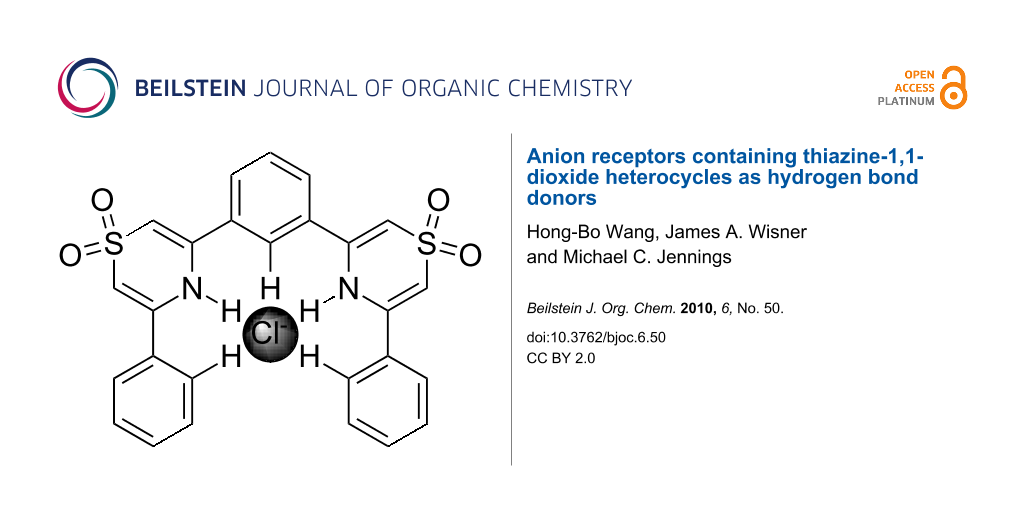
Abstract
The synthesis, X-ray crystal structures and anion recognition properties of two receptors containing thiazine-1,1-dioxide heterocycles as hydrogen bond donating subunits are reported. The newly synthesized receptors display much different anion selectivities in acetone-d6 than N,N′-diphenyl-1,3-disulfonamidobenzene that was used as a comparison. The selectivity exhibited by one of the new receptors for chloride anions can be attributed to greater steric demand in the cleft formed, in part, by its terminal phenyl rings; an effect that is absent in the comparison receptor.

Graphical Abstract
Introduction
The synthesis of neutral hosts and study of their anion recognition properties is an area of research that has grown in interest over the past several years owing to the potential use of such receptors in environmental, biomedical and materials applications [1,2]. The basic design methodology for these hosts has largely focused on the use of nitrogen-based hydrogen bond donor groups such as amides [3,4], ureas [5], pyrroles/indoles/carbazoles [6,7] and sulfonamides [8-23] to complex the anionic targets in a topologically complementary fashion. Sulfonamides are an interesting case as the hydrogen bond donor is often significantly more acidic (pKa approx. 11 for simple N-phenylaryl sulfonamides such as 3 (see below)) than that presented by other groups typically incorporated in these frameworks. The greater acidity of such a subunit can be an advantage by providing greater potential hydrogen bond donor strength with anionic guests. Alternatively, the possibility of deprotonation in some specific systems by basic anions such as carboxylates or fluoride can be employed as an indicator for these species. Regardless, the incorporation of sulfonamide functional groups has typically been realized synthetically by sulfonylation of an amine to form a sulfonamide product. This approach is somewhat limited, from a design perspective, in that the majority of examples to date consist of sulfonamides derived from a few commercially available starting materials such as benzenesulfonyl, toluenesulfonyl, dansyl, and benzenedisulfonyl chlorides [8-23]. We have recently investigated thiazine-1,1-dioxide heterocycles (Figure 1A) as hydrogen bond donor groups in the formation of double helical complexes [24]. The parent heterocycle can be viewed as a cyclic, vinylogous sulfonamide that presents a different spatial, conformational and electronic relationship between the sulfonyl and NH subunits than that of a typical sulfonamide function (Figure 1B). It is a simple matter to access many such derivatives with this framework using straightforward synthetic methods and inexpensive materials and reagents. Herein, we describe an illustrative synthesis of two anion hosts incorporating these heterocycles and compare their binding affinities with some common anionic guests to that of an analogous benzene disulfonamide anion receptor.
![[1860-5397-6-50-1]](/bjoc/content/figures/1860-5397-6-50-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of thiazine-1,1-dioxide heterocycle (A) and sulfonamide function (B).
Figure 1: Structures of thiazine-1,1-dioxide heterocycle (A) and sulfonamide function (B).
