Abstract
Background
The diastereoselectivity of a vinylogous Mukaiyama aldol reaction of a series of N-substituted 4-oxy-2-trimethylsilyloxypyrroles with a tartrate-based aldehyde has been explored as a model reaction for castanospermine synthesis.
Results
The study has revealed that the reaction is sensitive to the nature of the combination of N- and 4-oxy substituents. With a N-PMB or N-Bn and 4-methoxy combination, the reaction generates an aldol adduct with the correct absolute configurations for C-8 and C-8a of the indolizidine alkaloid castanospermine. The adduct was transformed to an indolizidine, whose ketal could not be transformed appropriately for the target alkaloid.
Conclusion
The first successful diastereoselective Mukaiyama aldol strategy for the C-8 and C-8a stereogenic centres of castanospermine is presented using silyloxypyrrole chemistry. The results suggest that a full enantioselective synthesis can be realized provided that C-1 functionalisation is accomplished early in the synthesis, post-coupling.

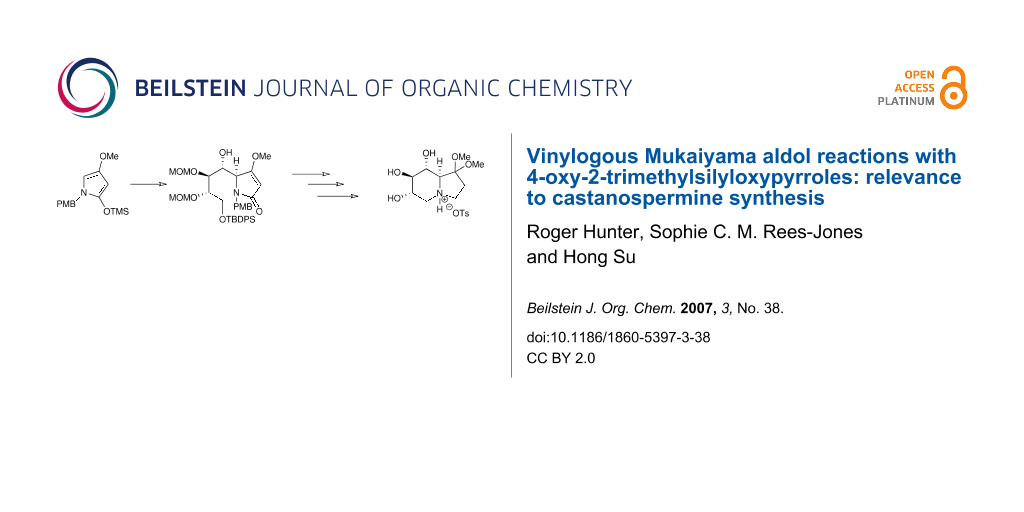
Graphical Abstract
Background
N-Protected silyloxypyrroles have emerged in recent years as powerful synthetic building blocks for synthesis, particularly of pyrrolizidine and indolizidine alkaloids.[1-3] Following the pioneering work of Casiraghi, N-(t-Boc)-2-(t-butyldimethylsilyloxy)pyrrole TBSOP 1 has established itself as the reagent of choice for promoting extended (vinylogous) Mukaiyama addition reactions to aldehydes,[4] imines [5] and conjugatively to enones [6,7] under Lewis-acid mediated dissociative reaction conditions. Many of these reactions reveal high diastereoselectivities, which has been exploited to access a range of natural products and their derivatives.[8,9] An example of Casiraghi's from the castanospermine repertoire showing the numbering system used in this article is shown in Scheme 1.[4]
![[1860-5397-3-38-i1]](/bjoc/content/inline/1860-5397-3-38-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: An example of a vinylogous Mukaiyama aldol in a natural product synthesis. Reagents and conditions: a) RCHO 2, SnCl4 (1.2 equiv), ether, -85°C.
Scheme 1: An example of a vinylogous Mukaiyama aldol in a natural product synthesis. Reagents and conditions:...
While TBSOP 1 can be prepared, isolated and stored (at low temperature) for general use, its synthesis involves usage of the relatively expensive TBSOTf. From the outset of this work, we were interested in developing a cost-effective alternative using TMS rather than TBS as the silylating source and changing the N-Boc group to Bn or PMB. As expected, such changes precluded isolation of the silyloxypyrrole and resulted in us developing a one-pot methodology involving its in situ generation. We have recently demonstrated the applicability of N-protected-4-methoxy-2-trimethylsilyloxy pyrroles 5 to the synthesis of key intermediates for the alkaloids lepadiformine [10] and castanospermine [11] using vinylogous Mukaiyama aldol reactions. For the latter, silyloxypyrrole 5a was shown to give adduct 6a with the correct stereogenicities at C-4 and C-5 (C-8a and C-8 respectively in the alkaloid) for castanospermine.
