Abstract
Supramolecular polymers are linear chains of low molar mass monomers held together by reversible and directional non-covalent interactions, which can form gels or highly viscous solutions if the self-assembled chains are sufficiently long and rigid. The viscosity of these solutions can be controlled by adding monofunctional compounds, which interact with the chain extremities: chain stoppers. We have synthesized new substituted ureas and thioureas and tested them as chain stoppers for a bis-urea based supramolecular polymer. In particular, the bis-thiourea analogue of the bis-urea monomer is shown not to form a supramolecular polymer, but a good chain stopper, because it is a strong hydrogen bond donor and a weak acceptor. Moreover, all substituted ureas tested reduce the viscosity of the supramolecular polymer solutions, but the best chain stopper is obtained when two hydrogen bond acceptors are placed in the same relative position as for the monomer and when no hydrogen bond donor is present.

Graphical Abstract
Introduction
Supramolecular polymers are linear chains of low molar mass monomers held together by reversible and highly directional non-covalent interactions [1-3]. Because of their macromolecular architecture, they can display polymer-like rheological properties, and they can, in particular, form gels if the self-assembled chains are sufficiently long and rigid [4-9]. Compared to the well-known organogelators formed by the entanglement of usually crystalline fibers [10-13], supramolecular polymers display some specific features. In particular, hydrogen-bonded supramolecular polymers are often dynamic at room temperature, which means that they do not need to be heated and then cooled to form a gel. Moreover, the gels formed are usually visco-elastic, meaning that they show an elastic response only at high frequencies.
The chain length of a supramolecular polymer depends on the strength of the association between the monomers, which is highly dependent on their concentration, the temperature, the solvent, i.e., environmental factors, but also on the presence of additives. Chain-stoppers are monofunctional monomers able to interact with monomers and therefore able to break polymer chains. They can be introduced in order to reduce the length of the supramolecular polymer (and thus reduce the viscosity of the solution) [14-16], but also in order to block the concentration dependence of the supramolecular polymers [17-19]. Chain stoppers can also be exploited to decorate the chain-ends with particular functional groups or labels [20,21]. The effectiveness of these schemes depends directly on the design of the chain-stopper: the interaction between chain-stopper and monomer should ideally be as strong as the interaction between monomers. It is therefore of interest to identify chain stoppers with an improved affinity toward a given supramolecular polymer. In this article, we investigate the efficiency of several new chain stoppers for a well-known bis-urea-based supramolecular polymer EHUT (Figure 1). This supramolecular polymer is particularly interesting, because it has been previously shown to self-assemble cooperatively into two competitive high molecular weight structures [22-24].
![[1860-5397-6-102-1]](/bjoc/content/figures/1860-5397-6-102-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of monomer EHUT and chain stoppers.
Figure 1: Structures of monomer EHUT and chain stoppers.
Results and Discussion
Design and synthesis
The bis-urea based monomer EHUT has been shown to self-assemble in non-polar solvents, into two supramolecular polymeric structures, the tube or the filament forms, which are in dynamic exchange [23,24]. The respective stability of each form depends on the solvent, the temperature and the concentration. The filament form contains a single molecule in its cross section [25-27], and is the most stable structure at concentrations above 10−3mol/L and at room temperature, in solvents such as chloroform [22], carbon tetrachloride [28] and 1,3,5-trimethylbenzene [29]. The tube form contains three molecules in its cross section [6,30,31], and is the most stable structure at concentrations above 10−5mol/L and at room temperature, in solvents such as toluene [32] and dodecane [5].
Chain stopper S1, with two NH groups replaced by N-butyl groups, was previously shown to be a good chain stopper for EHUT in carbon tetrachloride [17], i.e., a good chain stopper of the filament form. However, at high concentrations, the two remaining NH groups were shown to form hydrogen bonds [17], and therefore S1 can also behave to some extent as a co-monomer of EHUT: a small proportion of S1 molecules may be incorporated in the filament structure rather than at its extremities. Simple alkylation of the 2 remaining NH groups does not yield an efficient chain stopper [17]. This surprising result was tentatively attributed to the conformation of the tetrasubstituted urea group, which may be ill-adapted to form hydrogen bonds to the urea groups of EHUT (Figure 2).
![[1860-5397-6-102-2]](/bjoc/content/figures/1860-5397-6-102-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Substituted urea conformation. If R is alkyl, the most stable conformation is b); if R = H, the most stable conformation is a) [37].
Figure 2: Substituted urea conformation. If R is alkyl, the most stable conformation is b); if R = H, the mos...
