Universität Potsdam, Institut für Chemie, Organische Synthesechemie, Karl-Liebknecht-Straße 24–25, D-14476 Golm, Germany
Corresponding author email
Guest Editor: K. Grela Beilstein J. Org. Chem.2010,6, 1188–1198.https://doi.org/10.3762/bjoc.6.136 Received 10 Sep 2010,
Accepted 09 Nov 2010,
Published 15 Dec 2010
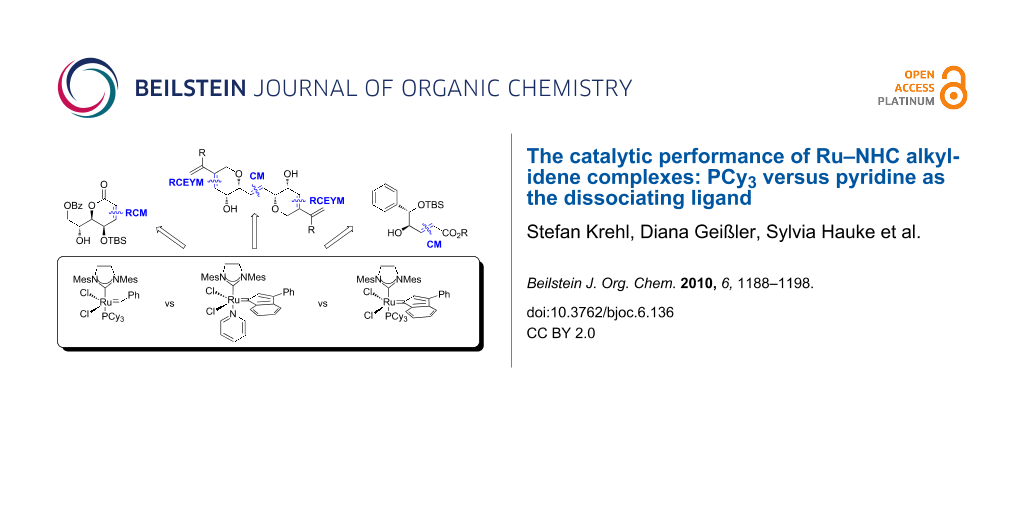
The catalytic performance of NHC-ligated Ru-indenylidene or benzylidene complexes bearing a tricyclohexylphosphine or a pyridine ligand in ring closing metathesis (RCM), cross metathesis, and ring closing enyne metathesis (RCEYM) reactions is compared. While the PCy3 complexes perform significantly better in RCM and RCEYM reactions than the pyridine complex, all catalysts show similar activity in cross metathesis reactions.
Over the past two decades, the olefin metathesis reaction became one of the most important C–C-bond forming reactions in organic synthesis [1]. The elucidation of the crucial role of metal carbenes by Chauvin [2] and the development of stable and defined precatalysts for homogeneously catalyzed reactions by Schrock [3] and Grubbs [4] paved the way for this development. Since then, molybdenum- [5] and ruthenium-based [6] catalysts have experienced extensive further developments and improvements. Due to their robustness towards air and moisture, and their comparatively low sensitivity towards functional groups, Ru-carbene complexes have attracted a particularly high degree of attention and “numerous variations on a theme by Grubbs”, as so accurately phrased by Fürstner [7], have been published. In the early stages of catalyst evolution improved methods for the introduction of the alkylidene ligand were the main focus. Thus, the original version of first generation Grubbs’ catalyst (A) [8], in which the alkylidene moiety originates from 2,2-diphenylcyclopropene, was soon replaced by the second version B[9], since the benzylidene ligand is more conveniently available from phenyldiazomethane. The obvious disadvantages of handling non-stabilized diazo compounds stimulated investigations into the use of propargylic alcohols as alkylidene precursors [10], which resulted in the synthesis of a first generation analogue C with an indenylidene ligand [11,12]. A landmark in the evolution of Ru-metathesis catalysts was the introduction of alkylidene complexes bearing N-heterocyclic carbenes (NHC) [13-15], in particular complex D, which became known as second generation Grubbs’ catalyst [16], and the Umicore M2 catalyst E[17,18]. The ligation of strongly σ-donating NHC’s leads to an improvement of catalytic activity, which sometimes equals the activity of Mo-based catalysts while maintaining the general robustness and tolerance towards several functional groups [19]. A third major topic in catalyst development has been the variation of the dissociating “placeholder”-ligand. In this respect, the introduction and further improvement of hemilabile benzylidene ligands by Hoveyda [20], Grela[21] and Blechert [22,23] have been important achievements. These phosphine-free catalysts of the general type F sometimes show higher activities due to enhanced catalyst lifetimes and have often been applied in cross metathesis reactions [24]. An alternative approach to phosphine-free Ru-metathesis catalysts uses pyridines as placeholders. Originally, complex G was synthesized as a precursor for mixed NHC-phosphine complexes other than D[25-28] or, very recently, for the synthesis of Ru-alkylidenes with two different NHC ligands [29]. Comparatively little information is available concerning the catalytic activity of these pyridine-NHC complexes. They were found to initiate ring opening metathesis polymerization reactions very rapidly [30,31] and show a certain preference for cross metathesis [32-35]. If the pyridine ring is attached to the alkylidene ligand [36], a latent catalyst results which might be a particularly useful property for metathesis polymerization reactions [37]. More recently, Ru-indenylidene complexes bearing one or two pyridine ligands were also synthesized [38,39], with the commercially available Umicore M31 catalyst H being a particularly interesting example. This catalyst is available from the M2 complex by ligand substitution [18] and shows high activity in living ROMP [40], whereas the reactivity in some RCM and CM reactions appears to be diminished [18,41].
During our research directed at the development of novel metathesis-non metathesis reaction sequences [42] catalyzed by a single site catalyst and initiated by organometallic transformations in situ, we became interested in the use of phosphine-free catalyst H. Unfortunately, only limited data is available concerning the catalytic efficiency of this catalyst in small molecule metathesis, in particular in comparison to the established phosphine containing catalysts D and E (Figure 1).
