Abstract
Reaction of tetrafluoropyridazine with catechol gives a tricyclic 9,10-dioxa-1,2-diaza-anthracene system by a sequential nucleophilic aromatic substitution ring annelation process, further extending the use of perfluoroheteroaromatic derivatives for the synthesis of unusual polyfunctional heterocyclic architectures. The tricyclic scaffold reacts with amines and sodium ethoxide providing a short series of functional 9,10-dioxa-1,2-diaza-anthracene systems.

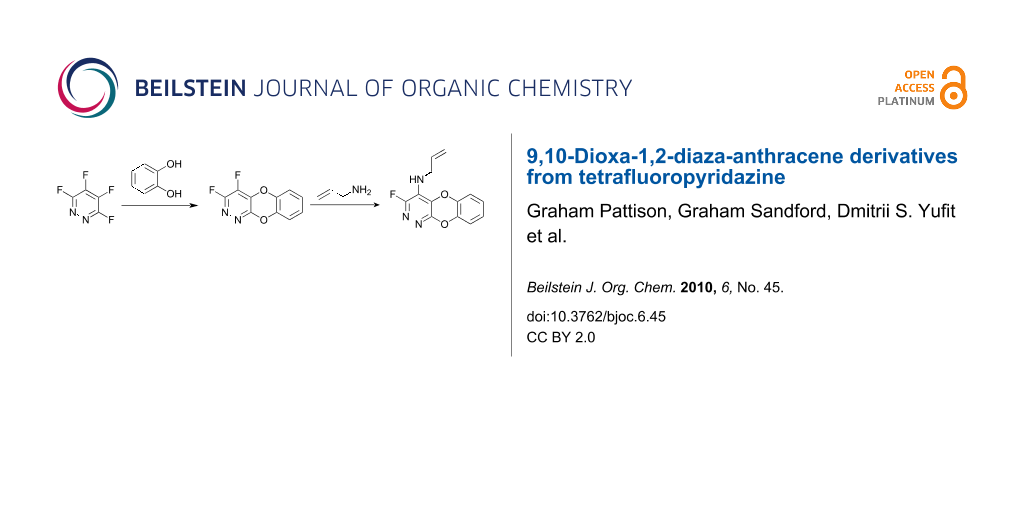
Graphical Abstract
Introduction
Drug discovery programmes are continually searching for viable synthetic routes to highly novel classes of heterocyclic compounds with the aim of exploring chemical ‘drug-like’ space [1] and uncovering valuable biological activity for hit-to-lead generation of new chemical entities by parallel synthesis techniques. The wide variety of relatively simple heterocyclic structural types that have not been synthesised [2], the relatively low level of structural diversity in all known organic structures [3] and, indeed, the perceived lack of structural diversity in pharmaceutical companies’ compound collections have often been suggested to be among the bottlenecks in drug discovery programmes [4]. Methodology for the ready synthesis of new organic frameworks is still required and, in this context, heterocyclic scaffolds based on novel molecular architecture that bear multiple functionality and can be rapidly processed into many analogues by parallel synthesis are particularly valuable [5,6].
In a continuing research programme, we have demonstrated that perfluorinated heteroaromatic derivatives are very useful starting scaffolds for the synthesis of a variety of heteroaromatic [7], [5,6] and [6,6]-bicyclic [8-11], and polycyclic heterocyclic systems [12]. Perfluoroheteroaromatic derivatives are either commercially available or can be accessed by halogen-exchange processes by reaction of the corresponding perchloroheteroaromatic system and potassium fluoride [13]. No special techniques for handling perfluoroheteroaromatic compounds are required, apart from the usual laboratory precautions, because these systems are generally volatile, colourless liquids. We established that highly novel tricyclic scaffolds, such as the relatively uncommon dipyrido[1,2-a:3′,4′-d]imidazole system 1, could be synthesised from pentafluoropyridine in a single step [12], exemplifying our general strategy for the synthesis of highly novel classes of polyfunctional heterocyclic compounds. Several dipyrido[1,2-a:3′,4′-d]imidazole analogues 2 were prepared by the displacement of the remaining ring fluorine atoms by nucleophilic aromatic substitution processes (Scheme 1).