Hence, we introduced cyclic urea groups in the structure of chain stopper S2, by the alkylation of EHUT with 1,3-dibromopropane [33]. The rigidity of the cyclic ureas forbids any conformational rearrangement and should make it possible to probe whether the presence of NH functions in S1 significantly affects the chain stopper efficiency. In order to see if both urea carbonyls in S2 interact cooperatively with EHUT assemblies ,the mono-urea stopper S3 was also prepared. Finally, chain stopper S2 can only interact with bis-urea assemblies as a hydrogen bond acceptor through its carbonyl groups. It is therefore of interest to try and design a potentially complementary chain stopper, which would interact with bis-urea assemblies as a hydrogen bond donor. For this purpose, we synthesized the bis-thiourea S4, from the corresponding bis-thioisocyanate, because thioureas are known to be strong hydrogen bond donors and weak hydrogen bond acceptors [34,35].
Before evaluating the chain stopper efficiency of these compounds, i.e., their interaction with EHUT, their self-association was probed.
Self-association of bis-thiourea
Chain stoppers S2 and S3 cannot self-associate because they contain only hydrogen bond acceptors, however this is not the case for S4, and it is of interest to determine the conditions under which S4 can be considered not to associate with itself. Figure 3a shows the FTIR spectra of S4 at several concentrations in chloroform. At concentrations below 53 mM, a single band is visible in the region corresponding to the N–H stretching vibration. This band (3405 cm−1) can be attributed to free NH groups. Only at a high concentration (0.5 mol/L) does a band characteristic for hydrogen bonded NH groups appear (3250 cm−1). The very weak hydrogen bonding propensity of bis-thiourea S4 is particularly obvious when compared to bis-urea EHUT (Figure 3b): at the same concentration (9 mM), the bis-urea is nearly fully associated, whereas the bis-thiourea is virtually not associated. The respective behaviour of the bis-urea and the bis-thiourea was also probed by Small Angle Neutron Scattering (SANS) in toluene. Figure 4 shows the previously established q−1 dependence of EHUT, which is characteristic for long and rigid fibrillar scatterers [23]. In contrast, the low intensity and flat profil for S4 at small angles is characteristic for small globular scatterers. A fit was performed with the form factor of a sphere, and yields a diameter of 22 Å, which is comparable to the largest dimension of the fully extended molecule (25 Å). In conclusion, bis-thiourea S4 does not self-assemble significantly at concentrations below 12 mM in toluene or 53 mM in chloroform.
![[1860-5397-6-102-3]](/bjoc/content/figures/1860-5397-6-102-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: FTIR spectra, at 20 °C, for CDCl3 solutions of S4 (a) or EHUT (b), at several concentrations.
Figure 3: FTIR spectra, at 20 °C, for CDCl3 solutions of S4 (a) or EHUT (b), at several concentrations.
![[1860-5397-6-102-4]](/bjoc/content/figures/1860-5397-6-102-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: SANS intensity versus scattering vector for 12 mM solutions of EHUT or S4 in d8-toluene, at 22 °C. The plain curves are fits according to a model for infinitely long rigid rods of diameter 26 Å (green), or for spheres of diameter 22 Å (black).
Figure 4: SANS intensity versus scattering vector for 12 mM solutions of EHUT or S4 in d8-toluene, at 22 °C. ...
Chain stopper effect on the EHUT filament structure
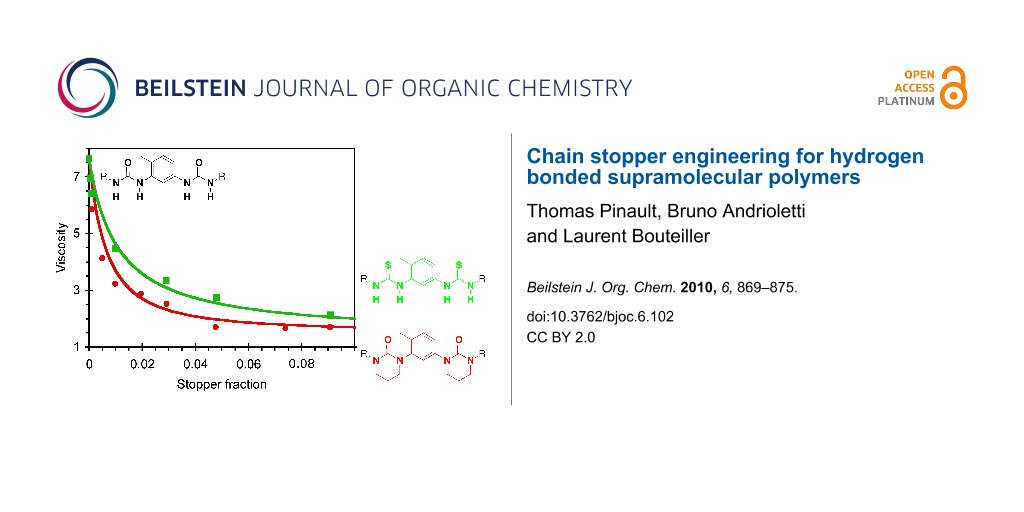
Viscosimetry is certainly the most sensitive technique to probe the efficiency of a chain stopper on supramolecular polymers. Therefore, we measured the viscosity of solutions of EHUT at a fixed concentration (20 mM) with increasing amounts of chain stopper. For this, 1,3,5-trimethylbenzene was chosen as the solvent because it is known to favor the formation of EHUT filaments at room temperature [29]. Figure 5 shows that all four compounds strongly reduce the relative viscosity of EHUT, which decreases from a value of 7.6 in the absence of stopper to a value close to 1 (i.e. the solution has approximately the same viscosity as the solvent) for a molar fraction ratio of stopper to monomer of 0.1. However, there are some significant differences between the stoppers: their efficiency increases in the order S1 < S3 ≈ S4 < S2. Several conclusions can be derived from this result. First, the lower viscosity of solutions containing S2 than those containing S1 means that the remaining two NH groups of S1 do participate in hydrogen bonding and reduce the efficiency of the stopper. Secondly, the lower viscosity of solutions containing S2 than those containing S3 indicates that both carbonyls are probably involved in the association between S2 and an EHUT filament. Finally, bis-thiourea S4 is a reasonably good chain stopper. The fact that it is not as good as S2 is perhaps due to some marginal hydrogen bonding involving the thiocarbonyl groups.