In this contribution, representative ring closing olefin, ring closing enyne and cross metathesis reactions of indenylidene complexes E and H and benzylidene complex D are compared.
Results and Discussion
Effects of solvent and catalyst loading
As a test reaction, we initially investigated the ring closing metathesis of allyl ether 1 to dihydropyran 2 (Scheme 1). To this end, conversion to the desired product in the presence of 5 mol % of catalyst H was determined after a reaction time of one hour at a slightly elevated temperature in seven different solvents, under otherwise identical conditions.
Scheme 1:
RCM of an allyl ether catalyzed by catalyst H.
Scheme 1:
RCM of an allyl ether catalyzed by catalyst H.
The results are summarized in Figure 2. Benzene, toluene, dichloromethane and 1,2-dichloroethane are commonly used solvents in Ru-catalyzed RCM reactions. However, for catalyst H only in dichloromethane was high conversion to the desired RCM product observed. In hexafluorobenzene, which has recently been shown to give highly impressive results, even in difficult metathesis reactions catalyzed by D, E, or F[43,44], the rate of conversion to dihydropyran 2 is quite similar to benzene or dichloroethane. Polar and, in particular, protic solvents would normally be considered inappropriate for metathesis reactions, because catalyst inhibition or degradation to Ru-hydrides might occur [45-47]. Nevertheless, such solvents have previously been investigated and useful results were obtained for esters [48] and – even more surprising – for acetic acid [49]. Therefore, we used ethyl acetate and acetic acid for our test reaction. While ethyl acetate gave a conversion of 75%, which is better than most of the classical solvents, nearly quantitative conversion to the RCM product was observed in acetic acid. Presumably, the pyridine ligand is protonated under these conditions which would result in a higher amount of the catalytically active 14-electron species. This interpretation is corroborated by the kinetic data obtained by Adjiman, Taylor et al. in their original investigation on solvent effects [49].
Figure 2:
Solvent screening for the RCM of 1 catalyzed by H (standardized conditions as denoted in Scheme 1).
Figure 2:
Solvent screening for the RCM of 1 catalyzed by H (standardized conditions as denoted in Scheme 1).
Next, we were interested to see how the performance of pyridine containing catalyst H compares with the more established phosphine complexes D and E. The test reaction depicted in Scheme 1 was therefore repeated in toluene and in acetic acid with a significantly lower catalyst loading, because we assumed that the highly active catalysts D and E would otherwise lead to full conversion in extremely short time (Figure 3).
Figure 3:
Comparison of catalysts D, E and H in toluene and acetic acid (standardized conditions as denoted in Scheme 1).
Figure 3:
Comparison of catalysts D, E and H in toluene and acetic acid (standardized conditions as denoted i...
Thus, with 1.0 mol % of catalyst under conditions identical to those given in Scheme 1, a quantitative conversion to the dihydropyran 2 was observed for both catalysts D and E in toluene. In accord with the results summarized in Figure 2, catalyst H gave only 34% conversion in this solvent after one hour, suggesting that initiation was rather slow. In acetic acid, the analogous phosphine containing indenylidene catalyst E displayed a slightly reduced conversion of 89%, which might be attributed to slow catalyst deactivation, whereas pyridine complex H showed significantly enhanced activity in acetic acid, with a conversion of 68% after one hour. This result suggests that by switching from toluene to acetic acid the balance of catalyst deactivation and enhanced initiation is shifted to the deactivation side for phosphine catalyst E, and to the enhanced initiation side for pyridine catalyst H. The most remarkable result from this set of experiments, however, is a collapse of conversion if benzylidene catalyst D is used in acetic acid. With D, a reproducible conversion of only 9% to the dihydropyran 2 was observed, compared to 89% conversion with the analogous indenylidene complex E. This result seems to contradict the observations by Adjiman, Taylor et al. who reported preparatively useful conversions and yields for second generation Grubbs’ catalyst D in acetic acid [49], however, their studies were conducted at ambient temperature and for different substrates, while our experiments were conducted at 40 °C. It is possible that our result suggests a higher robustness of indenylidene catalyst E compared to benzylidene catalyst D, at least under these rather unusual conditions.
To further evaluate the catalytic performance of pyridine catalyst H in acetic acid, we next wanted to determine the minimum amount of catalyst required to obtain a preparatively useful (>90%) rate of conversion. Consequently, it was demonstrated that, instead of using the conditions noted in Scheme 1, a catalyst loading of 2 mol % was sufficient to achieve a 92% conversion within one hour (Figure 4).
Figure 4:
Conversion vs catalyst loading for the RCM of 1 in acetic acid (standardized conditions as denoted in Scheme 1).
Figure 4:
Conversion vs catalyst loading for the RCM of 1 in acetic acid (standardized conditions as denoted ...
Having established the beneficial effect of acetic acid for the ring closing metathesis reaction of allyl ether 1 catalyzed by H, we were interested to see if a similar effect exists for other substrates. Therefore, acrylate 3a was subjected to the conditions of a ring closing metathesis reaction (Scheme 2 and Table 1). Not unexpectedly [50], significantly reduced initial substrate concentrations were required for useful rates of conversion to the desired α,β-unsaturated lactone 4a. It transpired, that preparatively useful yields could only be obtained with the phosphine containing catalysts D and E in toluene (Table 1, entries 1 and 2), whereas pyridine complex H gave a conversion of approximately 65% and an isolated yield of 41% of lactone 4a in this solvent (Table 1, entry 3). In contrast to the ring closing metathesis of allyl ether 1, the rate of conversion decreased significantly when catalyst H was used in acetic acid (Table 1, entry 4). Notably, the 1H NMR-spectra of the crude reaction mixtures did not indicate the presence of any decomposition products, thus, even the comparatively labile acrylate 3 appears to be quite stable in acetic acid at elevated temperatures for several hours.