This paper reports on the influence of substitution changes in 5 (R1 and R2) on the outcome of extended Mukaiyama reactions with aldehyde 7, and demonstrates how one of the adducts can be transformed into an advanced intermediate for castanospermine synthesis (Scheme 2).
![[1860-5397-3-38-i2]](/bjoc/content/inline/1860-5397-3-38-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Our vinylogous Mukaiyama aldol reaction with different substituents. Reagents and conditions: a) n-BuLi, (1.5 equiv), THF, -78°C, 30 mins; b) TMSCl (3 equiv), -78°C, 30 mins; c) 7 (0.7 equiv), THF, -78°C; d) SnCl4 (2 equiv), -78°C to -20°C.
Scheme 2: Our vinylogous Mukaiyama aldol reaction with different substituents. Reagents and conditions: a) n-...
Results and Discussion
Changing the N-protecting group
Synthesis of pyrrolinones 4a and 4b was straightforward involving condensing the appropriate amine (PMBNH2 or BnNH2 respectively) with ethyl E-4-chloro-3-methoxybut-2-enoate.[11] Reaction of 4b with n-BuLi as before followed by TMSCl to generate 5b, and reaction with aldehyde 7 (0.7 equiv) as limiting reagent using SnCl4 (2 equiv) as the Lewis-acid promoter with rapid stirring of the reaction gave a single crystalline diastereomer 6b as the major product (57%) together with a mixed fraction (33%) following chromatography. The latter revealed a complex array for the H-4 and H-5 signals in its 1H NMR spectrum, and no conclusive stereochemical assignments could be made. By comparison and as with 6a,[11] the key NMR signals of H-4 and H-5 in the 1H NMR and C-4 and C-5 in the 13C NMR of 6b suggested an identical stereochemistry to that of adduct 6a, and this was unambiguously confirmed for 6b as the 4,5-erythro-5,6-threo adduct by a single crystal X-ray determination (Figure 1). CCDC 653714 contains the Supplementary Crystallographic Data for compound 6b. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. Such a result confirmed the unimportance of the p-methoxy group of the N-PMB protecting group on the diastereoselectivity of the reaction. Given that tlc indicated that reaction only begins at around -50°C, and that the reaction is quenched at -20°C, we believe the major adduct to be the kinetic product. However, a more comprehensive study is needed to support this view beyond reasonable doubt.
![[1860-5397-3-38-1]](/bjoc/content/figures/1860-5397-3-38-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Attention was then turned to changing the N-protecting group to a carbamate in line with TBSOP 1. The pyrroline precursor 4 (R1 = H, R2 = OMe) was readily prepared via condensation of ethyl E-4-chloro-3-methoxybut-2-enoate with ammonia, and then transformed to carbamates 4c and 4d under the standard conditions of (Boc)2O/DMAP and NaH/CBzCl respectively. Each was independently subjected to our in situ Mukaiyama sequence involving n-BuLi followed by TMSCl, and then addition of aldehyde 7 and SnCl4. However, following the normal work-up, tlc analysis in each case revealed consumption of starting pyrrolinone with formation of a multitude of products. This was attributed to the instability of the pyrrolinone carbamates 4c and 4d towards n-BuLi, and indicated the need to resort to the Casiraghi conditions of 2,6-lutidine and TBSOTf for silyloxypyrrole generation.[12] However, in view of our objective of developing a cost-effective method, this option was not pursued.