![[1860-5397-6-45-i1]](/bjoc/content/inline/1860-5397-6-45-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of novel tricyclic heterocycles from pentafluoropyridine.
Scheme 1: Synthesis of novel tricyclic heterocycles from pentafluoropyridine.
We were interested in further expanding the use of highly fluorinated heterocycles for the preparation of novel heterocyclic structures and focussed upon the synthesis of ring fused systems that could be derived from the reaction of tetrafluoropyridazine (3) with catechol (4). In principle, two possible systems 5 and 6 may be formed depending upon the regioselectivity of the nucleophilic aromatic substitution processes (Scheme 2).
![[1860-5397-6-45-i2]](/bjoc/content/inline/1860-5397-6-45-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic route to dioxa-diaza-anthracene derivatives.
Scheme 2: Synthetic route to dioxa-diaza-anthracene derivatives.
Both 5 and 6 have ring fluorine atoms present that may, in principle, be displaced by nucleophiles which could lead to the synthesis of many analogues of these systems. The dioxa-1,2-diaza-anthracene (or 3,4-difluorobenzo[5,6][1,4]dioxino[2,3-c]pyridazine also referred to as benzodioxinopyridazine) systems are very rare heterocyclic structures and only a handful of analogues based upon this molecular skeleton have been synthesised, mainly by the reaction of chlorinated pyridazines with catechol [14-16].
In this paper, we describe the synthesis of dioxa-1,2-diaza-anthracene derivatives by the sequential reaction of commercially available tetrafluoropyridazine with catechol, and a short series of nucleophiles.
Results and Discussion
Initially, we carried out reactions of tetrafluoropyridazine (3) with one and two equivalents of sodium phenoxide as a model substrate for catechol (Scheme 3).
![[1860-5397-6-45-i3]](/bjoc/content/inline/1860-5397-6-45-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactions of tetrafluoropyridazine 3 with sodium phenoxide.
Scheme 3: Reactions of tetrafluoropyridazine 3 with sodium phenoxide.
Reaction of one equivalent of sodium phenoxide with (3) gave product 7 arising from substitution of fluorine located at the site para to activating ring nitrogen, consistent with earlier studies involving reactions between tetrafluoropyridazine and various nucleophiles [13]. Similarly, reaction of two equivalents of sodium phenoxide gave the 4,5-diphenoxy derivative 8 by displacement of both fluorine atoms that are attached to the sites para to ring nitrogen atoms.
In contrast, however, reaction of catechol (4) with tetrafluoropyridazine (3) under similar reaction conditions gave the tricyclic system 5 arising from displacement of the 3- and 4-fluorine atoms as the sole product according to a 19F NMR analysis of the crude reaction mixture (Scheme 4). The 19F NMR displays two resonances at −96.4 and −151.9 ppm in accord with structure 5, whereas if the symmetrical 4,5-disubstituted product 6 had been formed only one resonance in the 19F NMR spectrum at ca. −88 ppm (cf. 8) would have been observed.
![[1860-5397-6-45-i4]](/bjoc/content/inline/1860-5397-6-45-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of dioxa-1,2-diaza-anthracene scaffold 5.
Scheme 4: Synthesis of dioxa-1,2-diaza-anthracene scaffold 5.
It seems reasonable to assume that initial substitution occurs at the 4-position of 3, analogous to the reaction between 3 and phenoxide, to give intermediate 5a. At this point, we would expect cyclisation to occur at position 5 to give product 6, again by analogy to the outcome of reaction between 3 and excess phenoxide. However, since nucleophilic aromatic substitution reactions are frequently reversible [13], conversion of 6 must occur via intermediate 5a and lead to the most thermodynamically stable product 5 (Scheme 5).