Scheme 2:
Catalyst screening for RCM of acrylate 3a.
Scheme 2:
Catalyst screening for RCM of acrylate 3a.
aA catalyst loading of 5 mol % was used in all experiments. bRatio of product to starting material was determined from the 1H NMR-spectrum of the crude reaction mixture after three hours at 80 °C. cYield was not determined due to low conversion.
Because acetic acid did not lead to the expected improvement in acrylate metathesis reactions, only toluene was tested as a solvent in further experiments (Figure 5 and Table 2). Toluene was preferred over dichloromethane, because reactions are more conveniently conducted at elevated temperatures in this solvent.
The acrylates 3 investigated in ring closing metathesis reactions with catalysts D, E and H are listed in Figure 5, together with the resulting unsaturated lactones 4. Results for the ring closing metathesis of acrylates 3a–g are summarized in Table 2. Lactones 4b–f[50] are accessible in preparatively useful yields with catalyst loadings of 2.5 mol % to 5.0 mol % if catalysts D and E are used. Conversions observed with catalyst H under otherwise identical conditions are significantly lower and yields exceed 50% only in few cases such as 4b, 4e and 4f. A particularly difficult substrate for olefin metathesis reactions is acrylate 3g, which has recently been used by us as an intermediate in the synthesis of the natural product (–)-cleistenolide [51]. Acrylate 3g requires very high dilution, the addition of phenol as a rate accelerating additive [52], a rather high catalyst loading of 10 mol % and an even higher reaction temperature. Under these conditions, the best product to substrate ratio and the best isolated yield was obtained with the indenylidene complex E (Table 2, entry 6).
Table 2:
RCM reactions of acrylates to unsaturated lactones.a
Entry
3
4
c/mol·L−1
Catalyst loading
Ratiob4:3 (isolated yields) for
D
E
H
1
3b
4b
0.02
2.5 mol %
> 20:1 (86%)
>20:1 (90%)
1.8:1 (52%)
2
3c
4c
0.02
2.5 mol %
>20:1 (75%)
>20:1 (85%)
2.1:1 (43%)
3
3d
4d
0.02
2.5 mol %
7.1:1 (79%)
3.3:1 (41%)
0.6:1 (27%)
4
3e
4e
0.01
5.0 mol %
>10:1 (59%)
>20:1 (71%)
1.8:1 (56%)
5
3f
4f
0.02
2.5 mol %
>20:1 (80%)
>20:1 (53%)
4.5:1 (65%)
6c
3g
4g
0.002
10.0 mol %
1.1:1 (36%)
2.6:1 (58%)
0.3:1 (14%)
aReactions were run in toluene at 80 °C for 1 h, unless otherwise stated. bThe substrate to product ratio was determined from the 1H NMR-spectra of the crude reaction mixtures. cReaction was run in toluene at 110 °C for 3 h in the presence of 0.5 equiv of phenol.
Ring closing enyne metathesis
Imahori et al. have recently discovered that allylic hydroxy groups significantly enhance the rate of ring closing enyne metathesis reactions [53,54]. In these cases, addition of an ethylene atmosphere [55] which is normally considered to be mandatory for good results, is not required. We have recently investigated the highly selective enyne metathesis of substrates such as 5a,b to dihydropyrans 6a,b in the presence of first generation catalysts B and C[56]. Notably, no dihydrofurans or other isomers were observed. Sometimes, the selectivity decreases when the more reactive second generation catalysts are used [57,58], and we were therefore interested to test NHC-ligated catalysts in this transformation (Scheme 3).
Scheme 3:
Ring closing enyne metathesis reactions.
Scheme 3:
Ring closing enyne metathesis reactions.
The results for ring closing enyne metathesis reactions with indenylidene catalysts E and H are summarized in Table 3. Enyne 5a reacted to afford the expected dihydropyran 6a with high conversion and high selectivity in toluene at 80 °C and at 110 °C in the presence of phosphine complex E (Table 3, entries 1 and 2). The dimerization product 7a was not detected, and although the 1H NMR spectra of the crude reaction mixtures showed only signals of 6a and trace amounts of the starting material 5a, the isolated yields are mediocre which can be attributed to a significant loss of material during purification. Significantly lower conversions of 50% and 80% were observed with the pyridine complex H at 80 °C or 110 °C, respectively. Again, 1H NMR-spectra of the crude reaction mixtures showed only signals for 6a and the starting material 5a and not even trace amounts of a dimerization product 7a (Table 3, entries 3 and 4) were detected. Different results were found for the methyl substituted enyne precursor 5b. Remarkably, with both catalysts and both reaction temperatures apparently identical conversions and product distributions were observed: In all cases (Table 3, entries 5–8) the starting material was fully consumed and the 1H NMR-spectra of the crude reaction mixtures revealed only the presence of dihydropyran 6b and its dimer 7b in roughly a 1:1 ratio. This ratio is reflected nicely in the isolated yields, which were determined for one example (Table 3, entry 8). From these results, it can be concluded that benzyloxy-substituted enyne 5a is significantly less reactive in enyne metathesis reactions which becomes more obvious if the pyridine complex H is used as a catalyst. For other examples, we have previously observed that benzyl ether moieties in close proximity to a C–C-multiple bond retard or inhibit metathesis reactions [50]. Presumably, partial catalyst deactivation by coordination of the benzyloxy group to the metal plays a role, and this might also explain why no dimerization product 7a was observed, whereas 7b, the self-metathesis product of 6b, was isolated in significant quantities, which might suggest a remarkable residual activity of the catalysts after completion of the enyne metathesis.
Table 3:
Ring closing enyne metathesis reactions catalyzed with E and H.
Entry
5
–R
Catalysta
T
Conversionb
Ratio of 6:7b
Product (Yield)
1
5a
–CH2OBn
E
80 °C
95%
>20:1
6a (50%)
2
5a
–CH2OBn
E
110 °C
95%
>20:1
6a (44%)
3
5a
–CH2OBn
H
80 °C
50%
>20:1
6a (29%)
5a (21%)
4
5a
–CH2OBn
H
110 °C
80%
>20:1
6a (34%)
5
5b
–CH3
E
80 °C
100%
10:10.7
n. d.