Changing the 4-oxy substituent
In terms of synthetic design, the 4-methoxy substituent was envisaged as having implications for reactivity of the silyloxypyrrole as well as relevance to installation of the C-1 hydroxyl in a convergent castanospermine synthesis using a Mukaiyama aldol reaction as a key step. Thus it was decided to investigate the influence of changing both the O-protecting group as well as substituting the 4-oxy substituent with hydrogen. Deprotection of a methyl ether to its hydroxyl group requires aggressive conditions that can present problems in end-game aspects of total synthesis. Thus it was decided to change methyl to the more deprotection-friendly benzyl group. Synthesis of benzyl pyrrolinone 4e was readily achieved by heating 4a with excess benzyl alcohol at 80°C in an acid-catalysed (p-TsOH) exchange with in vacuo (water pressure) removal of methanol as it formed. Thus 4e was isolated in 65% yield after chromatography. Subjecting 4e to the standard silyloxypyrrole formation conditions as before to afford 5e in situ followed by reaction with aldehyde 7 resulted in isolation of a major diastereoisomeric adduct in 63% yield following conventional work-up and chromatography. Although an X-ray structure determination was not carried out, the tlc and spectral characteristics provided strong evidence that adduct 6e had the same C4/5 stereochemistry as 6a, b. For example, the coupling constant between H-5/H-6 of 6e of 6.0 Hz compared to 7.2 and 7.3 Hz for 6a and 6b respectively indicated the same relative 5/6 stereochemistry (threo)[9] as in 6a/b. Unfortunately, the signals for H-4 and H-5 were part of a complex signal and thus ascertaining the relative stereochemistry at H-4 was more difficult. However, the [α]D found in this case had a positive value, as it was for 6a and 6b. Casiraghi has demonstrated that the sign of the rotation is dependent on C-4 absolute configuration,[4] and it is likely that the same holds here, thus since the 13C values of 6e for C-4, C-5 and C-6 were similar within 1 ppm to those for 6a and 6b, the absolute stereochemistries at C-4 and C-5 of 6e were taken as likely to be the same as those for 6a and 6b.
Finally, the role of the C-4 methoxy group was investigated. Thus, pyrrolinone 4f (R2 = H) was prepared from the known 4-hydroxy lactam 8 [13-15] via elimination of its mesylate (Scheme 3). Subsequent to this we became aware of a much shorter sequence for realising 4f via condensation of dimethoxydihydrofuran with benzylamine [16] or via a ring-closing metathesis reaction.[17]
![[1860-5397-3-38-i3]](/bjoc/content/inline/1860-5397-3-38-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of N-benzyl-3-pyrrolin-2-one 4f. Reagents and conditions: a) MsCl, Et3N, DMAP, 90%; b) Et3N, THF, Δ, 70%.
Scheme 3: Synthesis of N-benzyl-3-pyrrolin-2-one 4f. Reagents and conditions: a) MsCl, Et3N, DMAP, 90%; b) Et3...
Pyrrolinone 4f was then subjected to the standard Mukaiyama aldol sequence (Scheme 2), but tlc indicated that no reaction to form an aldol adduct had taken place. The reaction sequence was repeated and each time starting pyrrolinone was recovered. Assuming that formation of the silyloxypyrrole occurred in this sequence, which we feel to be likely in this case, this outcome came as an interesting and unexpected result, particularly since chiral N-alkyl silyloxypyrroles are known to undergo asymmetric vinylogous Mukaiyama aldol reactions with simple aldehydes.[3,18] It would appear that in our case in the N-benzyl series, a C-4 methoxy group is essential for reactivity by raising the energy of the silyloxypyrrole HOMO. However, this still leaves the question of why Casiraghi's TBSOP 1 with an N-carbamate protecting group is so effective compared to our case 5f with the N-benzyl group in which the nitrogen lone pair is more delocalized into the pyrrole ring. Possible interference of the Lewis-acid at the nitrogen of 5f (not likely in TBSOP) is a possible explanation, which presumably changes in the presence of the methoxy group of 5a. The latter possibility is in line with the transition-state model recently postulated by us in which it was suggested that the methoxy group plays a coordinating role in the extended Mukaiyama aldol addition resulting in an endo-like transition state.[11] In the known cases [3,18] of N-alkyl silyloxypyrroles successfully reacting with simple aldehydes just mentioned, a more hindered α-asymmetric benzyl centre on nitrogen was used which would have presented steric hindrance at nitrogen towards the Lewis-acid.
We have also previously reported on the use of 4-methoxy-2-trimethylsilyloxypyrroles for generating C-5 quaternary centres using trimethyl orthoformate as the electrophile and BF3·OEt2 as the promoter.[10] Together with the present study, this confirms the usefulness of N-benzyl-4-oxy-2-trimethylsilyloxypyrroles in vinylogous Mukaiyama aldol reactions.
As a demonstration of the usefulness of the methodology described herein, adduct 6a was transformed into an advanced intermediate for the synthesis of (+)-castanospermine 9 (Figure 2). The latter is an indolizidine alkaloid that has received significant attention from the synthetic organic community in view of its potent biological activity as an α- and β-glycosidase inhibitor with promising anti-diabetic,[19] anti-cancer,[20] anti-viral [21] and anti-AIDS activity.[22] The early castanospermine syntheses were carbohydrate-based, and used glucose or mannose as their starting materials. However, in more recent times there has been a trend towards using other building blocks from the chiral pool to introduce some of the chiral centres.[23,24] Of these approaches, a number have utilized a convergent stategy via a C-8/C-8a disconnection (Figure 3). Thus, Gallagher,[25] Martin [26,27] and Casiraghi [4] have all attempted syntheses involving either a carbanion at C-8a or a Lewis-acid promoted aldol reaction as in Scheme 1 to form the C-8/C-8a bond. All the syntheses have suffered from incorrect diastereoselectivity in the aldol step as well as a lack of provision for C-1 hydroxyl group installation, and none of the syntheses based on this disconnection to date have successfully synthesized the target 9. Given our success [9] in synthesizing the 4,5-erythro-5,6-threo adduct 6a (Scheme 2) with correct absolute configurations for castanospermine (C-8/C-8a), we sought to transform 6a into the target alkaloid, and in this paper report on the synthesis of an advanced intermediate towards this goal.