![[1860-5397-6-45-i5]](/bjoc/content/inline/1860-5397-6-45-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
The utility of the dioxa-1,2-diaza-anthracene system 5 as a scaffold for array synthesis was assessed in representative reactions with a short series of nucleophiles (Scheme 6).
![[1860-5397-6-45-i6]](/bjoc/content/inline/1860-5397-6-45-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reactions of dioxa-1,2-diaza-anthracene scaffold 5 with nucleophiles.
Scheme 6: Reactions of dioxa-1,2-diaza-anthracene scaffold 5 with nucleophiles.
Nucleophilic substitution of fluorine at the 4-position occurs regiospecifically to afford products 9a–c according to 19F NMR analysis of the corresponding reaction mixtures. The 19F NMR resonances located at ca. −90 ppm are characteristic of fluorine atoms located at sites ortho to a ring nitrogen atom. X-ray crystallography of the allylamino derivative 9b (Figure 1), and a comparison of NMR spectral data, confirms the structures of these analogues.
![[1860-5397-6-45-1]](/bjoc/content/figures/1860-5397-6-45-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of 4-allylamino-3-fluoro-9,10-dioxa-1,2-diaza-anthracene (9b).
Figure 1: Molecular structure of 4-allylamino-3-fluoro-9,10-dioxa-1,2-diaza-anthracene (9b).
The geometrical parameters of the molecule 9b are close to expected values. The molecules of 9b in the crystal are linked together by N–H···N hydrogen bonds in chains, parallel to the [101] direction and π···π stacking interactions (shortest interatomic distance C5···C3 is 3.336 Å), and short C–H···O contacts (C···O 3.387 Å) bind adjacent chains in the [100] and [010] directions, respectively.
Again, the regiospecificity of these reaction processes occurs because of the activating effect of ring nitrogen directly opposite the site of nucleophilic substitution.
Conclusions
A small range of dioxa-1,2-diaza-anthracene analogues 5 and 9 have been synthesised from tetrafluoropyridazine in two efficient steps, further expanding the application of highly fluorinated heterocycles for the synthesis of rare heterocyclic architectures.
Experimental
Synthetic procedures for the preparation of all the new compounds described in this paper are given below.
Reactions of tetrafluoropyridazine (3) with sodium phenoxide
3,4,6-Trifluoro-5-phenoxypyridazine (7)
Phenol (0.17 g, 1.81 mmol) was dissolved in THF (20 mL) and added to sodium hydride (0.07 g, 1.8 mmol, 60% dispersion in mineral oil) which was cooled to 0 °C and stirred. Tetrafluoropyridazine (3) (0.25 g, 1.64 mmol) was added slowly and the mixture stirred at 0 °C for 8 h. The solvent was evaporated and the crude material partitioned between dichloromethane (25 mL) and water (25 mL). The organic layer was separated and the aqueous layer extracted with dichloromethane (3 × 25 mL). The combined organic extracts were then dried (MgSO4), filtered and evaporated in vacuo to provide a crude yellow material. Column chromatography on silica gel using hexane:ethyl acetate (4:1) as elutant gave 3,4,6-trifluoro-5-phenoxypyridazine (7) (0.25 g, 68%) as a colourless oil; Anal. Calcd for C10H5F3N2O: C, 53.1; H, 2.2; N, 12.4%. Found: C, 53.2; H, 2.5; N, 12.2. 1H NMR (200 MHz, CDCl3, δH): 7.04–7.42 (5H, m, ArH); 19F NMR (188 MHz, CDCl3, δF): −86.2 (1F, dd, 5JFF = 31.2 Hz, 4JFF = 23.7 Hz, F-6), −94.5 (1F, dd, 5JFF = 31.2 Hz, 3JFF = 23.7 Hz, F-3), −140.4 (1F, dd, 3JFF = 23.7 Hz, 4JFF = 23.7 Hz, F-4); MS (ES+) m/z: 227 ([MH]+, 100%).