6
5b
–CH3
E
110 °C
100%
10:10.3
n. d.
7
5b
–CH3
H
80 °C
100%
10:10.2
n. d.
8
5b
–CH3
H
110 °C
100%
10:9.6
6b (24%)
7b (45%)
a2.5 mol % of catalyst, toluene as a solvent and an initial substrate concentration of 0.1 mol/L were used in all experiments. bRates of conversion and monomer/dimer ratios were determined from the 1H NMR-spectra of the crude reaction mixtures, which showed only signals of starting materials 5, dihydropyrans 6 and dimers 7.
The results discussed above for the less reactive enyne 5a suggest that pyridine complex H is less active than E in enyne metathesis reactions, however, the results for the apparently more reactive substrate 5b may indicate that the gap in catalytic activity between E and H is much smaller for ring closing enyne reactions than for ring closing acrylate metathesis reactions. However, the remarkably high amount of homodimerization product 7b points to considerable activity of H in cross metathesis reactions, a peculiarity which has previously been noted for the bis(pyridine) complex G[33-35].
Cross metathesis reactions
Allylic alcohol 8[59] was chosen to test the catalysts investigated in this study for cross metathesis activity, because allylic alcohols are known to undergo undesired “redox isomerization” in the presence of Ru metathesis catalysts in some cases with the formation of ethyl ketones [47,60]. Therefore, 8 can be considered as a rather challenging substrate. As partners in the cross metathesis reactions, three acrylates 9a–c were chosen. In addition, homodimerization of 8 to diol 11 was also investigated (Scheme 4).
Scheme 4:
Cross metathesis reactions with allylic alcohol 8.
Scheme 4:
Cross metathesis reactions with allylic alcohol 8.
The results are summarized in Table 4. With methyl acrylate 9a in dichloromethane at 40 °C, only the benzylidene catalyst D showed a satisfactory rate of conversion (Table 4, entry 1). A significantly lower conversion was observed under these conditions for pyridine complex H (Table 4, entry 3), however, the isolated yields were similar in both cases. Surprisingly, with catalyst E conversion was incomplete and a considerable amount of dimer 11 was formed (Table 4, entry 2). Compound 11 is an inseparable mixture of diastereomers, because 10a was used as a racemate. Raising the temperature to 80 °C in toluene solved the problems of incomplete conversion and competing dimerization for all three catalysts (Table 4, entries 4–6) and rac-10a was isolated in comparable yields of between 83% and 86%. Phenyl acrylate (9b) is a less reactive cross metathesis partner, and all three catalysts led to incomplete conversion (Table 4, entries 7–9). The best result in this series was achieved with catalyst H, which gave a 2:1 ratio of product 10b and starting material 8, and an isolated yield of 60% (Table 4, entry 9). Methyl methacrylate (9c) did not react in cross metathesis reactions with 8 using either D or H. Instead, dimerization to compound 11 occurred. We [50] and others [61] have recently observed a considerable reduction of isomerization side reactions if acrylates are present during a metathesis reaction. Therefore, 8 was subjected to olefin metathesis conditions in the absence of acrylates to check if any isomerization occurred, or if dimer 11 was the preferred or sole product. With all three catalysts similar results were obtained: The starting material was rapidly and almost completely consumed, and the two diastereomers of 11 were the only detectable products in the reaction mixture (Table 4, entries 12–14).
Table 4:
Cross metathesis reactions catalyzed by D, E, and H.
Entry
Acrylate
Solvent
T
Catalysta
Ratio of
10:8:11b
Product (Yield)c
1
9a
CH2Cl2
40 °C
D
16:1:0
rac-10a (72%)
2
9a
CH2Cl2
40 °C
E
1.4:1.3:1
n. d.
3
9a
CH2Cl2
40 °C
H
3.7:1:0
rac-10a (65%)
4
9a
toluene
80 °C
D
1:0:0
rac-10a (84%)
5
9a
toluene
80 °C
E
1:0:0
rac-10a (83%)
6
9a
toluene
80 °C
H
1:0:0
rac-10a (86%)
7
9b
toluene
80 °C
D
1.2:1:0
rac-10b (53%)
8
9b
toluene
80 °C
E
1:1:0
rac-10b (47%)
9
9b
toluene
80 °C
H
2:1:0
rac-10b (60%)
10
9c
toluene
80 °C
D
0:0:1
11 (n. d.)
11
9c
toluene
80 °C
H
0:0:1
11 (n. d.)
12
—
toluene
80 °C
D
—:1:30
11 (68%)
13
—
toluene
80 °C
E
—:1:9
11 (n. d.)
14
—
toluene
80 °C
H
—:1:16
11 (n. d.)
aA catalyst loading of 5.0 mol % was used in all experiments. bRatios were determined from the 1H NMR-spectra of crude reaction mixtures. cAll cross metathesis products were obtained as single E-isomers.
These results demonstrate that the novel catalyst H, albeit significantly less active than D or E in the ring closing metathesis of acrylates, appears to be competitive in cross metathesis reactions.
Conclusion
In conclusion, we have evaluated and compared the catalytic performance of two indenylidene NHC-metathesis catalysts and the well established second generation Grubbs catalyst in various small molecule metathesis reactions. The activity of the mixed NHC-phosphine catalysts D and E appears to be similar in most applications. Some results hint at a somewhat faster reaction with benzylidene complex D, while E apparently performs slightly better in “slow” metathesis reactions, presumably since it is more robust. The novel pyridine complex H was also tested in several olefin metathesis reactions. While this catalyst gives rather unsatisfactory results in the ring closing metathesis of acrylates, its performance in ring closing enyne metathesis reactions is only slightly lower than the phosphine complexes D and E. However, the activity of H in cross metathesis reactions is similar to D and E. Furthermore, initial studies concerning the use of unconventional solvents revealed that H might be quite active, at least for some applications, in acetic acid.