![[1860-5397-3-38-2]](/bjoc/content/figures/1860-5397-3-38-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of (+)-castanospermine 9.
Figure 2: Structure of (+)-castanospermine 9.
![[1860-5397-3-38-3]](/bjoc/content/figures/1860-5397-3-38-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: C-8/C-8a disconnection stategy for castanospermine synthesis.
Figure 3: C-8/C-8a disconnection stategy for castanospermine synthesis.
Thus, adduct 6a underwent oxidative cleavage of the PMB protecting group under acid conditions with CAN to give lactam 10 in 82% yield with retention of the MOM groups (Scheme 4). The vinyl ether 10 was then converted to a ketal in order to preclude any epimerization occurring at C-4, and this was achieved by subjecting 10 to bromine in the presence of methanol to give bromoketal 11 in 91% yield as a mixture of diastereomers (4:1). The diastereomeric mixture of 11 then underwent zinc reduction in the presence of methanol/THF/ammonium chloride [28] to give ketal 12 in 90% yield as a single diastereomer as evidenced by 1H and 13C NMR spectra. Having developed C-1 functionality in a protected form, attention turned to lactam carbonyl group removal and cyclization. In order to avoid interference during cyclization from the C-5 hydroxyl group as well as to link up with the lactam reduction methodology, the C-5 hydroxyl group and lactam N-H were both converted to their Boc derivatives using standard conditions ((Boc)2O (4 equiv), THF, DMAP) to give 13 in 90% yield as a crystalline solid. N-Boc lactam 13 was then reduced to lactol 14 with DIBAL-H to afford a mixture of diastereomers in 86% yield, which was reduced with triethylsilane in the presence of BF3·OEt2 to give carbamate 15 in 91% yield. This was then desilylated with TBAF at 10°C for 5 days to give alcohol 16 in 84% yield. These conditions were chosen, as by-products formed at higher temperature.
![[1860-5397-3-38-i4]](/bjoc/content/inline/1860-5397-3-38-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of Adduct 16 from adduct 6a. Reagents and conditions: a) CAN, aq CH3CN, -20°C to rt, 5 h, 82%; b) Br2, MeOH, -20°C, 30 mins, 91%; c) Zn, aq NH4Cl, THF, MeOH, RT, 30 mins, 90%; d) (Boc)2O (4 equiv), THF, DMAP (cat), rt, 18 h, 90%; e) DIBAL-H, THF, -78°C to -20°C, 2 h, 86%; f) Et3SiH, BF3·OEt2, DCM, -70°C, 1 h, 91%; g) TBAF, THF, 10°C, 5 days, 84%.
Scheme 4: Synthesis of Adduct 16 from adduct 6a. Reagents and conditions: a) CAN, aq CH3CN, -20°C to rt, 5 h,...
Adduct 16 was crystallized from ethyl acetate and hexane and used to obtain a single-crystal X-ray structure (Figure 4). CCDC 653715 contains the Supplementary Crystallographic Data for compound 16. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. The structure revealed the tartrate-derived centres C-6 and C-7 to be in their correct absolute configurations as derived from L-tartrate (both S-), and thus established the C-4 and C-5 absolute configurations to be S-, and R- respectively as shown in Scheme 4 and correct for castanospermine synthesis. The result also confirmed that the Mukaiyama adduct 6a had not epimerized during this end-game sequence. Interestingly, the N-Boc group appears in its s-trans form, with the carbonyl oxygen pointing away from C-4. Compound 16 was then converted in high yield to tosylate 17 for the final cyclization, hydrolysis and reduction sequence to target (Scheme 5).
![[1860-5397-3-38-i5]](/bjoc/content/inline/1860-5397-3-38-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Conversion of adduct 16 to indolizidine 18. Reagents and conditions a) (1.5 equiv), Et3N (2 equiv), DMAP (cat), DCM, 27°C, 24 h, 100%; b) i) TFA: DCM (1:4), 0°C 2 h; ii) Hünig's base (4 equiv), DCM, 0°C, 18 h, 56%.