3,6-Difluoro-4,5-diphenoxypyridazine (8)
Using the procedure described above, phenol (0.32 g, 3.45 mmol), sodium hydride (0.138 g, 3.45 mmol, 60% dispersion in mineral oil), tetrafluoropyridazine (0.25 g, 1.64 mmol) and THF (20 mL) gave 3,6-difluoro-4,5-diphenoxypyridazine (8) (0.29 g, 59%) as a white solid; mp 123–124 °C; Anal. Calcd for C16H10F2N2O2: C, 64.0; H, 3.4; N, 9.3%. Found: C, 63.7; H, 3.5; N, 9.2. 1H NMR (700 MHz, CDCl3, δH): 6.80 (2H, d, 3JHH = 7.7, H-2′), 7.09 (1H, t, 3JHH = 7.7, H-4′), 7.23 (2H, t, 3JHH = 7.7, H-3′); 13C NMR (175 MHz, CDCl3, δC): 116.4 (s, C-2′), 124.9 (s, C-4′), 129.7 (s, C-3′), 137.2 (dd, 2JCF = 20.6, 3JCF = 12.9, C-4), 155.2 (s, C-1′), 160.1 (dd, 1JCF = 251.2, 4JCF = 6.8, C-3); 19F NMR (658 MHz, CDCl3 δF): −88.2 (s); MS (ES+) m/z: 301 ([MH]+, 100%).
Synthesis of 3,4-difluoro-9,10-dioxa-1,2-diaza-anthracene (5)
Catechol (0.80 g, 7.2 mmol) was dissolved in THF (20 mL) at 0 °C under an argon atmosphere with stirring and added to sodium hydride (0.35 g, 14.5 mmol, 60% dispersion in mineral oil). Tetrafluoropyridazine (1.00 g, 6.6 mmol) was added dropwise and the mixture stirred at 0 °C for 8 h. After this period, the solvent was evaporated, and the crude material redissolved in dichloromethane (25 mL) and water (25 mL). The organic layer was separated and the aqueous layer extracted with dichloromethane (3 × 25 mL). The combined organic extracts were then dried (MgSO4), filtered and evaporated to provide a crude yellow material. Crystallisation from acetonitrile gave 3,4-difluoro-9,10-dioxa-1,2-diaza-anthracene (5) (1.09 g, 75%) as white solid; mp 146–148 °C; Anal. Calcd for C8H11FN4O: C, 54.1; H, 1.8; N, 12.6%. Found: C, 54.0; H, 1.9; N, 12.6. IR, νmax/cm−1: 1015, 1035, 1094, 1115, 1260, 1416, 1464, 1490, 1568, 1654; 1H NMR (400 MHz, CDCl3, δH): 7.13–7.06 (4 H, m, ArH); 13C NMR (100 MHz, CDCl3, δC): 117.0 (s, C-5), 118.1 (s, C-6), 126.0 (s, C-7), 126.8 (s, C-8), 133.4 (dd, 2JCF = 6.3 Hz, 3JCF = 6.3 Hz, C-4a), 136.8 (dd, 1JCF = 278.0 Hz, 2JCF = 30.0 Hz, C-4), 138.6 (s, C-8a), 140.3 (s, C-9a), 154.5 (s, C-10a), 156.9 (dd, 1JCF = 290 Hz, 2JCF = 8.0 Hz, C-3); 19F NMR (376 MHz, CDCl3, δF): −96.4 (1F, d, 3JFF = 25.8 Hz, F-3), −151.9 (1F, d, 3JFF = 25.9 Hz, F-4); MS (EI+) m/z: 222 ([M]+, 10%), 138 (43), 74 (66), 63 (67), 50 (100).