![[1860-5397-6-102-5]](/bjoc/content/figures/1860-5397-6-102-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Relative viscosity for solutions of EHUT (20 mM) in 1,3,5-trimethylbenzene at 25 °C, with increasing molar fraction of chain stoppers. The lines are a guide for the eye only.
Figure 5: Relative viscosity for solutions of EHUT (20 mM) in 1,3,5-trimethylbenzene at 25 °C, with increasin...
Chain stopper effect on the EHUT tube structure
For the above, toluene was chosen as the solvent, because it is known to favor the formation of EHUT tubes at room temperature [32] and has a similar polarity as 1,3,5-trimethylbenzene. Figure 6 shows that all four compounds also reduce the relative viscosity of EHUT in toluene, but the situation is more complex than in trimethylbenzene. If we consider first the part of the curves with a stopper to monomer fraction lower than 0.05, the efficiency of the chain stoppers increases in the order S3 < S1 ≈ S4 < S2. Therefore, the same conclusions for the interactions with the EHUT tubes can be derived as for the interactions with the EHUT filaments: i) the lower viscosity of solutions containing S2 than those containing S1 means that the remaining two NH groups of S1 participate in hydrogen bonding and reduce the efficiency of the stopper; ii) the lower viscosity of solutions containing S2 than those containing S3 indicates that both carbonyl groups are involved in the association between S2 and an EHUT tube; and iii) bis-thiourea S4 is a reasonably good chain stopper, but not as good as S2 probably due to some marginal hydrogen bonding involving the thiocarbonyls. If we consider now the part of the curves with a stopper to monomer fraction larger than 0.05, it is surprising to see that instead of the value decreasing to 1, the relative viscosity reaches a plateau at a value of 5 and 4 in the cases of S1 and S2, respectively. To our knowledge, such a saturating effect is unprecedented, and may indicate that an additional mechanism is involved in the interaction between the bis-urea tubes and S1 or S2. For example, we can hypothesize that at sufficiently high concentrations, S1 or S2 do not only interact with the extremities of the tubes, but also anywhere along them, without breaking them. However, additional characterizations will be required to test this interpretation [36].
![[1860-5397-6-102-6]](/bjoc/content/figures/1860-5397-6-102-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Relative viscosity for solutions of EHUT (1.1 mM) in toluene at 25 °C, with increasing molar fraction of chain stoppers. The lines are a guide for the eye only.
Figure 6: Relative viscosity for solutions of EHUT (1.1 mM) in toluene at 25 °C, with increasing molar fracti...
Conclusion
We have synthesized new substituted ureas and thioureas and tested them as chain stoppers for a bis-urea based supramolecular polymer. Depending on the solvent used, the bis-urea either forms filaments with a single monomer in the cross-section or tubes with three monomers in the cross-section. For both supramolecular architectures, similar conclusions can be derived: while all compounds tested reduce the viscosity of the supramolecular polymer solutions, the best chain stopper is obtained when two hydrogen bond acceptors are placed in the same relative position as for the monomer, and when no hydrogen bond donor is present.
Moreover, we have shown that a bis-thiourea with the same structure as the bis-urea monomer does not to form a supramolecular polymer, but acts as a good chain stopper, because it is a strong hydrogen bond donor and a weak acceptor.
Experimental
Synthesis
The synthesis of EHUT [32] and chain stopper S1 [17] have previously been reported.