Scheme 5: Conversion of adduct 16 to indolizidine 18. Reagents and conditions a) (1.5 equiv), Et3N (2 equiv),...
![[1860-5397-3-38-4]](/bjoc/content/figures/1860-5397-3-38-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Exposure of 17 to TFA at 0°C followed by addition of Hünig's base resulted in cyclization to indolizidine 18, which was isolated chromatographically as a tosylate salt in 56% yield. Compound 18 was identified from its NMR data. In particular, C-3(H-3), C-5(H-5) and C-8a(H-8a) resonated ≈ 10 ppm (C) and 1 ppm (H) downfield respectively compared to the signals in castanospermine as a result of deshielding by the quaternised nitrogen atom. Indolizidine 18 indicated concomitant hydrolysis of both MOM ethers to have occurred. A minor, less polar fraction was identified as a partially protected (C-6 or C-7) indolizidine (26%) that could independently be transformed into 18 by treatment with HCl. Unfortunately, attempts to hydrolyse the C-1 ketal of 18 to its carbonyl function under a variety of concentrations of acid (HCl) and at different temperatures all failed. Prolonged treatment led to decomposition. We attribute this unreactivity to destabilization of the intermediate oxocarbenium ion by the adjacent α-ammonium cation.
Conclusion
In summary, the present work has laid the foundation for a full enantioselective synthesis of castanospermine using a C-8/C-8a disconnection strategy. Future work will focus on C-1 transformation earlier in the sequence.
Experimental
See Supporting Information File 1 for full experimental data. Figure 5 describes the numbering system used in the experimental section.
![[1860-5397-3-38-5]](/bjoc/content/figures/1860-5397-3-38-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Structure of 6a with numbering to demonstrate numbering system used in this section.
Figure 5: Structure of 6a with numbering to demonstrate numbering system used in this section.
References
-
Langlois, N.; Le Nguyen, B. K.; Retailleau, P.; Tarnus, C.; Salomon, E. Tetrahedron: Asymmetry 2006, 17, 53–60. doi:10.1016/j.tetasy.2005.11.017
Return to citation in text: [1] -
Rassu, G.; Carta, P.; Pinna, L.; Battistini, L.; Zanardi, F.; Acquotti, D.; Casiraghi, G. Eur. J. Org. Chem. 1999, 6, 1395–1400. doi:10.1002/(SICI)1099-0690(199906)1999:6<1395::AID-EJOC1395>3.0.CO;2-T
Return to citation in text: [1] -
Dudot, B.; Micouin, L.; Baussanne, I.; Royer, J. Synthesis 1999, 688–694. doi:10.1055/s-1999-3434
Return to citation in text: [1] [2] [3] -
Casiraghi, G.; Ulgheri, F.; Spanu, P.; Rassu, G.; Pinna, L.; Fava, G. G.; Ferrari, M. B.; Pelosi, G. J. Chem. Soc., Perkin Trans. 1 1993, 2991–2997. doi:10.1039/p19930002991
Return to citation in text: [1] [2] [3] [4] -
DeGoey, D. A.; Chen, H. J.; Flosi, W. J.; Grampovnik, D. J.; Yeung, C. M.; Klein, L. L.; Kempf, D. J. J. Org. Chem. 2002, 67, 5445–5453. doi:10.1021/jo0162890
Return to citation in text: [1] -
Arroyo, Y.; de Paz, M.; Rodriguez, J. F.; Sanz-Tejedor, M. A.; Ruano, J. L. G. J. Org. Chem. 2002, 67, 5638–5643. doi:10.1021/jo016239v
Return to citation in text: [1] -
