meek, S. J. J. Am. Chem. Soc. 2017, 139, 14061–14064. doi:10.1021/jacs.7b09309
Return to citation in text: [1] -
Liu, X.; Deaton, T. M.; Haeffner, F.; Morken, J. P. Angew. Chem. 2017, 129, 11643–11647. doi:10.1002/ange.201705720
Return to citation in text: [1] -
Fang, H.; García-Camacho, A.; Hwang, H. S.; Sudsamart, A.; Daniliuc, C. G.; Grygorenko, O. O.; Funes-Ardoiz, I.; Molloy, J. J. J. Am. Chem. Soc. 2025, 147, 47730–47741. doi:10.1021/jacs.5c17624
Return to citation in text: [1] -
He, Q.; Zhang, J.; Deng, D.; Fan, Y.; Wang, Q.; Hua, Y.; Jing, S.; Hu, J. Org. Lett. 2026, 28, 746–751. doi:10.1021/acs.orglett.5c04923
Return to citation in text: [1] -
Liu, J.; Zhang, Z.; Troya, D.; Chen, M. J. Am. Chem. Soc. 2026, 148, 4899–4912. doi:10.1021/jacs.5c06735
Return to citation in text: [1] -
Rosario-Collazo, J. L.; Keita, H.; Velo, M.; Meek, S. J. Org. Lett. 2025, 27, 12092–12097. doi:10.1021/acs.orglett.5c03667
Return to citation in text: [1] -
Kim, M.; Kim, G.; Kim, D.; Cho, S. H. J. Am. Chem. Soc. 2024, 146, 34861–34869. doi:10.1021/jacs.4c14150
Return to citation in text: [1] -
Coombs, J. R.; Zhang, L.; Morken, J. P. J. Am. Chem. Soc. 2014, 136, 16140–16143. doi:10.1021/ja510081r
Return to citation in text: [1] -
Takahashi, F.; Nogi, K.; Sasamori, T.; Yorimitsu, H. Org. Lett. 2019, 21, 4739–4744. doi:10.1021/acs.orglett.9b01622
Return to citation in text: [1] -
Jin, H.; Han, J.; Liu, X.; Feng, C.; Zhan, M. Org. Lett. 2023, 25, 4168–4172. doi:10.1021/acs.orglett.3c01465
Return to citation in text: [1] -
Matteson, D. S. J. Org. Chem. 2013, 78, 10009–10023. doi:10.1021/jo4013942
Return to citation in text: [1] -
Stymiest, J. L.; Bagutski, V.; French, R. M.; Aggarwal, V. K. Nature 2008, 456, 778–782. doi:10.1038/nature07592
Return to citation in text: [1] -
Xu, N.; Holmgren, J. L., III; Morken, J. P. Angew. Chem., Int. Ed. 2024, 63, e202408436. doi:10.1002/anie.202408436
Return to citation in text: [1] -
Scott, H. K.; Aggarwal, V. K. Chem. – Eur. J. 2011, 17, 13124–13132. doi:10.1002/chem.201102581
Return to citation in text: [1] -
Clement, H. A.; Hall, D. G. Recent advances in the stereoselective additions of allylic boronates to carbonyl compounds. In In PATAI'S Chemistry of Functional Groups; Gandelman, M.; Marek, I., Eds.; John Wiley & Sons: New York, NY, USA, 2020; pp 243–328. doi:10.1002/9780470682531.pat0968
Return to citation in text: [1] -
Pierrot, D.; Marek, I. Angew. Chem., Int. Ed. 2020, 59, 20434–20438. doi:10.1002/anie.202010135
Return to citation in text: [1] -
Pavlickova, T.; Orbach, N.; Kaushansky, A.; Marek, I. J. Am. Chem. Soc. 2025, 147, 41204–41209. doi:10.1021/jacs.5c12433
Return to citation in text: [1] [2] [3] [4] -
Parra, A.; Amenós, L.; Guisán-Ceinos, M.; López, A.; García Ruano, J. L.; Tortosa, M. J. Am. Chem. Soc. 2014, 136, 15833–15836. doi:10.1021/ja510419z
Return to citation in text: [1] -
Augustin, A. U.; Di Silvio, S.; Marek, I. J. Am. Chem. Soc. 2022, 144, 16298–16302. doi:10.1021/jacs.2c07394