Reaction of 3,4-difluoro-9,10-dioxa-1,2-diaza-anthracene (5) with morpholine
3-Fluoro-4-(morpholin-4-yl)-9,10-dioxa-1,2-diaza-anthracene (9a)
A mixture of 3,4-difluoro-9,10-dioxa-1,2-diaza-anthracene (5) (0.20 g, 0.90 mmol), morpholine (0.16 mL, 1.80 mmol) and acetonitrile (2 mL) were placed in a 0.5–2 ml microwave vial under an argon atmosphere and subjected to microwave irradiation at 150 °C for 20 min. The mixture was partitioned between dichloromethane (20 mL) and water (20 mL) and the organic layer separated. The aqueous layer was then extracted with dichloromethane (3 × 20 mL) to give a crude yellow material. Column chromatography on silica gel using hexane:ethyl acetate (2:1) as eluent gave 3-fluoro-4-(morpholin-4-yl)-9,10-dioxa-1,2-diaza-anthracene (9a) (0.18 g, 71%) as white crystals; mp 207–208 °C; Found: [MH]+, 290.09345. C14H12FN3O3 requires: [MH]+, 290.09355; 1H NMR (700 MHz, CDCl3, δH): 3.44 (4H, t, 3JHH = 4.4 Hz, H-2′), 3.84 (4H, t, 3JHH = 4.4 Hz, H-3′), 6.94 (1H, d, 3JHH = 7.6 Hz, ArH), 7.01 (1H, tm, 3JHH = 7.6 Hz, ArH), 7.06 (2H, m, ArH); 13C NMR (175 MHz, CDCl3, δC): 50.4 (d, 4JCF = 4.0 Hz, C-2′), 67.3 (s, C-3′), 116.5 (s, C-5), 117.7 (s, C-8), 125.2 (s, C-6), 125.9 (s, C-7), 126.0 (d, 2JCF = 25.4 Hz, C-4), 134.6 (d, 3JCF = 8.9 Hz, C-4a), 139.4 (s, C-8a), 140.9 (s, C-10a), 153.7 (s, C-9a), 159.0 (d, 1JCF = 237.7 Hz, C-3); 19F NMR (658 MHz, CDCl3, δF): −86.4 (s); MS (ES+) m/z: 290 ( [MH]+, 100%).
Reaction of 3,4-difluoro-9,10-dioxa-1,2-diaza-anthracene (5) with allylamine
4-Allylamino-3-fluoro-9,10-dioxa-1,2-diaza-anthracene (9b)
Using the procedure described above, 3,4-difluoro-9,10-dioxa-1,2-diaza-anthracene (5) (0.15 g, 0.67 mmol), allylamine (0.10 mL, 1.35 mmol) and acetonitrile (2 mL) gave 4-allylamino-3-fluoro-9,10-dioxa-1,2-diaza-anthracene (9b) (0.14 g, 80%) as white crystals; mp 175–177 °C; Anal Calcd for C13H10FN3O2: C, 60.2; H, 3.9; N, 16.2%. Found: C, 60.3; H, 4.0; N, 16.3. 1H NMR (500 MHz, DMSO-d6, δH): 4.04 (2H, t, 3JHH = 5.1 Hz, NCH2), 5.10 (1H, dd, 3JHH = 10.3 Hz, 2JHH = 1.5 Hz, =CH2), 5.17 (1H, dd, 3JHH = 17.2 Hz, 2JHH = 1.5 Hz, =CH2), 5.94 (1H, ddt, 3JHH = 17.2 Hz, 10.2, 5.1, -CH=), 6.96 (1H, br t, 3JHH = 5.1 Hz, NH), 7.07 (3H, m, ArH), 7.12 (1H, m, ArH); 13C NMR (125 MHz, DMSO-d6, δC): 45.7 (d, 4JCF = 2.5 Hz, NCH2), 115.3 (s, =CH2), 116.4 (s, C-5), 117.0 (s, C-6), 124.6 (d, 2JCF = 28.2 Hz, C-4), 125.1 (s, C-7), 125.2 (s, C-8), 127.2 (d, 3JCF = 9.6 Hz, C-4a), 136.1 (s, CH=), 139.4 (s, C-8a), 140.5 (s, C-10a), 152.3 (s, C-9a), 155.2 (d, 1JCF = 230.4 Hz, C-3); 19F NMR (470 MHz, DMSO-d6, δF) −93.7 (s); MS (ES+) m/z: 323 ([M+MeCN+Na]+, 100%), 260 ([MH]+, 68), 219 (69).