Experimental
All experiments were conducted in dry reaction vessels under an atmosphere of dry argon. Solvents were purified by standard procedures. All yields and conversions reported herein are average values of at least two experiments. 1H NMR spectra were obtained at 300 MHz in CDCl3 with CHCl3 (δ = 7.26 ppm) as the internal standard. Coupling constants (J) are given in Hz. 13C NMR spectra were recorded at 75 MHz in CDCl3 with CDCl3 (δ = 77.0 ppm) as the internal standard. IR spectra were recorded as films on NaCl or KBr plates or as KBr discs. Wavenumbers (ν) are given in cm–1. Mass spectra were obtained at 70 eV. Whenever known compounds were used as starting materials, reagents or catalysts, they were either purchased or were synthesized following literature procedures: 1[62], 3a[63], 3b–3e[50], 3g[51], 5a,b[56]. Catalyst D was purchased from Aldrich and used without further purification. Catalysts E and H were donated by Umicore, Hanau, Germany, and also used without further purification. The following products have previously been synthesized via olefin metathesis reactions under different conditions: 2[62], 4a[63], 4b–4e[50], 4f[64], 4g[51], 6a,b[56].
General procedure for the RCM of 1: variation of solvent, catalyst and catalyst loading
Allyl ether 1 (94.0 mg, 0.5 mmol) was dissolved in the appropriate dry and degassed solvent (5.0 mL). Catalyst D (4.2 mg for 1.0 mol %), E (4.7 mg for 1.0 mol %) or H (3.7 mg for 1.0 mol %, 7.4 mg for 2.0 mol %, 11.2 mg for 3.0 mol %, 15.0 mg for 4.0 mol % or 18.8 mg for 5.0 mol %) was then added. Immediately after addition of the catalyst, the reaction vessel was immersed in an oil bath preheated to 40 °C (electronic temperature control) for a period of time between 60 and 62 min. After this time, the reaction vessel was allowed to cool to ambient temperature, the solvent was removed by evaporation, and the residue immediately subjected to NMR spectroscopy. The ratio of dihydropyran 2 to allyl ether 1 was determined by integration of characteristic, baseline separated signals. Each experiment was repeated at least two times. The reported rates of conversion are average values.
Ring closing metathesis of acrylates
General procedure for the synthesis of furanones 4b–4f by RCM: To a solution of the appropriate acrylate 3 (1.0 mmol) in dry and degassed toluene (50 mL for 0.02 mol·L–1 or 100 mL for 0.01 mol·L–1), was added either catalyst D (21.2 mg for 2.5 mol % or 42.4 mg for 5.0 mol %), E (23.7 mg for 2.5 mol % or 47.4 mg for 5.0 mol %) or H (18.7 mg for 2.5 mol % or 37.4 mg for 5.0 mol %). The solution was heated to 80 °C for 90 min. The solvent was then removed by evaporation and the residue purified by flash column chromatography on silica to give the corresponding lactones 4. The ratio of lactone 4 to acrylate 3 was determined by integration of characteristic, baseline separated signals in the 1H NMR-spectrum of the crude reaction mixture. Representative example: 5-phenylfuran-2(5H)-one (4f). This compound was obtained as a colourless oil from 3f (189 mg, 1.0 mmol) following the general procedure. Yield of 4f using catalyst D: 128 mg (0.80 mmol, 80%). Yield of 4f using catalyst E: 85 mg (0.53 mmol, 53%). Yield of 4f using catalyst H: 104 mg (0.65 mmol, 65%). 1H NMR (300 MHz, CDCl3) δ 7.53 (dd, J = 5.7, 1.7, 1H), 7.45–7.35 (3H), 7.30–7.23 (2H), 6.23 (dd, J = 5.7, 2.1, 1H), 6.01 (t (br), J = 1.9, 1H); 13C NMR (75 MHz, CDCl3) δ 173.0 (0), 155.8 (1), 134.4 (0), 129.3 (1), 129.0 (1), 126.5 (1), 121.0 (1), 84.3 (1); HRMS (ESI) calcd for C10H8O2Na [M+Na]+: 183.0422, found: 183.0439.
Procedure for the synthesis of 6-phenyl-5,6-dihydropyran-2-one (4a): The acrylate 3a (201 mg, 1.0 mmol) was dissolved in dry and degassed toluene (100 mL). After adding the catalyst (D: 42.4 mg B: 47.4 mg H: 37.4 mg, 0.05 mmol, 5 mol %) the solution was stirred for 3 h at 80 °C. After cooling the solution to ambient temperature, all volatiles were evaporated. The residue was purified by chromatography on silica (hexane/MTBE 2:1). Yield of 4a using catalyst D: 139 mg (0.80 mmol, 80%). Yield of 4a using catalyst E: 160 mg (0.92 mmol, 92%). Yield of 4a using catalyst H: 170 mg (0.41 mmol, 41%). The ratios of lactone 4a to acrylate 3a were determined by integration of characteristic, baseline separated signals in the 1H NMR-spectrum of the crude reaction mixture. mp 58–59 °C; 1H NMR (300 MHz, CDCl3) δ 7.43–7.33 (5H), 6.97 (ddd, J = 9.7, 5.7, 2.6, 1H), 6.13 (ddd, J = 9.7, 1.1, 1.1, 1H), 5.45 (dd, J = 11.2, 4.8, 1H), 2.70–2.57 (2H); 13C NMR (75 MHz, CDCl3) δ 164.0 (0), 144.8 (1), 138.4 (0), 128.6 (1), 128.6 (1), 126.0 (1), 121.7 (1), 79.2 (1), 31.6 (2); IR (neat) ν 3064 (w), 2904 (w), 1716 (s), 1381 (m), 1242 (s), 1059 (m), 1020 (m), 909 (m); EIMS (%) m/z 174 (M+, 6), 77 (13), 68 (100), 39 (63); HRMS (EI) calcd. for C11H10O2 [M+]: 174.0675, found 174.0689; Anal. calcd for C11H10O2: C, 75.8, H, 5.8; found: C, 75.6, H, 5.8.