Return to citation in text: [1] -
Pavlíčková, T.; Stöckl, Y.; Marek, I. Org. Lett. 2022, 24, 8901–8906. doi:10.1021/acs.orglett.2c03756
Return to citation in text: [1] -
Burns, A. S.; Dooley, C., III; Carlson, P. R.; Ziller, J. W.; Rychnovsky, S. D. Org. Lett. 2019, 21, 10125–10129. doi:10.1021/acs.orglett.9b04133
Return to citation in text: [1] -
Namirembe, S.; Gao, C.; Wexler, R. P.; Morken, J. P. Org. Lett. 2019, 21, 4392–4394. doi:10.1021/acs.orglett.9b01663
Return to citation in text: [1]
| 1. | Maza, R. J.; Fernández, E.; Carbó, J. J. Chem. – Eur. J. 2021, 27, 12352–12361. doi:10.1002/chem.202101464 |
| 2. | Maza, R. J.; Carbó, J. J.; Fernández, E. Adv. Synth. Catal. 2021, 363, 2274–2289. doi:10.1002/adsc.202100192 |
| 34. | Pavlickova, T.; Orbach, N.; Kaushansky, A.; Marek, I. J. Am. Chem. Soc. 2025, 147, 41204–41209. doi:10.1021/jacs.5c12433 |
| 28. | Matteson, D. S. J. Org. Chem. 2013, 78, 10009–10023. doi:10.1021/jo4013942 |
| 29. | Stymiest, J. L.; Bagutski, V.; French, R. M.; Aggarwal, V. K. Nature 2008, 456, 778–782. doi:10.1038/nature07592 |
| 30. | Xu, N.; Holmgren, J. L., III; Morken, J. P. Angew. Chem., Int. Ed. 2024, 63, e202408436. doi:10.1002/anie.202408436 |
| 31. | Scott, H. K.; Aggarwal, V. K. Chem. – Eur. J. 2011, 17, 13124–13132. doi:10.1002/chem.201102581 |
| 32. | Clement, H. A.; Hall, D. G. Recent advances in the stereoselective additions of allylic boronates to carbonyl compounds. In In PATAI'S Chemistry of Functional Groups; Gandelman, M.; Marek, I., Eds.; John Wiley & Sons: New York, NY, USA, 2020; pp 243–328. doi:10.1002/9780470682531.pat0968 |
| 33. | Pierrot, D.; Marek, I. Angew. Chem., Int. Ed. 2020, 59, 20434–20438. doi:10.1002/anie.202010135 |
| 25. | Coombs, J. R.; Zhang, L.; Morken, J. P. J. Am. Chem. Soc. 2014, 136, 16140–16143. doi:10.1021/ja510081r |
| 26. | Takahashi, F.; Nogi, K.; Sasamori, T.; Yorimitsu, H. Org. Lett. 2019, 21, 4739–4744. doi:10.1021/acs.orglett.9b01622 |
| 27. | Jin, H.; Han, J.; Liu, X.; Feng, C.; Zhan, M. Org. Lett. 2023, 25, 4168–4172. doi:10.1021/acs.orglett.3c01465 |
| 3. | Zhang, C.; Hu, W.; Morken, J. P. ACS Catal. 2021, 11, 10660–10680. doi:10.1021/acscatal.1c02496 |
| 4. | Lee, Y.; Han, S.; Cho, S. H. Acc. Chem. Res. 2021, 54, 3917–3929. doi:10.1021/acs.accounts.1c00455 |
| 5. | Hong, K.; Liu, X.; Morken, J. P. J. Am. Chem. Soc. 2014, 136, 10581–10584. doi:10.1021/ja505455z |
| 6. | McDonald, T. R.; Rousseaux, S. A. L. Chem. Sci. 2023, 14, 963–969. doi:10.1039/d2sc06088d |
| 7. | McDonald, T. R.; Turner, J. A.; Gabbey, A. L.; Balasubramanian, P.; Rousseaux, S. A. L. Org. Lett. 2024, 26, 3822–3827. doi:10.1021/acs.orglett.4c00987 |
| 8. | Chen, X.-Y.; Gao, F.-C.; Ning, P.-F.; Wei, Y.; Hong, K. Angew. Chem., Int. Ed. 2023, 62, e202302638. doi:10.1002/anie.202302638 |
| 9. | Gu, H.-Y.; Chen, X.-Y.; Yang, Y.-F.; Hong, K. Org. Lett. 2025, 27, 4208–4213. doi:10.1021/acs.orglett.5c00886 |