Chain stopper S2. NaH (9 g) was placed in a three necked round bottomed flask and washed with pentane (25 mL) under a nitrogen atmosphere. An EHUT solution (4.32 g, 10 mmol) in dry THF (400 mL) was added and the mixture stirred for 1 h. 1,3-Dibromopropane (20.5 mL, 200 mmol) in dry THF (100 mL) was then added, and the solution heated under reflux for 24 h. After cooling, ice was slowly added and the solvent evaporated. Chloroform (200 mL) was added and the organic phase washed successively with brine (300 mL) and water (2 × 300 mL), dried over magnesium sulfate and concentrated. Silica gel column chromatography (ethyl acetate) followed by recrystallization from pentane afforded 1.9 g of a white solid (37%). 1H NMR (200 MHz, DMSO-d6): δ (ppm) = 7.2 (d, J = 1.5 Hz, 1H, Ar-H), 7.12 (d, J = 8.1 Hz, 1H, Ar-H), 7.06 (dd, J = 8.1 Hz, J = 1.5, 1H, Ar-H), 3.74–3.02 (m, 12H, N-CH2), 2.21 (s, 3H, Ar-CH3), 2.07 (m, 4H, CH2), 1.66 (m, 2H, CH), 1.36 (m, 16H, CH2), 0.92 (t, 12H, CH3). 13C NMR (50 MHz, DMSO-d6): δ (ppm) = 153.7/153.2 (C=O), 136.3/136.2/129.9/129.7/108.3/103.2 (Ar), 51.7/51.5/50.3/50.2/47.7/46.9 (N-CH2), 37.4 (CH), 31.5/31.3/27.9/27.7/24.2/24.1/22.3 (CH2), 17.2 (Ar-CH3), 14/13.8/11.5/11.4 (CH3).
Chain stopper S3. NaH (1.5 g) was placed in a three necked round bottomed flask and washed with pentane (5 mL) under a nitrogen atmosphere. A solution of N-(2-ethylhexyl)-N'-(4-methylphenyl)urea [22] (1 g, 3.8 mmol) in dry THF (25 mL) was added and the mixture stirred for 1 h. 1,3-Dibromopropane (3.9 mL, 38 mmol) in dry THF (50 mL) was then added, and the solution heated under reflux for 24 h. After cooling, ice was slowly added and the solvent evaporated. Chloroform (50 mL) was added and the organic phase washed successively with brine (70 mL) and water (2 × 70 mL), dried over magnesium sulfate and concentrated. Silica gel column chromatography (ethyl acetate/dichloromethane and then ethyl acetate) followed by recrystallization from pentane afforded 0.66 g of a white solid (57%). 1H NMR (200 MHz, DMSO-d6): δ (ppm) = 7.42/7.17 (2d, 4H, Ar-H), 3.21 (m, 6H, N-CH2), 2.15 (s, 3H, Ar-CH3), 1.78 (m, 3H, CH2(cycle) + CH) , 1.32 (m, 8H, CH2), 0.93 (t, 6H, CH3).
Chain stopper S4. 2-Ethylhexylamine (8.8 mL, 52 mmol) in dichloromethane (50 mL) was added slowly under a nitrogen atmosphere to a stirred solution of 2,4-toluene diisothiocyanate (5.06 g, 24.5 mmol) in dichloromethane (200 mL, distilled over calcium hydride). After 24 h, the solvent was evaporated. Recrystallization from ethanol/water afforded 7.74 g of a white solid (68%). 1H NMR (250 MHz, DMSO-d6, δ (ppm)): 9.47/9.04 (2s, 2H, Ar-NH), 7.52/7.37 (2s, 2H, CH2-NH), 7.32 (s, 1H, Ar-H), 7.21–7.11 (m, 2H, Ar-H), 3.40 (m, 4H, N-CH2), 2.14 (s, 3H, Ar-CH3), 1.59 (m, 2H, CH), 1.25 (m, 16H, CH2), 0.84 (m, 12H, CH3). 13C NMR (62.5 MHz, DMSO-d6, δ (ppm)): 181.2/180.4 (C=S), 137.0/130.6/130.0/122.6/121.1 (Ar), 47.4/47.1 (N-CH2), 38.4/38.3 (CH), 30.5/28.4/23.8/22.6 (CH2), 17.1 (Ar-CH3), 14.0/10.7 (CH3). MS (ESI) = [M-H] 463.4
Viscometry
Solutions were prepared by stirring at room temperature for at least 1 day prior to use. Capillary viscosity was measured at 25 ± 0.1 °C with an automatic Anton-Paar AMVn viscometer (capillary internal diameter 1.8 mm; ball diameter 1.5 mm). The measurements were performed with an angle of 20° and repeated six times.
FTIR spectroscopy
Infrared spectra were recorded on a Nicolet Avatar 320 spectrometer in KBr cells of 0.3 to 2.5 cm path length.
SANS
Measurements were made at the LLB (Saclay, France) on the Paxy instrument, at three distance-wavelength combinations to cover the 3 10−3 to 0.3 Å−1 q-range, where the scattering vector q is defined as usual, assuming elastic scattering, as q = (4π/λ)sin(θ/2), where θ is the angle between incident and scattered beam. The sample diaphragm was 7.6 mm. Collimation was achieved with a diaphragm of 22 mm for a sample – detector distance of 1.5 m, or 16 mm for a sample – detector distance of 3.2 and 6.7 m. Data were corrected for the empty cell signal and the solute and solvent incoherent background. A light water standard was used to normalize the scattered intensities into cm−1 units.