Results and Discussion
The two receptor structures 1 and 2 (Figure 2) were chosen with the intent of evaluating their efficacy in comparison to the known anion host N,N′-diphenyl-1,3-disulfonamidobenzene 3 (Figure 2) [21]. Originally investigated for anion recognition by Crabtree and coworkers, 3 was considered a representative comparator given the similar stereochemical arrangement of the two NH donors and the 1,3-benzenediyl spacer. The incorporation of a pyridyl spacer in 2 was chosen to examine the possible effect the ring nitrogen atom might have on the preorganization and anion recognition properties of the resulting host in comparison to 1. It is well known that structurally related 2,6-dicarboxamidopyridine containing hosts have markedly different properties compared to their analogous isophthalamide derivatives in these regards as well [25,26]. It is an indication of the potential versatility of the synthetic method described here that the elusive 2,6-disulfonamidopyridine host 4 (Figure 2) that would provide a more direct comparison to 2 is at present unknown and likely synthesized only with some difficulty.
![[1860-5397-6-50-2]](/bjoc/content/figures/1860-5397-6-50-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of anion receptors 1–4.
Figure 2: Structures of anion receptors 1–4.
The syntheses of receptors 1 and 2 are summarized in Scheme 1. α,α′-Dibromo-1,3-diacetylbenzene (5) and α,α′-dibromo-2,6-diacetylpyridine (6) are both simply generated by bromination of the corresponding diacylarenes. The reaction of either dibromide with α-mercaptoacetophenone in the presence of 2,6-lutidine yields dithioether intermediates 7 and 8. Oxidation of these dithioethers to the disulfones 9 and 10 with urea-hydrogen peroxide (UHP) and trifluoroacetic anhydride (TFAA) in acetonitrile at room temperature proceeds in high yields. The final products 1 and 2 were obtained by the cyclization and dehydration of these intermediate disulfones with ammonium acetate in refluxing glacial acetic acid. Overall, the yields of receptors 1 and 2 are 62% and 72% respectively, from the dibromides. The simplicity and mild nature of these transformations make them easily applicable to the derivatization of most α-bromoacyl functional groups should one desire the installation of this subunit in a potential host.
![[1860-5397-6-50-i1]](/bjoc/content/inline/1860-5397-6-50-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Syntheses of 1 and 2. Reaction conditions: (a) (X = CH) NBS, TsOH, CH3CN, reflux or (X = N) Br2, AlCl3, Et2O, 0 °C; (b) 2,6-lutidine, α-mercaptoacetophenone (2 equiv), CH2Cl2; (c) UHP/TFAA, CH3CN; (d) NH4OAc, AcOH, reflux.
Scheme 1: Syntheses of 1 and 2. Reaction conditions: (a) (X = CH) NBS, TsOH, CH3CN, reflux or (X = N) Br2, Al...
The solid-state structures of both newly synthesized receptors were confirmed by X-ray diffraction analysis of single crystals grown by the slow diffusion of isopropyl ether into concentrated DMSO solutions of each (Figure 3). Unfortunately, attempts to co-crystallize the receptors with anionic guests in a number of organic solvents were unsuccessful. The conformations of the receptors in the solid state are surprisingly different given the similarity in molecular structure; the two receptors differ only in the replacement of an aryl CH in 1 for N in 2. The structure of 1 is in an extended, approximately anti-anti conformation [27] where each of the NH groups is hydrogen bonded to a different DMSO solvent molecule in the lattice (N···O = 2.821(3) and 2.770(3) Å). In contrast, 2 crystallizes in an approximately cleft-shaped syn-syn conformation where both NH groups are hydrogen bonded to a single DMSO solvent molecule (N···O = 2.971(2) and 2.950(3) Å). The difference may be rationalized by the presence of a weak intramolecular N-H···N hydrogen bond between the central pyridine ring and each of the two thiazine-1,1-dioxide rings in 2 that is necessarily absent in 1. This conclusion is supported by the approximate 0.5 ppm chemical shift difference of the two receptor NH resonances in acetone-d6 (2 > 1) and similar shift differences observed in analogous hosts that are even larger when measured in the less competitive solvent CDCl3 [28].
![[1860-5397-6-50-3]](/bjoc/content/figures/1860-5397-6-50-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Stick representations of the X-ray crystal structures of (a) receptor 1 and (b) receptor 2. Non-acidic hydrogen atoms omitted for clarity. Red = oxygen, blue = nitrogen, yellow = sulfur, grey = carbon. Hydrogen bonds denoted by dotted orange lines.
Figure 3: Stick representations of the X-ray crystal structures of (a) receptor 1 and (b) receptor 2. Non-aci...