Brimble, M. A.; Burgess, C.; Halim, R.; Petersson, M.; Ray, J. Tetrahedron 2004, 60, 5751–5758. doi:10.1016/j.tet.2004.05.008
Return to citation in text: [1] -
Casiraghi, G.; Rassu, G. Synthesis 1995, 607–626. doi:10.1055/s-1995-3983
Return to citation in text: [1] -
Rassu, G.; Zanardi, F.; Battistini, L.; Casiraghi, G. Chem. Soc. Rev. 2000, 20, 109–118. doi:10.1039/a900200f
Return to citation in text: [1] [2] [3] -
Hunter, R.; Richards, P. Synlett 2003, 271–275. doi:10.1055/s-2003-36782
Return to citation in text: [1] [2] -
Hunter, R.; Rees-Jones, S. C. M.; Su, H. Tetrahedron Lett. 2007, 48, 2819–2822. doi:10.1016/j.tetlet.2007.02.100
Return to citation in text: [1] [2] [3] [4] -
Casiraghi, G.; Rassu, G.; Spanu, P.; Pinna, L. J. Org. Chem. 1992, 57, 3760–3763. doi:10.1021/jo00040a006
Return to citation in text: [1] -
Louwrier, S.; Ostendorf, A. B.; Hiemstra, H.; Speckamp, W. N. Tetrahedron 1996, 52, 2603–2628. doi:10.1016/0040-4020(95)01085-8
Return to citation in text: [1] -
Tomori, H.; Shibutani, K.; Ogura, K. Bull. Chem. Soc. Jpn. 1996, 69, 207–215. doi:10.1246/bcsj.69.207
Return to citation in text: [1] -
Huang, P. Q.; Zheng, X.; Wang, S. L.; Ye, J. L.; Jin, L. R.; Chen, Z. Tetrahedron: Asymmetry 1999, 10, 3309–3317. doi:10.1016/S0957-4166(99)00321-3
Return to citation in text: [1] -
Baussanne, I.; Chiaroni, A.; Husson, H.-P.; Riche, C.; Royer, J. Tetrahedron Lett. 1994, 35, 3931–3934. doi:10.1016/S0040-4039(00)76705-0
We thank Professor Jacques Royer of the CNRS in Paris for this information as well as alerting us to reference 18.
Return to citation in text: [1] -
Vo-Thanh, G.; Boucard, V.; Sauriat-Dorizon, H.; Guibe, F. Synlett 2001, 37–40.
Return to citation in text: [1] -
Uno, H.; Baldwin, J. E.; Russell, A. T. J. Am. Chem. Soc. 1994, 116, 2139–2140. doi:10.1021/ja00084a062
Return to citation in text: [1] [2] -
Nojima, H.; Kimura, I.; Chen, G.-J.; Sugihara, Y.; Haruno, M.; Kato, A.; Asano, N. J. Nat. Prod. 1998, 61, 397–400. doi:10.1021/np970277l
Return to citation in text: [1] -
Pili, R.; Chang, J.; Partis, R. A.; Mueller, R. A.; Chrest, F. J.; Passaniti, A. Cancer Res. 1995, 55, 2920–2926.
Return to citation in text: [1] -
Sunkara, P. S.; Bowlin, T. L.; Liu, P. S.; Sjoerdsma, A. Biochem. Biophys. Res. Commun. 1987, 148, 206–210. doi:10.1016/0006-291X(87)91096-5
Return to citation in text: [1] -
Fleet, G. W. J.; Karpas, A.; Dwek, R. A.; Fellows, L. E.; Tyms, A. S.; Petursson, S.; Namgoong, S. K.; Ramsden, N. G.; Smith, P. W.; Son, J. C.; Wilson, F.; Witty, D. R.; Jacobs, G. S.; Rademacher, T. W. FEBS Lett. 1988, 237, 128–132. doi:10.1016/0014-5793(88)80185-6
Return to citation in text: [1] -
Somfai, P.; Marchand, P.; Torsell, S.; Lindström, U. M. Tetrahedron 2003, 59, 1293–1299. doi:10.1016/S0040-4020(02)01660-5
Return to citation in text: [1] -
Kang, S. H.; Kim, J. S. Chem. Commun. 1998, 1353–1354. doi:10.1039/a802741b
Return to citation in text: [1] -
Gallagher, T.; Giles, M.; Subramanian, R. S.; Hadley, M. S. J. Chem. Soc., Chem. Commun. 1992, 166–168. doi:10.1039/c39920000166
Return to citation in text: [1] -
Martin, S. F.; Chen, H. J.; Yang, C. P. J. Org. Chem. 1993, 58, 2867–2873. doi:10.1021/jo00062a035