References
-
Ciferri, A., Ed. Supramolecular Polymers; Marcel Dekker: New York, 2005.
Return to citation in text: [1] -
Brunsveld, L.; Folmer, B. J. B.; Meijer, E. W.; Sijbesma, R. P. Chem. Rev. 2001, 101, 4071–4097. doi:10.1021/cr990125q
Return to citation in text: [1] -
Bouteiller, L. Adv. Polym. Sci. 2007, 207, 79–112. doi:10.1007/12_2006_110
Return to citation in text: [1] -
van der Gucht, J.; Besseling, N. A. M.; Knoben, W.; Bouteiller, L.; Cohen Stuart, M. A. Phys. Rev. B 2003, 67, 051106. doi:10.1103/PhysRevE.67.051106
Return to citation in text: [1] -
Ducouret, G.; Chassenieux, C.; Martins, S.; Lequeux, F.; Bouteiller, L. J. Colloid Interface Sci. 2007, 310, 624–629. doi:10.1016/j.jcis.2007.01.059
Return to citation in text: [1] [2] -
Shikata, T.; Nishida, T.; Isare, B.; Linares, M.; Lazzaroni, R.; Bouteiller, L. J. Phys. Chem. B 2008, 112, 8459–8465. doi:10.1021/jp800495v
Return to citation in text: [1] [2] -
van Gorp, J. J.; Vekemans, J. A. J. M.; Meijer, E. W. J. Am. Chem. Soc. 2002, 124, 14759–14769. doi:10.1021/ja020984n
Return to citation in text: [1] -
Ogata, D.; Shikata, T.; Hanabusa, K. J. Phys. Chem. B 2004, 108, 15503–15510. doi:10.1021/jp0486604
Return to citation in text: [1] -
Sakamoto, A.; Ogata, D.; Shikata, T.; Hanabusa, K. Macromolecules 2005, 38, 8983–8986. doi:10.1021/ma051489p
Return to citation in text: [1] -
Terech, P.; Weiss, R. G. Chem. Rev. 1997, 97, 3133–3159. doi:10.1021/cr9700282
Return to citation in text: [1] -
van Esch, J. H.; Feringa, B. L. Angew. Chem., Int. Ed. 2000, 39, 2263–2266. doi:10.1002/1521-3773(20000703)39:13<2263::AID-ANIE2263>3.0.CO;2-V
Return to citation in text: [1] -
Sangeetha, N. M.; Maitra, U. Chem. Soc. Rev. 2005, 34, 821–836. doi:10.1039/b417081b
Return to citation in text: [1] -
George, M.; Weiss, R. G. Acc. Chem. Res. 2006, 39, 489–497. doi:10.1021/ar0500923
Return to citation in text: [1] -
Folmer, B. J. B.; Cavini, E.; Sijbesma, R. P.; Meijer, E. W. Chem. Commun. 1998, 1846–1848.
Return to citation in text: [1] -
Ercolani, G. Chem. Commun. 2001, 1416–1417. doi:10.1039/b101678b
Return to citation in text: [1] -
Pinault, T.; Cannizzo, C.; Andrioletti, B.; Ducouret, G.; Lequeux, F.; Bouteiller, L. Langmuir 2009, 25, 8404–8407. doi:10.1021/la804138u
Return to citation in text: [1] -
Lortie, F.; Boileau, S.; Bouteiller, L.; Chassenieux, C.; Lauprêtre, F. Macromolecules 2005, 38, 5283–5287. doi:10.1021/ma050168a
Return to citation in text: [1] [2] [3] [4] [5] -
Knoben, W.; Besseling, N. A. M.; Cohen Stuart, M. A. Macromolecules 2006, 39, 2643–2653. doi:10.1021/ma0518914
Return to citation in text: [1] -
Knoben, W.; Besseling, N. A. M.; Bouteiller, L.; Cohen Stuart, M. A. Phys. Chem. Chem. Phys. 2005, 7, 2390–2398. doi:10.1039/b503463a
Return to citation in text: [1] -
Hirschberg, J. H. K.; Ramzi, A.; Sijbesma, R. P.; Meijer, E. W. Macromolecules 2003, 36, 1429–1432. doi:10.1021/ma025723c