Procedure for the synthesis of (R)-2-((2R,3R)-3-(tert-butyldimethylsilyloxy)-6-oxo-3,6-dihydro-2H-pyran-2-yl)-2-hydroxyethyl benzoate (4g): The acrylate 3g (330 mg, 0.78 mmol) and phenol (37 mg, 0.39 mmol) were dissolved in dry and degassed toluene (390 mL). After adding the catalyst (D: 66.1 mg B: 73.9 mg H: 58.3 mg, 0.078 mmol, 10 mol %), the solution was stirred for 3 h at 80 °C. The solution was cooled to ambient temperature and all volatiles were evaporated. The residue was purified by chromatography on silica. Yield of 4g using catalyst D: 110 mg (0.28 mmol, 36%). Yield of 4g using catalyst E: 177 mg (0.45 mmol, 58%). Yield of 4g using catalyst H: 43 mg (0.11 mmol, 14%). The ratios of lactone 4g to acrylate 3g were determined by integration of characteristic, baseline separated signals in the 1H NMR-spectrum of the crude reaction mixture. All analytical data were identical to those reported previously in the literature [51].
Ring closing enyne metathesis reactions
General procedure: To a solution of the corresponding precursor 5 (2.0 mmol) in toluene (40 mL), was added catalyst E (47.4 mg, 2.5 mol %) or H (37.4 mg, 2.5 mol %). The solution was heated to the appropriate temperature (80 °C or 110 °C) for 20 h, then cooled to ambient temperature and the solvent evaporated. The crude product was analyzed by 1H and 13C NMR spectroscopy. Analytically pure samples were obtained by column chromatography on silica. Representative example: Ring closing enyne metathesis of 5b. Following the general procedure, 5b (330 mg, 2.0 mmol) was treated with catalyst H (37.4 mg, 2.5 mol %) at 80 °C. After column chromatography, 6b (80 mg, 0.48 mmol, 24%) and 7b (274 mg, 0.90 mmol, 45%) were isolated. The rate of conversion and the ratios of 6b to 7b were determined by integration of characteristic, baseline separated signals in the 1H NMR-spectrum of the crude reaction mixture. All analytical data of 6b were identical to those reported previously in the literature.[56] Analytical data for (2R,2'R,3R,3'R,E)-2,2'-(ethene-1,2-diyl)bis(5-(prop-1-en-2-yl)-3,6-dihydro-2H-pyran-3-ol (7b): [α]D25: –390.0 (c 0.13, CH2Cl2); 1H NMR (300 MHz, CDCl) δ 6.05 (d, J = 5.4, 2H); 5.96 (d, J = 1.7, 2H); 4.93 (s, 1H); 4.83 (s, 2H); 4.49 (d, J = 15.6, 2H); 4.25 (d, J = 15.5, 2H); 4.04 (s, 2H); 3.99 (d, J = 5.5, 1H); 3.32 (s, 2H); 1.87 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 139.5 (0), 138.5 (0); 129.1 (2), 122.8 (1); 112.3, (2); 77.3 (1), 66.2 (2); 64.2 (1); 20.1 (3); IR ν 3297 (s); 3087 (m); 2945 (m); 2828 (m); 1606 (m); 1371 (w); 1124 (s); 1103 (m); 1055 (m), 882 (s), 827 (s); HRMS (EI) calcd for C18H24O4 [M]+: 304.1675, found: 304.1683; Anal. calcd for C18H24O4: C, 71.0, H, 8.0, found: C, 70.8, H, 7.7.
Cross-metathesis reactions
General procedure: A solution of the allylic alcohol rac-8 (139 mg, 0.5 mmol) and the corresponding acrylate 9 (5 mmol) in dry and degassed toluene (1.0 mL for 0.5 mol·L–1, 5.0 mL for 0.1 mol·L–1) was warmed to approximately 45 °C. Then the catalyst (0.025 mmol, 5 mol %, D: 42.4 mg E: 47.4 mg H: 18.7 mg) was added to the solution and stirred for 90 min at 80 °C. After cooling to ambient temperature all volatiles were evaporated and the residue purified by chromatography on silica. The rates of conversion and the ratios of 10 to 8 to 11 were determined by integration of characteristic, baseline separated signals in the 1H NMR-spectrum of the crude reaction mixture.
Dimerization of rac-8 (Compound 11). The catalyst (5 mol %, 0.029 mmol, D: 24.2 mg, E: 27.0 mg, H: 21.3 mg) was added to a solution of alcohol rac-8 (150 mg, 0.57 mmol) in dry and degassed toluene (1.13 mL, 0.5 M). The reaction mixture was stirred for 90 min at 80 °C. After cooling to room temperature all volatiles were evaporated. The crude product 11 was purified by chromatography on silica. Compound 11 was isolated as a partially separable mixture of diastereomers (combined yield for catalyst D: 107 mg, 0.39 mmol, 68%). The rates of conversion were determined by integration of characteristic, baseline separated signals in the 1H NMR-spectrum of the crude reaction mixture. Analytical data for diastereomer 11a: 1H NMR (300 MHz, CDCl3) δ 7.30–7.24 (3H), 7.11–7.06 (2H), 5.44 (dd, J = 2.6, 0.9, 1H), 4.20 (d, J = 7.5, 1H), 4.00 (dm, J = 7.4, 1 H), 2.83 (d, J = 2.4, 1H), 0.87 (s, 9H), 0.01 (s, 3H), −0.22 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 141.0 (0), 129.9 (1), 128.0 (1), 127.7 (1), 127.3 (1), 79.5 (1), 76.7 (1), 25.8 (3), 18.1 (0), −4.5 (3), −5.1 (3); HRMS (ESI) calcd for C30H48O4Si2Na [M+Na]+: 551.2989, found: 551.3010; EIMS (%) m/z = 283 (4), 189 (4), 157 (56), 129 (16), 103 (22), 99 (100). Analytical data for diastereomer 11b: 1H NMR (300 MHz, CDCl3) δ 7.33–7.17 (5H), 5.52 (dd, J = 2.6, 0.9, 1H), 4.38 (d, J = 6.6, 1 H), 4.03 (ddm, J = 6.0, 3.0, 1H), 2.74 (d, J = 3.4, 1H), 0.89 (s, 9H), 0.03 (s, 3H), −0.19 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 141.0 (0), 130.3 (1), 128.0 (1), 127.7 (1), 127.1 (1), 79.0 (1), 76.4 (1), 25.8 (3), 18.1 (0), −4.6 (3), −5.1 (3).