The three receptors were each titrated in acetone-d6 with a number of TBA (tetrabutylammonium) salts of common anionic guests and the shifts of their 1H NMR resonances were observed as a function of anion concentration. In the majority of cases the downfield shift of the NH protons of the receptors was used to determine the stability constants. However, the addition of less than a half an equivalent of either acetate or dihydrogen phosphate anions to any of the three receptors resulted in the disappearance of the receptor NH proton resonances in their NMR spectra. In these cases, either the upfield shift of a CH resonance on the thiazine-1,1-dioxide rings (1 and 2) or the downfield shift of the 2-CH proton on the central phenyl ring of the receptor (3) was used to determine the stability constants. Non-linear least squares fitting of the data using the program EQNMR [29] yielded the complex stability constants in all cases. All of the titrations were fit to a 1:1 (receptor:anion) binding model except for the titration of receptor 1 with TBA chloride. In this case the data fit a 1:1 binding model that included a much weaker 2:1 (receptor:anion) component. It should also be noted that the data from titration of 3 with acetate had a binding constant that was too large to be reliably fit by this method. The titration results are summarized in Table 1.
Table 1: Stability constants (Ka) determined by 1H NMR in acetone-d6 solution at 298 K for receptors 1–3 with a variety of anionic guests.
| Aniona | Receptor 1 | Receptor 2 | Receptor 3 |
|---|---|---|---|
| Cl− |
(2:1) 300
(1:1) 59000 |
300 | 4300 |
| Br− | 380 | 83 | 740 |
| I− | 53 | —b | 86 |
| HSO4− | 220 | 130 | 560 |
| AcO− | 12500 | 480 | >105 |
| H2PO4− | 540 | 360 | 79000 |
aAdded as their tetrabutylammonium salts. Errors are estimated to be <10%.
bNo change was observed in the 1H NMR of the receptor upon anion addition.
Receptor 1 exhibits a clear preference for a chloride anion guest over the other anions tested. Chloride is likely an excellent steric match to the cleft formed by the NH protons, the 2-CH proton of the central aryl ring and two of the ortho-protons of the terminal phenyl rings of 1 in a planar syn-syn binding conformation similar to that observed in the solid state structure of 2 with DMSO (Figure 1B). This conclusion is supported by the observation of significant downfield shifts of all three of these protons (Figure 2) in the 1H NMR spectrum upon chloride complexation (Δδmax = 0.15 (Ha), 1.91 (Hb), 0.17 (Hc) ppm). The progressively reduced affinity of 1 for bromide and iodide can be attributed both to their inability to fit into this idealized cleft conformation and their respectively decreasing efficacies as hydrogen bond acceptors. In fact, this observation is common to 1 and 2 and mirrors the behaviour of similar acyclic isophthalamide hosts studied previously by Crabtree and coworkers in CD2Cl2 [21]. Receptor 1 shows a preference for the complexation of chloride over acetate (5:1) and a distinct discrimination against the dihydrogen phosphate (>100:1 Cl:H2PO4) guest. Presumably, the larger size of the dihydrogen phosphate anion prevents complexation by 1 in a coplanar syn-syn conformation. The distortion of the receptor from a low energy coplanar binding geometry should reduce its affinity for such guests, despite their greater basicity. This supposition is supported in the case of dihydrogen phosphate by a very small Δδmax for Ha observed when 1 is titrated with this anion (0.01 ppm) though Δδmax of protons Hc remains significant (0.15 ppm) indicating their continued participation in the binding event. The titration of 1 with the less basic but similarly sized and shaped HSO4− anion displays an upfield shift of Ha (Δδmax = −0.05 ppm) and a reduced downfield shift of Hc (Δδmax = 0.07 ppm).
Replacement of the central phenyl ring spacer of 1 for pyridine in 2 results in a significant reduction of the association constants for all of the anions tested. This result was expected as a consequence of repulsion of anionic guests by the lone pair of the pyridine ring nitrogen atom upon binding in the cleft of the receptor. The other outcome of this replacement is a loss of any selectivity for chloride and, in fact, a slight preference by receptor 2 for acetate and dihydrogen phosphate.