Return to citation in text: [1] -
Dudek, S. P.; Pouderoijen, M.; Abbel, R.; Schenning, A. P. H. J.; Meijer, E. W. J. Am. Chem. Soc. 2005, 127, 11763–11768. doi:10.1021/ja052054k
Return to citation in text: [1] -
Simic, V.; Bouteiller, L.; Jalabert, M. J. Am. Chem. Soc. 2003, 125, 13148–13154. doi:10.1021/ja037589x
Return to citation in text: [1] [2] [3] -
Bouteiller, L.; Colombani, O.; Lortie, F.; Terech, P. J. Am. Chem. Soc. 2005, 127, 8893–8898. doi:10.1021/ja0511016
Return to citation in text: [1] [2] [3] -
Bellot, M.; Bouteiller, L. Langmuir 2008, 24, 14176–14182. doi:10.1021/la802367r
Return to citation in text: [1] [2] -
Vonau, F.; Suhr, D.; Aubel, D.; Bouteiller, L.; Reiter, G.; Simon, L. Phys. Rev. Lett. 2005, 94, 066103. doi:10.1103/PhysRevLett.94.066103
Return to citation in text: [1] -
Vonau, F.; Aubel, D.; Bouteiller, L.; Reiter, G.; Simon, L. Phys. Rev. Lett. 2007, 99, 086103. doi:10.1103/PhysRevLett.99.086103
Return to citation in text: [1] -
Vonau, F.; Linares, M.; Isare, B.; Aubel, D.; Habar, M.; Bouteiller, L.; Reiter, G.; Geskin, V.; Zerbetto, F.; Lazzaroni, R.; Simon, L. J. Phys. Chem. C 2009, 113, 4955–4959. doi:10.1021/jp809552j
Return to citation in text: [1] -
Boileau, S.; Bouteiller, L.; Lauprêtre, F.; Lortie, F. New J. Chem. 2000, 24, 845–848. doi:10.1039/b006742n
Return to citation in text: [1] -
Pinault, T.; Isare, B.; Bouteiller, L. ChemPhysChem 2006, 7, 816–819. doi:10.1002/cphc.200500636
Return to citation in text: [1] [2] -
Isare, B.; Linares, M.; Lazzaroni, R.; Bouteiller, L. J. Phys. Chem. B 2009, 113, 3360–3364. doi:10.1021/jp810236z
Return to citation in text: [1] -
Isare, B.; Linares, M.; Zargarian, L.; Fermandjian, S.; Miura, M.; Motohashi, S.; Vanthuyne, N.; Lazzaroni, R.; Bouteiller, L. Chem.–Eur. J. 2010, 16, 173–177. doi:10.1002/chem.200902399
Return to citation in text: [1] -
Lortie, F.; Boileau, S.; Bouteiller, L.; Chassenieux, C.; Demé, B.; Ducouret, G.; Jalabert, M.; Lauprêtre, F.; Terech, P. Langmuir 2002, 18, 7218–7222. doi:10.1021/la0255166
Return to citation in text: [1] [2] [3] -
Katz, H. E.; Cram, D. J. J. Am. Chem. Soc. 1984, 106, 4977–4987. doi:10.1021/ja00329a058
Return to citation in text: [1] -
Laurence, C.; Berthelot, M.; Le Questel, J.-Y.; El Ghomari, M. J. J. Chem. Soc., Perkin Trans. 2 1995, 2075–2079. doi:10.1039/p29950002075
Return to citation in text: [1] -
Masunov, A.; Dannenberg, J. J. J. Phys. Chem. B 2000, 104, 806–810. doi:10.1021/jp993078e
Return to citation in text: [1] -
SANS experiments have been performed for EHUT solutions in d8-toluene, in the presence of chain stopper S1 or S2 ([EHUT] = 11 mM; [stopper]/[EHUT] = 0.2). In the q-range investigated (3.16 10−3 – 3.14 10−1 Å−1), the scattering curves are virtually the same as without chain-stopper (data not shown). This indicates that the local structure is not significantly affected.