Supporting Information
Supporting Information File 1:
Experimental procedures and characterization data for starting materials which have previously not been described in the literature; representative illustrations for determination of conversions; copies of spectra for new compounds.
We thank Umicore, Hanau (Germany) for a generous donation of various Umicore M olefin metathesis catalysts, including Umicore M2 and Umicore M31 used in this study.
References
Grubbs, R. H., Ed. Handbook of metathesis; Wiley-VCH: Weinheim, 2003.
Return to citation in text:
[1]
Schrock, R. R.; Murdzek, J. S.; Bazan, G. C.; Robbins, J.; DiMare, M.; O'Regan, M. J. Am. Chem. Soc.1990,112, 3875–3886. doi:10.1021/ja00166a023
Return to citation in text:
[1]
Fu, G. C.; Nguyen, S. T.; Grubbs, R. H. J. Am. Chem. Soc.1993,115, 9856–9857. doi:10.1021/ja00074a085
Return to citation in text:
[1]
Dragutan, V.; Dragutan, I.; Delaude, L.; Demonceau, A. Coord. Chem. Rev.2007,251, 765–794. doi:10.1016/j.ccr.2006.09.002
Return to citation in text:
[1]
Fürstner, A. Angew. Chem., Int. Ed.2000,39, 3013–3043.
Return to citation in text:
[1]
Nguyen, S. T.; Johnson, L. K.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc.1992,114, 3974–3975. doi:10.1021/ja00036a053
Return to citation in text:
[1]
Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc.1996,118, 100–110. doi:10.1021/ja952676d
Return to citation in text:
[1]
Bruneau, C.; Dixneuf, P. H. Acc. Chem. Res.1999,32, 311–323. doi:10.1021/ar980016i
Return to citation in text:
[1]
Harlow, K. J.; Hill, A. F.; Wilton-Ely, J. D. E. T. J. Chem. Soc., Dalton Trans.1999, 285–291.
Return to citation in text:
[1]
Fürstner, A.; Liebl, M.; Hill, A. F.; Wilton-Ely, J. D. E. T. Chem. Commun.1999, 601–602. doi:10.1039/a900187e
Return to citation in text:
[1]
Weskamp, T.; Kohl, F. J.; Herrmann, W. A. J. Organomet. Chem.1999,582, 362–365. doi:10.1016/S0022-328X(99)00163-1
Return to citation in text:
[1]
Huang, J.; Stevens, E. D.; Nolan, S. P.; Petersen, J. L. J. Am. Chem. Soc.1999,121, 2674–2678. doi:10.1021/ja9831352
Return to citation in text:
[1]
Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett.1999,1, 953–956. doi:10.1021/ol990909q
Return to citation in text:
[1]
Jafarpour, L.; Schanz, H.-J.; Stevens, E. D.; Nolan, S. P. Organometallics1999,18, 5416–5419. doi:10.1021/om990587u
Return to citation in text:
[1]
Monsaert, S.; Drozdzak, R.; Dragutan, V.; Dragutan, I.; Verpoort, F. Eur. J. Inorg. Chem.2008, 432–440. doi:10.1002/ejic.200700879
Return to citation in text:
[1]
[2]
[3]
Schmidt, B.; Hermanns, J. Top. Organomet. Chem.2004,13, 223–267.
Return to citation in text:
[1]
Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc.2000,122, 8168–8179. doi:10.1021/ja001179g
Return to citation in text:
[1]
Michrowska, A.; Bujok, R.; Harutyunyan, S.; Sashuk, V.; Dolgonos, G.; Grela, K. J. Am. Chem. Soc.2004,126, 9318–9325. doi:10.1021/ja048794v
Return to citation in text:
[1]
Gessler, S.; Randl, S.; Blechert, S. Tetrahedron Lett.2000,41, 9973–9976. doi:10.1016/S0040-4039(00)01808-6
Return to citation in text:
[1]
Connon, S. J.; Blechert, S. Angew. Chem., Int. Ed.2003,42, 1900–1923. doi:10.1002/anie.200200556
Return to citation in text:
[1]
Sanford, M. S.; Love, J. A.; Grubbs, R. H. Organometallics2001,20, 5314–5318. doi:10.1021/om010599r
Return to citation in text:
[1]
Love, J. A.; Sanford, M. S.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc.2003,125, 10103–10109. doi:10.1021/ja027472t
Return to citation in text:
[1]
Zhang, W.-Z.; He, R.; Zhang, R. Eur. J. Inorg. Chem.2007, 5345–5352. doi:10.1002/ejic.200700631
Return to citation in text:
[1]
Williams, J. E.; Harner, M. J.; Sponsler, M. B. Organometallics2005,24, 2013–2015. doi:10.1021/om050040h
Return to citation in text:
[1]
Bantreil, X.; Randall, R. A. M.; Slawin, A. M. Z.; Nolan, S. P. Organometallics2010,29, 3007–3011. doi:10.1021/om100310f
Return to citation in text:
[1]
Choi, T.-L.; Grubbs, R. H. Angew. Chem., Int. Ed.2003,42, 1743–1746. doi:10.1002/anie.200250632
Return to citation in text:
[1]
P'Pool, S. J.; Schanz, H.-J. J. Am. Chem. Soc.2007,129, 14200–14212. doi:10.1021/ja071938w
Return to citation in text:
[1]