Crystal data for 9b: C13H10FN3O2, M = 259.24, monoclinic, space group P21/n, a = 4.9065(1), b = 19.5663(4), c = 11.8180(2) Å, β = 94.25(1)°, U = 1131.44(4) Å3, F(000) = 536, Z = 4, Dc = 1.5220 mg·m−3, μ = 0.117 mm−1 (Mo Kα, λ = 0.71073 Å), T = 120.0(2) K. 14166 reflections were collected on a Bruker SMART 6000 diffractometer (ω-scan, 0.3°/frame) yielding 2875 unique data (Rmerg = 0.0615). The structure was solved by direct method and refined by full-matrix least squares on F2 for all data using Olex2 software. All non-hydrogen atoms were refined with anisotropic displacement parameters, H-atoms were located on the difference map and refined isotropically. Final wR2(F2) = 0.1275 for all data (212 refined parameters), conventional R(F) = 0.0439 for 1918 reflections with I ≥ 2σ, GOF = 0.985. Crystallographic data for the structure have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC-764716.
Reaction of 3,4-difluoro-9,10-dioxa-1,2-diaza-anthracene (5) with sodium ethoxide
4-Ethoxy-3-fluoro-9,10-dioxa-1,2-diaza-anthracene (9c)
Using the procedure described above, 3,4-difluoro-9,10-dioxa-1,2-diaza-anthracene (5) (0.10 g, 0.45 mmol), sodium ethoxide (0.06 g, 0.90 mmol) and ethanol (2 mL) gave 4-ethoxy-3-fluoro-9,10-dioxa-1,2-diaza-anthracene (9c) (0.07 g, 66%), as white crystals; mp 131–133 °C; Anal Calcd for C12H9FN2O3: C, 58.1; H, 3.7; N, 11.3%. Found: C, 58.0; H, 3.7; N, 11.2. 1H NMR (500 MHz, CDCl3, δH): 1.49 (3H, t, 3JHH = 7.0 Hz, CH3), 4.52 (2H, qd, 3JHH = 7.0 Hz, 5JHF = 1.4 Hz, OCH2), 7.00–7.11 (4H, m, ArH); 13C NMR (125 MHz, DMSO-d6, δC): 15.7 (s, CH3), 70.6 (d, 4JCF = 3.9 Hz, OCH2), 116.7 (s, C-5), 117.8 (s, C-6), 125.4 (s, C-7), 126.1 (s, C-8), 133.6 (d, 2JCF = 27.2 Hz, C-4), 135.1 (d, 3JCF = 8.2 Hz, C-4a), 139.2 (s, C-8a), 140.7 (s, C-10a), 154.0 (d, 4JCF = 1.5 Hz, C-9a), 158.2 (d, 1JCF = 239.5 Hz, C-3); 19F NMR (470 MHz, CDCl3, δF) −92.4 (s); MS (ES+) m/z: (249 ([MH]+), 100%).