| 10. | Gava, R.; Fernández, E. Chem. – Eur. J. 2019, 25, 8013–8017. doi:10.1002/chem.201901406 |
| 11. | Sakai, M.; Saito, S.; Kanai, G.; Suzuki, A.; Miyaura, N. Tetrahedron 1996, 52, 915–924. doi:10.1016/0040-4020(95)00950-7 |
| 12. | Joannou, M. V.; Moyer, B. S.; Goldfogel, M. J.; Meek, S. J. Angew. Chem., Int. Ed. 2015, 54, 14141–14145. doi:10.1002/anie.201507171 |
| 13. | Nallagonda, R.; Karimov, R. R. ACS Catal. 2021, 11, 248–254. doi:10.1021/acscatal.0c04474 |
| 14. | Park, J.; Lee, Y.; Kim, J.; Cho, S. H. Org. Lett. 2016, 18, 1210–1213. doi:10.1021/acs.orglett.6b00376 |
| 15. | Pereira, S.; Srebnik, M. Tetrahedron Lett. 1995, 36, 1805–1808. doi:10.1016/0040-4039(95)00148-6 |
| 16. | Knochel, P. J. Am. Chem. Soc. 1990, 112, 7431–7433. doi:10.1021/ja00176a074 |
| 17. | Schwenzer, M.; Studer, A. Adv. Synth. Catal. 2025, 367, e70117. doi:10.1002/adsc.70117 |
| 18. | Murray, S. A.; Liang, M. Z.; Meek, S. J. J. Am. Chem. Soc. 2017, 139, 14061–14064. doi:10.1021/jacs.7b09309 |
| 19. | Liu, X.; Deaton, T. M.; Haeffner, F.; Morken, J. P. Angew. Chem. 2017, 129, 11643–11647. doi:10.1002/ange.201705720 |
| 20. | Fang, H.; García-Camacho, A.; Hwang, H. S.; Sudsamart, A.; Daniliuc, C. G.; Grygorenko, O. O.; Funes-Ardoiz, I.; Molloy, J. J. J. Am. Chem. Soc. 2025, 147, 47730–47741. doi:10.1021/jacs.5c17624 |
| 21. | He, Q.; Zhang, J.; Deng, D.; Fan, Y.; Wang, Q.; Hua, Y.; Jing, S.; Hu, J. Org. Lett. 2026, 28, 746–751. doi:10.1021/acs.orglett.5c04923 |
| 22. | Liu, J.; Zhang, Z.; Troya, D.; Chen, M. J. Am. Chem. Soc. 2026, 148, 4899–4912. doi:10.1021/jacs.5c06735 |
| 23. | Rosario-Collazo, J. L.; Keita, H.; Velo, M.; Meek, S. J. Org. Lett. 2025, 27, 12092–12097. doi:10.1021/acs.orglett.5c03667 |
| 24. | Kim, M.; Kim, G.; Kim, D.; Cho, S. H. J. Am. Chem. Soc. 2024, 146, 34861–34869. doi:10.1021/jacs.4c14150 |
| 34. | Pavlickova, T.; Orbach, N.; Kaushansky, A.; Marek, I. J. Am. Chem. Soc. 2025, 147, 41204–41209. doi:10.1021/jacs.5c12433 |
| 34. | Pavlickova, T.; Orbach, N.; Kaushansky, A.; Marek, I. J. Am. Chem. Soc. 2025, 147, 41204–41209. doi:10.1021/jacs.5c12433 |
| 38. | Burns, A. S.; Dooley, C., III; Carlson, P. R.; Ziller, J. W.; Rychnovsky, S. D. Org. Lett. 2019, 21, 10125–10129. doi:10.1021/acs.orglett.9b04133 |
| 35. | Parra, A.; Amenós, L.; Guisán-Ceinos, M.; López, A.; García Ruano, J. L.; Tortosa, M. J. Am. Chem. Soc. 2014, 136, 15833–15836. doi:10.1021/ja510419z |
| 36. | Augustin, A. U.; Di Silvio, S.; Marek, I. J. Am. Chem. Soc. 2022, 144, 16298–16302. doi:10.1021/jacs.2c07394 |
| 37. | Pavlíčková, T.; Stöckl, Y.; Marek, I. Org. Lett. 2022, 24, 8901–8906. doi:10.1021/acs.orglett.2c03756 |
| 34. | Pavlickova, T.; Orbach, N.; Kaushansky, A.; Marek, I. J. Am. Chem. Soc. 2025, 147, 41204–41209. doi:10.1021/jacs.5c12433 |
| 39. | Namirembe, S.; Gao, C.; Wexler, R. P.; Morken, J. P. Org. Lett. 2019, 21, 4392–4394. doi:10.1021/acs.orglett.9b01663 |
© 2026 Pavlíčková et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.