Return to citation in text: [1] -
Nowick, J. S.; Powell, N. A.; Martinez, E. J.; Smith, E. M.; Noronha, G. J. Org. Chem. 1992, 57, 3763–3765. doi:10.1021/jo00040a007
Return to citation in text: [1]
| 22. | Simic, V.; Bouteiller, L.; Jalabert, M. J. Am. Chem. Soc. 2003, 125, 13148–13154. doi:10.1021/ja037589x |
| 1. | Ciferri, A., Ed. Supramolecular Polymers; Marcel Dekker: New York, 2005. |
| 2. | Brunsveld, L.; Folmer, B. J. B.; Meijer, E. W.; Sijbesma, R. P. Chem. Rev. 2001, 101, 4071–4097. doi:10.1021/cr990125q |
| 3. | Bouteiller, L. Adv. Polym. Sci. 2007, 207, 79–112. doi:10.1007/12_2006_110 |
| 17. | Lortie, F.; Boileau, S.; Bouteiller, L.; Chassenieux, C.; Lauprêtre, F. Macromolecules 2005, 38, 5283–5287. doi:10.1021/ma050168a |
| 18. | Knoben, W.; Besseling, N. A. M.; Cohen Stuart, M. A. Macromolecules 2006, 39, 2643–2653. doi:10.1021/ma0518914 |
| 19. | Knoben, W.; Besseling, N. A. M.; Bouteiller, L.; Cohen Stuart, M. A. Phys. Chem. Chem. Phys. 2005, 7, 2390–2398. doi:10.1039/b503463a |
| 5. | Ducouret, G.; Chassenieux, C.; Martins, S.; Lequeux, F.; Bouteiller, L. J. Colloid Interface Sci. 2007, 310, 624–629. doi:10.1016/j.jcis.2007.01.059 |
| 14. | Folmer, B. J. B.; Cavini, E.; Sijbesma, R. P.; Meijer, E. W. Chem. Commun. 1998, 1846–1848. |
| 15. | Ercolani, G. Chem. Commun. 2001, 1416–1417. doi:10.1039/b101678b |
| 16. | Pinault, T.; Cannizzo, C.; Andrioletti, B.; Ducouret, G.; Lequeux, F.; Bouteiller, L. Langmuir 2009, 25, 8404–8407. doi:10.1021/la804138u |
| 17. | Lortie, F.; Boileau, S.; Bouteiller, L.; Chassenieux, C.; Lauprêtre, F. Macromolecules 2005, 38, 5283–5287. doi:10.1021/ma050168a |
| 10. | Terech, P.; Weiss, R. G. Chem. Rev. 1997, 97, 3133–3159. doi:10.1021/cr9700282 |
| 11. | van Esch, J. H.; Feringa, B. L. Angew. Chem., Int. Ed. 2000, 39, 2263–2266. doi:10.1002/1521-3773(20000703)39:13<2263::AID-ANIE2263>3.0.CO;2-V |
| 12. | Sangeetha, N. M.; Maitra, U. Chem. Soc. Rev. 2005, 34, 821–836. doi:10.1039/b417081b |
| 13. | George, M.; Weiss, R. G. Acc. Chem. Res. 2006, 39, 489–497. doi:10.1021/ar0500923 |
| 6. | Shikata, T.; Nishida, T.; Isare, B.; Linares, M.; Lazzaroni, R.; Bouteiller, L. J. Phys. Chem. B 2008, 112, 8459–8465. doi:10.1021/jp800495v |
| 30. | Isare, B.; Linares, M.; Lazzaroni, R.; Bouteiller, L. J. Phys. Chem. B 2009, 113, 3360–3364. doi:10.1021/jp810236z |
| 31. | Isare, B.; Linares, M.; Zargarian, L.; Fermandjian, S.; Miura, M.; Motohashi, S.; Vanthuyne, N.; Lazzaroni, R.; Bouteiller, L. Chem.–Eur. J. 2010, 16, 173–177. doi:10.1002/chem.200902399 |
| 4. | van der Gucht, J.; Besseling, N. A. M.; Knoben, W.; Bouteiller, L.; Cohen Stuart, M. A. Phys. Rev. B 2003, 67, 051106. doi:10.1103/PhysRevE.67.051106 |
| 5. | Ducouret, G.; Chassenieux, C.; Martins, S.; Lequeux, F.; Bouteiller, L. J. Colloid Interface Sci. 2007, 310, 624–629. doi:10.1016/j.jcis.2007.01.059 |
| 6. | Shikata, T.; Nishida, T.; Isare, B.; Linares, M.; Lazzaroni, R.; Bouteiller, L. J. Phys. Chem. B 2008, 112, 8459–8465. doi:10.1021/jp800495v |
| 7. | van Gorp, J. J.; Vekemans, J. A. J. M.; Meijer, E. W. J. Am. Chem. Soc. 2002, 124, 14759–14769. doi:10.1021/ja020984n |
| 8. | Ogata, D.; Shikata, T.; Hanabusa, K. J. Phys. Chem. B 2004, 108, 15503–15510. doi:10.1021/jp0486604 |
| 9. | Sakamoto, A.; Ogata, D.; Shikata, T.; Hanabusa, K. Macromolecules 2005, 38, 8983–8986. doi:10.1021/ma051489p |
| 32. | Lortie, F.; Boileau, S.; Bouteiller, L.; Chassenieux, C.; Demé, B.; Ducouret, G.; Jalabert, M.; Lauprêtre, F.; Terech, P. Langmuir 2002, 18, 7218–7222. doi:10.1021/la0255166 |