Supporting Information
Supporting Information with 1H NMR and 13C NMR spectra for 3,4,6-trifluoro-5-phenoxypyridazine (7), 1H NMR, 13C NMR and 19F NMR spectra for 3,6-difluoro-4,5-diphenoxypyridazine (8), 3,4-difluoro-9,10-dioxa-1,2-diaza-anthracene (5), 3-fluoro-4-(morpholin-4-yl)-9,10-dioxa-1,2-diaza-anthracene (9a), 4-allylamino-3-fluoro-9,10-dioxa-1,2-diaza-anthracene (9b), 4-ethoxy-3-fluoro-9,10-dioxa-1,2-diaza-anthracene (9c).
| Supporting Information File 1: NMR spectra of all synthesized compounds 7, 8, 5 and 9a–9c | ||
| Format: PDF | Size: 331.9 KB | Download |
References
-
Lipinski, C.; Hopkins, A. Nature 2004, 432, 855–861. doi:10.1038/nature03193
Return to citation in text: [1] -
Pitt, W. R.; Parry, D. M.; Perry, B. G.; Groom, C. R. J. Med. Chem. 2009, 52, 2952–2963. doi:10.1021/jm801513z
Return to citation in text: [1] -
Lipkus, A. H.; Yuan, Q.; Lucas, K. A.; Funk, S. A.; Bartelt, W. F.; Schenck, R. J.; Trippe, A. J. J. Org. Chem. 2008, 73, 4443–4451. doi:10.1021/jo8001276
Return to citation in text: [1] -
Burke, M. D.; Berger, E. M.; Schreiber, S. L. Science 2003, 302, 613–618. doi:10.1126/science.1089946
Return to citation in text: [1] -
Collins, I. J. Chem. Soc., Perkin Trans. 1 2000, 2845–2861. doi:10.1039/a904715h
Return to citation in text: [1] -
Collins, I. J. Chem. Soc., Perkin Trans. 1 2002, 1921–1940. doi:10.1039/b108580h
Return to citation in text: [1] -
Pattison, G.; Sandford, G.; Yufit, D. S.; Howard, J. A. K.; Christopher, J. A.; Miller, D. D. J. Org. Chem. 2009, 74, 5533–5540. doi:10.1021/jo9006943
Return to citation in text: [1] -
Sandford, G.; Hargreaves, C. A.; Slater, R.; Yufit, D. S.; Howard, J. A. K.; Vong, A. Tetrahedron 2007, 63, 5204–5211. doi:10.1016/j.tet.2007.03.164
Return to citation in text: [1] -
Sandford, G.; Slater, R.; Yufit, D. S.; Howard, J. A. K.; Vong, A. J. Org. Chem. 2005, 70, 7208–7216. doi:10.1021/jo0508696
Return to citation in text: [1] -
Baron, A.; Sandford, G.; Slater, R.; Yufit, D. S.; Howard, J. A. K.; Vong, A. J. Org. Chem. 2005, 70, 9377–9381. doi:10.1021/jo051453v
Return to citation in text: [1] -
Cartwright, M. W.; Sandford, G.; Bousbaa, J.; Yufit, D. S.; Howard, J. A. K.; Christopher, J. A.; Miller, D. D. Tetrahedron 2007, 63, 7027–7035. doi:10.1016/j.tet.2007.05.016
Return to citation in text: [1] -
Cartwright, M. W.; Convery, L.; Kraynck, T.; Sandford, G.; Yufit, D. S.; Howard, J. A. K.; Christopher, J. A.; Miller, D. D. Tetrahedron 2010, 66, 519–529. doi:10.1016/j.tet.2009.11.036
Return to citation in text: [1] [2] -
Brooke, G. M. J. Fluorine Chem. 1997, 86, 1–76. doi:10.1016/S0022-1139(97)00006-7
Return to citation in text: [1] [2] [3] -
Oishi, E.; Sugiyama, C.; Iwamoto, K.; Kato, I. Heterocycles 2004, 63, 591–608. doi:10.3987/COM-03-9961