Kang, B.; Kim, D.; Do, Y.; Chang, S. Org. Lett.2003,5, 3041–3043. doi:10.1021/ol035014z
Return to citation in text:
[1]
Ung, T.; Hejl, A.; Grubbs, R. H.; Schrodi, Y. Organometallics2004,23, 5399–5401. doi:10.1021/om0493210
Return to citation in text:
[1]
Monsaert, S.; Vila, A. L.; Drozdzak, R.; van der Voort, P.; Verpoort, F. Chem. Soc. Rev.2009,38, 3360–3372. doi:10.1039/b902345n
Return to citation in text:
[1]
Clavier, H.; Petersen, J. L.; Nolan, S. P. J. Organomet. Chem.2006,691, 5444–5447. doi:10.1016/j.jorganchem.2006.08.007
Return to citation in text:
[1]
Boeda, F.; Clavier, H.; Nolan, S. P. Chem. Commun.2008, 2726–2740. doi:10.1039/b718287b
Return to citation in text:
[1]
Burtscher, D.; Lexer, C.; Mereiter, K.; Winde, R.; Karch, R.; Slugovc, C. J. Polym. Sci., Part A: Polym. Chem.2008,46, 4630–4635. doi:10.1002/pola.22763
Return to citation in text:
[1]
Monsaert, S.; De Canck, E.; Drozdzak, R.; van der Voort, P.; Verpoort, F.; Martins, J. C.; Hendrickx, P. M. S. Eur. J. Org. Chem.2009, 655–665. doi:10.1002/ejoc.200800973
Return to citation in text:
[1]
Miao, X.; Fischmeister, C.; Bruneau, C.; Dixneuf, P. H. ChemSusChem2008,1, 813–816. doi:10.1002/cssc.200800074
Return to citation in text:
[1]
Adjiman, C. S.; Clarke, A. J.; Cooper, G.; Taylor, P. C. Chem. Commun.2008, 2806–2808. doi:10.1039/b802921k
Return to citation in text:
[1]
[2]
[3]
Schmidt, B.; Geißler, D. ChemCatChem2010,2, 423–429. doi:10.1002/cctc.200900282
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
Schmidt, B.; Kunz, O.; Biernat, A. J. Org. Chem.2010,75, 2389–2394. doi:10.1021/jo1002642
Return to citation in text:
[1]
[2]
[3]
[4]
Forman, G. S.; McConnell, A. E.; Tooze, R. P.; van Rensburg, W. J.; Meyer, W. H.; Kirk, M. M.; Dwyer, C. L.; Serfontein, D. W. Organometallics2005,24, 4528–4542. doi:10.1021/om0503848
Return to citation in text:
[1]
Imahori, T.; Ojima, H.; Tateyama, H.; Mihara, Y.; Takahata, H. Tetrahedron Lett.2008,49, 265–268. doi:10.1016/j.tetlet.2007.11.098
Return to citation in text:
[1]
Imahori, T.; Ojima, H.; Yoshimura, Y.; Takahata, H. Chem.–Eur. J.2008,14, 10762–10771. doi:10.1002/chem.200801439
Return to citation in text:
[1]
Mori, M.; Sakakibara, N.; Kinoshita, A. J. Org. Chem.1998,63, 6082–6083. doi:10.1021/jo980896e
Return to citation in text:
[1]
Schmidt, B.; Staude, L. J. Org. Chem.2009,74, 9237–9240. doi:10.1021/jo9018649
Return to citation in text:
[1]
[2]
[3]
[4]
Schmidt, B.; Nave, S. Chem. Commun.2006, 2489–2491. doi:10.1039/b604359c
Return to citation in text:
[1]
Schmidt, B.; Nave, S. Adv. Synth. Catal.2007,349, 215–230. doi:10.1002/adsc.200600473
Return to citation in text:
[1]
Lombardo, M.; Licciulli, S.; Trombini, C. Tetrahedron: Asymmetry2004,15, 289–292. doi:10.1016/j.tetasy.2003.10.026
Return to citation in text:
[1]
Monsaert, S.; Drozdzak, R.; Dragutan, V.; Dragutan, I.; Verpoort, F. Eur. J. Inorg. Chem.2008, 432–440. doi:10.1002/ejic.200700879
41.
Monsaert, S.; De Canck, E.; Drozdzak, R.; van der Voort, P.; Verpoort, F.; Martins, J. C.; Hendrickx, P. M. S. Eur. J. Org. Chem.2009, 655–665. doi:10.1002/ejoc.200800973
Forman, G. S.; McConnell, A. E.; Tooze, R. P.; van Rensburg, W. J.; Meyer, W. H.; Kirk, M. M.; Dwyer, C. L.; Serfontein, D. W. Organometallics2005,24, 4528–4542. doi:10.1021/om0503848


![[1860-5397-6-136-1]](/bjoc/content/figures/1860-5397-6-136-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-6-136-i1]](/bjoc/content/inline/1860-5397-6-136-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-6-136-2]](/bjoc/content/figures/1860-5397-6-136-2.svg?scale=1.6&max-width=1024&background=FFFFFF)
![[1860-5397-6-136-3]](/bjoc/content/figures/1860-5397-6-136-3.svg?scale=1.6&max-width=1024&background=FFFFFF)
![[1860-5397-6-136-4]](/bjoc/content/figures/1860-5397-6-136-4.svg?scale=1.6&max-width=1024&background=FFFFFF)
![[1860-5397-6-136-i2]](/bjoc/content/inline/1860-5397-6-136-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-6-136-5]](/bjoc/content/figures/1860-5397-6-136-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-6-136-i3]](/bjoc/content/inline/1860-5397-6-136-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-6-136-i4]](/bjoc/content/inline/1860-5397-6-136-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)