| 25. | Vonau, F.; Suhr, D.; Aubel, D.; Bouteiller, L.; Reiter, G.; Simon, L. Phys. Rev. Lett. 2005, 94, 066103. doi:10.1103/PhysRevLett.94.066103 |
| 26. | Vonau, F.; Aubel, D.; Bouteiller, L.; Reiter, G.; Simon, L. Phys. Rev. Lett. 2007, 99, 086103. doi:10.1103/PhysRevLett.99.086103 |
| 27. | Vonau, F.; Linares, M.; Isare, B.; Aubel, D.; Habar, M.; Bouteiller, L.; Reiter, G.; Geskin, V.; Zerbetto, F.; Lazzaroni, R.; Simon, L. J. Phys. Chem. C 2009, 113, 4955–4959. doi:10.1021/jp809552j |
| 28. | Boileau, S.; Bouteiller, L.; Lauprêtre, F.; Lortie, F. New J. Chem. 2000, 24, 845–848. doi:10.1039/b006742n |
| 23. | Bouteiller, L.; Colombani, O.; Lortie, F.; Terech, P. J. Am. Chem. Soc. 2005, 127, 8893–8898. doi:10.1021/ja0511016 |
| 24. | Bellot, M.; Bouteiller, L. Langmuir 2008, 24, 14176–14182. doi:10.1021/la802367r |
| 29. | Pinault, T.; Isare, B.; Bouteiller, L. ChemPhysChem 2006, 7, 816–819. doi:10.1002/cphc.200500636 |
| 22. | Simic, V.; Bouteiller, L.; Jalabert, M. J. Am. Chem. Soc. 2003, 125, 13148–13154. doi:10.1021/ja037589x |
| 23. | Bouteiller, L.; Colombani, O.; Lortie, F.; Terech, P. J. Am. Chem. Soc. 2005, 127, 8893–8898. doi:10.1021/ja0511016 |
| 24. | Bellot, M.; Bouteiller, L. Langmuir 2008, 24, 14176–14182. doi:10.1021/la802367r |
| 20. | Hirschberg, J. H. K.; Ramzi, A.; Sijbesma, R. P.; Meijer, E. W. Macromolecules 2003, 36, 1429–1432. doi:10.1021/ma025723c |
| 21. | Dudek, S. P.; Pouderoijen, M.; Abbel, R.; Schenning, A. P. H. J.; Meijer, E. W. J. Am. Chem. Soc. 2005, 127, 11763–11768. doi:10.1021/ja052054k |
| 22. | Simic, V.; Bouteiller, L.; Jalabert, M. J. Am. Chem. Soc. 2003, 125, 13148–13154. doi:10.1021/ja037589x |
| 37. | Nowick, J. S.; Powell, N. A.; Martinez, E. J.; Smith, E. M.; Noronha, G. J. Org. Chem. 1992, 57, 3763–3765. doi:10.1021/jo00040a007 |
| 17. | Lortie, F.; Boileau, S.; Bouteiller, L.; Chassenieux, C.; Lauprêtre, F. Macromolecules 2005, 38, 5283–5287. doi:10.1021/ma050168a |
| 17. | Lortie, F.; Boileau, S.; Bouteiller, L.; Chassenieux, C.; Lauprêtre, F. Macromolecules 2005, 38, 5283–5287. doi:10.1021/ma050168a |
| 32. | Lortie, F.; Boileau, S.; Bouteiller, L.; Chassenieux, C.; Demé, B.; Ducouret, G.; Jalabert, M.; Lauprêtre, F.; Terech, P. Langmuir 2002, 18, 7218–7222. doi:10.1021/la0255166 |
| 17. | Lortie, F.; Boileau, S.; Bouteiller, L.; Chassenieux, C.; Lauprêtre, F. Macromolecules 2005, 38, 5283–5287. doi:10.1021/ma050168a |
| 32. | Lortie, F.; Boileau, S.; Bouteiller, L.; Chassenieux, C.; Demé, B.; Ducouret, G.; Jalabert, M.; Lauprêtre, F.; Terech, P. Langmuir 2002, 18, 7218–7222. doi:10.1021/la0255166 |
| 36. | SANS experiments have been performed for EHUT solutions in d8-toluene, in the presence of chain stopper S1 or S2 ([EHUT] = 11 mM; [stopper]/[EHUT] = 0.2). In the q-range investigated (3.16 10−3 – 3.14 10−1 Å−1), the scattering curves are virtually the same as without chain-stopper (data not shown). This indicates that the local structure is not significantly affected. |
| 23. | Bouteiller, L.; Colombani, O.; Lortie, F.; Terech, P. J. Am. Chem. Soc. 2005, 127, 8893–8898. doi:10.1021/ja0511016 |
| 29. | Pinault, T.; Isare, B.; Bouteiller, L. ChemPhysChem 2006, 7, 816–819. doi:10.1002/cphc.200500636 |
| 33. | Katz, H. E.; Cram, D. J. J. Am. Chem. Soc. 1984, 106, 4977–4987. doi:10.1021/ja00329a058 |
| 34. | Laurence, C.; Berthelot, M.; Le Questel, J.-Y.; El Ghomari, M. J. J. Chem. Soc., Perkin Trans. 2 1995, 2075–2079. doi:10.1039/p29950002075 |
| 35. | Masunov, A.; Dannenberg, J. J. J. Phys. Chem. B 2000, 104, 806–810. doi:10.1021/jp993078e |
© 2010 Pinault et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)