Return to citation in text: [1] -
Chupp, J. P.; Jones, C. R.; Dahl, M. L. J. Heterocycl. Chem. 1993, 30, 789–798. doi:10.1002/jhet.5570300330
Return to citation in text: [1] -
Ames, D. E.; Ward, R. J. J. Chem. Soc., Perkin Trans. 1 1975, 534–538. doi:10.1039/P19750000534
Return to citation in text: [1]
| 5. | Collins, I. J. Chem. Soc., Perkin Trans. 1 2000, 2845–2861. doi:10.1039/a904715h |
| 6. | Collins, I. J. Chem. Soc., Perkin Trans. 1 2002, 1921–1940. doi:10.1039/b108580h |
| 4. | Burke, M. D.; Berger, E. M.; Schreiber, S. L. Science 2003, 302, 613–618. doi:10.1126/science.1089946 |
| 3. | Lipkus, A. H.; Yuan, Q.; Lucas, K. A.; Funk, S. A.; Bartelt, W. F.; Schenck, R. J.; Trippe, A. J. J. Org. Chem. 2008, 73, 4443–4451. doi:10.1021/jo8001276 |
| 13. | Brooke, G. M. J. Fluorine Chem. 1997, 86, 1–76. doi:10.1016/S0022-1139(97)00006-7 |
| 2. | Pitt, W. R.; Parry, D. M.; Perry, B. G.; Groom, C. R. J. Med. Chem. 2009, 52, 2952–2963. doi:10.1021/jm801513z |
| 13. | Brooke, G. M. J. Fluorine Chem. 1997, 86, 1–76. doi:10.1016/S0022-1139(97)00006-7 |
| 14. | Oishi, E.; Sugiyama, C.; Iwamoto, K.; Kato, I. Heterocycles 2004, 63, 591–608. doi:10.3987/COM-03-9961 |
| 15. | Chupp, J. P.; Jones, C. R.; Dahl, M. L. J. Heterocycl. Chem. 1993, 30, 789–798. doi:10.1002/jhet.5570300330 |
| 16. | Ames, D. E.; Ward, R. J. J. Chem. Soc., Perkin Trans. 1 1975, 534–538. doi:10.1039/P19750000534 |
| 12. | Cartwright, M. W.; Convery, L.; Kraynck, T.; Sandford, G.; Yufit, D. S.; Howard, J. A. K.; Christopher, J. A.; Miller, D. D. Tetrahedron 2010, 66, 519–529. doi:10.1016/j.tet.2009.11.036 |
| 13. | Brooke, G. M. J. Fluorine Chem. 1997, 86, 1–76. doi:10.1016/S0022-1139(97)00006-7 |
| 8. | Sandford, G.; Hargreaves, C. A.; Slater, R.; Yufit, D. S.; Howard, J. A. K.; Vong, A. Tetrahedron 2007, 63, 5204–5211. doi:10.1016/j.tet.2007.03.164 |
| 9. | Sandford, G.; Slater, R.; Yufit, D. S.; Howard, J. A. K.; Vong, A. J. Org. Chem. 2005, 70, 7208–7216. doi:10.1021/jo0508696 |
| 10. | Baron, A.; Sandford, G.; Slater, R.; Yufit, D. S.; Howard, J. A. K.; Vong, A. J. Org. Chem. 2005, 70, 9377–9381. doi:10.1021/jo051453v |
| 11. | Cartwright, M. W.; Sandford, G.; Bousbaa, J.; Yufit, D. S.; Howard, J. A. K.; Christopher, J. A.; Miller, D. D. Tetrahedron 2007, 63, 7027–7035. doi:10.1016/j.tet.2007.05.016 |
| 7. | Pattison, G.; Sandford, G.; Yufit, D. S.; Howard, J. A. K.; Christopher, J. A.; Miller, D. D. J. Org. Chem. 2009, 74, 5533–5540. doi:10.1021/jo9006943 |
| 12. | Cartwright, M. W.; Convery, L.; Kraynck, T.; Sandford, G.; Yufit, D. S.; Howard, J. A. K.; Christopher, J. A.; Miller, D. D. Tetrahedron 2010, 66, 519–529. doi:10.1016/j.tet.2009.11.036 |
© 2010 Pattison et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)