Return to citation in text: [1] -
Martin, S. F.; Chen, H. J.; Lynch, V. M. J. Org. Chem. 1995, 60, 276–278. doi:10.1021/jo00106a051
Return to citation in text: [1] -
Cheung, M. J.; Harris, P. A.; Lackey, K. E. Tetrahedron Lett. 2001, 42, 999–1001. doi:10.1016/S0040-4039(00)02219-X
Return to citation in text: [1] -
McCaig, A. E.; Meldrum, K. P.; Wightman, R. H. Tetrahedron 1998, 54, 9429–9446. doi:10.1016/S0040-4020(98)00572-9
-
Kochhar, K. S.; Carson, H. J.; Clouser, K. A.; Elling, J. W.; Gramnes, L. A.; Parry, J. L.; Sherman, H. L.; Bratt, K.; Pinnick, H. W. Tetrahedron Lett. 1984, 25, 1871–1874. doi:10.1016/S0040-4039(01)90063-2
-
Kochhar, K. S.; Pinnick, H. W. J. Org. Chem. 1984, 49, 3222–3224. doi:10.1021/jo00191a038
-
Duc, L.; McGarrity, J. F.; Meul, T.; Warm, A. Synthesis 1992, 391–394. doi:10.1055/s-1992-26120
| 4. | Casiraghi, G.; Ulgheri, F.; Spanu, P.; Rassu, G.; Pinna, L.; Fava, G. G.; Ferrari, M. B.; Pelosi, G. J. Chem. Soc., Perkin Trans. 1 1993, 2991–2997. doi:10.1039/p19930002991 |
| 9. | Rassu, G.; Zanardi, F.; Battistini, L.; Casiraghi, G. Chem. Soc. Rev. 2000, 20, 109–118. doi:10.1039/a900200f |
| 28. | Cheung, M. J.; Harris, P. A.; Lackey, K. E. Tetrahedron Lett. 2001, 42, 999–1001. doi:10.1016/S0040-4039(00)02219-X |
| 1. | Langlois, N.; Le Nguyen, B. K.; Retailleau, P.; Tarnus, C.; Salomon, E. Tetrahedron: Asymmetry 2006, 17, 53–60. doi:10.1016/j.tetasy.2005.11.017 |
| 2. | Rassu, G.; Carta, P.; Pinna, L.; Battistini, L.; Zanardi, F.; Acquotti, D.; Casiraghi, G. Eur. J. Org. Chem. 1999, 6, 1395–1400. doi:10.1002/(SICI)1099-0690(199906)1999:6<1395::AID-EJOC1395>3.0.CO;2-T |
| 3. | Dudot, B.; Micouin, L.; Baussanne, I.; Royer, J. Synthesis 1999, 688–694. doi:10.1055/s-1999-3434 |
| 8. | Casiraghi, G.; Rassu, G. Synthesis 1995, 607–626. doi:10.1055/s-1995-3983 |
| 9. | Rassu, G.; Zanardi, F.; Battistini, L.; Casiraghi, G. Chem. Soc. Rev. 2000, 20, 109–118. doi:10.1039/a900200f |
| 16. |
Baussanne, I.; Chiaroni, A.; Husson, H.-P.; Riche, C.; Royer, J. Tetrahedron Lett. 1994, 35, 3931–3934. doi:10.1016/S0040-4039(00)76705-0
We thank Professor Jacques Royer of the CNRS in Paris for this information as well as alerting us to reference 18. |
| 6. | Arroyo, Y.; de Paz, M.; Rodriguez, J. F.; Sanz-Tejedor, M. A.; Ruano, J. L. G. J. Org. Chem. 2002, 67, 5638–5643. doi:10.1021/jo016239v |
| 7. | Brimble, M. A.; Burgess, C.; Halim, R.; Petersson, M.; Ray, J. Tetrahedron 2004, 60, 5751–5758. doi:10.1016/j.tet.2004.05.008 |
| 17. | Vo-Thanh, G.; Boucard, V.; Sauriat-Dorizon, H.; Guibe, F. Synlett 2001, 37–40. |
| 5. | DeGoey, D. A.; Chen, H. J.; Flosi, W. J.; Grampovnik, D. J.; Yeung, C. M.; Klein, L. L.; Kempf, D. J. J. Org. Chem. 2002, 67, 5445–5453. doi:10.1021/jo0162890 |
| 4. | Casiraghi, G.; Ulgheri, F.; Spanu, P.; Rassu, G.; Pinna, L.; Fava, G. G.; Ferrari, M. B.; Pelosi, G. J. Chem. Soc., Perkin Trans. 1 1993, 2991–2997. doi:10.1039/p19930002991 |
| 4. | Casiraghi, G.; Ulgheri, F.; Spanu, P.; Rassu, G.; Pinna, L.; Fava, G. G.; Ferrari, M. B.; Pelosi, G. J. Chem. Soc., Perkin Trans. 1 1993, 2991–2997. doi:10.1039/p19930002991 |
| 13. | Louwrier, S.; Ostendorf, A. B.; Hiemstra, H.; Speckamp, W. N. Tetrahedron 1996, 52, 2603–2628. doi:10.1016/0040-4020(95)01085-8 |
| 14. | Tomori, H.; Shibutani, K.; Ogura, K. Bull. Chem. Soc. Jpn. 1996, 69, 207–215. doi:10.1246/bcsj.69.207 |
| 15. | Huang, P. Q.; Zheng, X.; Wang, S. L.; Ye, J. L.; Jin, L. R.; Chen, Z. Tetrahedron: Asymmetry 1999, 10, 3309–3317. doi:10.1016/S0957-4166(99)00321-3 |
| 11. | Hunter, R.; Rees-Jones, S. C. M.; Su, H. Tetrahedron Lett. 2007, 48, 2819–2822. doi:10.1016/j.tetlet.2007.02.100 |
| 12. | Casiraghi, G.; Rassu, G.; Spanu, P.; Pinna, L. J. Org. Chem. 1992, 57, 3760–3763. doi:10.1021/jo00040a006 |
| 11. | Hunter, R.; Rees-Jones, S. C. M.; Su, H. Tetrahedron Lett. 2007, 48, 2819–2822. doi:10.1016/j.tetlet.2007.02.100 |
| 9. | Rassu, G.; Zanardi, F.; Battistini, L.; Casiraghi, G. Chem. Soc. Rev. 2000, 20, 109–118. doi:10.1039/a900200f |
| 4. | Casiraghi, G.; Ulgheri, F.; Spanu, P.; Rassu, G.; Pinna, L.; Fava, G. G.; Ferrari, M. B.; Pelosi, G. J. Chem. Soc., Perkin Trans. 1 1993, 2991–2997. doi:10.1039/p19930002991 |
| 11. | Hunter, R.; Rees-Jones, S. C. M.; Su, H. Tetrahedron Lett. 2007, 48, 2819–2822. doi:10.1016/j.tetlet.2007.02.100 |
| 3. | Dudot, B.; Micouin, L.; Baussanne, I.; Royer, J. Synthesis 1999, 688–694. doi:10.1055/s-1999-3434 |
| 18. | Uno, H.; Baldwin, J. E.; Russell, A. T. J. Am. Chem. Soc. 1994, 116, 2139–2140. doi:10.1021/ja00084a062 |
| 3. | Dudot, B.; Micouin, L.; Baussanne, I.; Royer, J. Synthesis 1999, 688–694. doi:10.1055/s-1999-3434 |
| 18. | Uno, H.; Baldwin, J. E.; Russell, A. T. J. Am. Chem. Soc. 1994, 116, 2139–2140. doi:10.1021/ja00084a062 |
| 11. | Hunter, R.; Rees-Jones, S. C. M.; Su, H. Tetrahedron Lett. 2007, 48, 2819–2822. doi:10.1016/j.tetlet.2007.02.100 |
| 25. | Gallagher, T.; Giles, M.; Subramanian, R. S.; Hadley, M. S. J. Chem. Soc., Chem. Commun. 1992, 166–168. doi:10.1039/c39920000166 |
| 26. | Martin, S. F.; Chen, H. J.; Yang, C. P. J. Org. Chem. 1993, 58, 2867–2873. doi:10.1021/jo00062a035 |
| 27. | Martin, S. F.; Chen, H. J.; Lynch, V. M. J. Org. Chem. 1995, 60, 276–278. doi:10.1021/jo00106a051 |
| 22. | Fleet, G. W. J.; Karpas, A.; Dwek, R. A.; Fellows, L. E.; Tyms, A. S.; Petursson, S.; Namgoong, S. K.; Ramsden, N. G.; Smith, P. W.; Son, J. C.; Wilson, F.; Witty, D. R.; Jacobs, G. S.; Rademacher, T. W. FEBS Lett. 1988, 237, 128–132. doi:10.1016/0014-5793(88)80185-6 |
| 23. | Somfai, P.; Marchand, P.; Torsell, S.; Lindström, U. M. Tetrahedron 2003, 59, 1293–1299. doi:10.1016/S0040-4020(02)01660-5 |
| 24. | Kang, S. H.; Kim, J. S. Chem. Commun. 1998, 1353–1354. doi:10.1039/a802741b |
| 20. | Pili, R.; Chang, J.; Partis, R. A.; Mueller, R. A.; Chrest, F. J.; Passaniti, A. Cancer Res. 1995, 55, 2920–2926. |
| 21. | Sunkara, P. S.; Bowlin, T. L.; Liu, P. S.; Sjoerdsma, A. Biochem. Biophys. Res. Commun. 1987, 148, 206–210. doi:10.1016/0006-291X(87)91096-5 |
| 19. | Nojima, H.; Kimura, I.; Chen, G.-J.; Sugihara, Y.; Haruno, M.; Kato, A.; Asano, N. J. Nat. Prod. 1998, 61, 397–400. doi:10.1021/np970277l |
© 2007 Hunter et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)