The simple disulfonamide receptor 3 exhibits very different complexation behaviour than 1 and 2 with the anions investigated. Receptor 3 displays a strong preference for acetate and dihydrogen phosphate over all of the other anions investigated in this solvent. The binding constant for chloride is reduced by an order of magnitude but is still preferred over bromide even though the affinity of 3 for the latter guest has approximately doubled in comparison to receptor 1. No change in the 1H NMR spectrum of 3 is observed upon the addition of iodide. We believe that the differences in anion binding between these two receptors can be satisfactorily explained by the differences in their cleft geometries. The central three atoms (NH and CHa) that define the binding cleft in both 1 and 3 circumscribe a very similar meridian. In fact, we manipulated the single crystal molecular structures of 1 and the 4,4′-di-t-butyl derivative of 3 [30] (available from the Cambridge Crystallographic Database #1003/6124) by rotating the two relevant dihedral angles to bring the NH groups into plane with their central aryl rings in an idealized syn-syn conformation. Measurement of these “closest approach” N···N distances in the two models yields values of 4.76 and 4.77 Å for 1 and the derivative of 3, respectively; a difference of 0.01 Å. The terminal phenyl rings of 3 do not, however, occlude this central cleft like those of 1 and 2. Rather, they form a divergent “V”-shaped geometry upon chelation of anionic species by the two NH groups of 3, regardless of whether coplanarity is maintained with the central benzene ring. The ortho-protons of these terminal rings (Hc) are certainly too far away to contribute to the stability of the halide anion complexes that presumably form in this manner. The general result of this relaxation of the steric requirements for anion complexation by 3, in comparison to 1, is a marked increase in binding strength for all of the larger anions. Thus, the binding affinities of 3 for these larger anions follow the trend of their aqueous basicities (pKa conj. acid): AcO− (4.75), H2PO4− (2.12), HSO4− (−3), Br− (−9), I− (−10) [31].
Conclusions
We have presented a simple synthetic route for the incorporation of thiazine-1,1-dioxide heterocycles as hydrogen bond donating subunits in two new acyclic anion receptors. The two new receptors 1 and 2, were titrated with a number of anions and displayed very different complexation behaviour to the known disulfonamide receptor 3 that was used as a comparison. The difference can be attributed to the differing steric demands of the terminal phenyl rings in the two different receptor geometries despite the similar character of their central binding clefts. The steric effect of these rings in receptor 1 generates significant selectivity by the receptor for chloride versus the other, larger anions studied. The replacement of the central 1,3-benzenediyl spacer in 1 for 2,6-pyridinediyl in 2 greatly reduces the affinity of the resulting receptor for all of the anions examined and eliminates any selectivity for chloride. The synthetic approach described here can be easily adapted to the synthesis of oligomeric analogues of these two receptors that we expect will display an even greater selectivity for chloride anions and operate in more competitive solvent environments.
Supporting Information
Details of synthetic procedures, characterization data for intermediates and final products, and binding isotherms for receptors 1 and 2 with TBA salts of anions.
| Supporting Information File 1: Experimental details. | ||
| Format: PDF | Size: 349.6 KB | Download |
| Supporting Information File 2: X-ray crystal data for receptor 1. | ||
| Format: CIF | Size: 24.3 KB | Download |
| Supporting Information File 3: X-ray crystal data for receptor 2. | ||
| Format: CIF | Size: 23.3 KB | Download |
References
-
Sessler, J. L.; Gale, P. A.; Cho, W.-S. Anion Receptor Chemistry; Royal Society of Chemistry: Cambridge, U.K., 2006.
Return to citation in text: [1] -
Katayev, E. A.; Kolesnikov, G. V.; Sessler, J. L. Chem. Soc. Rev. 2009, 38, 1572–1586. doi:10.1039/b806468g
Return to citation in text: [1] -
Kubik, S. Chem. Soc. Rev. 2009, 38, 585–605. doi:10.1039/b810531f
Return to citation in text: [1] -
Kang, S. O.; Begum, R. A.; Bowman-James, K. Angew. Chem., Int. Ed. 2006, 45, 7882–7894. doi:10.1002/anie.200602006
Return to citation in text: [1] -
Zhang, Z.; Schreiner, P. R. Chem. Soc. Rev. 2009, 38, 1187–1198. doi:10.1039/b801793j
Return to citation in text: [1] -
Cho, W.-S.; Sessler, J. L. Artificial pyrrole-based anion receptors. In Functional Synthetic Receptors; Schrader, T.; Hamilton, A. D., Eds.; Wiley-VCH: Weinheim, Germany, 2005; pp 165–256. doi:10.1002/352760572X.ch4
Return to citation in text: [1] -
Gale, P. A. Chem. Commun. 2008, 4525–4540. doi:10.1039/b809508f
Return to citation in text: [1] -
López, M. V.; Bermejo, M. R.; Vásquez, M. E.; Taglietti, A.; Zaragoza, G.; Pedrido, R.; Martínez-Calvo, M. Org. Biomol. Chem. 2010, 8, 357–362. doi:10.1039/b916040j
Return to citation in text: [1] [2] -
Warr, R. J.; Westra, A. N.; Bell, K. J.; Chartres, J.; Ellis, R.; Tong, C.; Simmance, T. G.; Gadzhieva, A.; Blake, A. J.; Tasker, P. A.; Schröder, M. Chem.–Eur. J. 2009, 15, 4836–4850. doi:10.1002/chem.200802377
Return to citation in text: [1] [2] -
Kakuchi, R.; Kodama, T.; Shimada, R.; Tago, Y.; Sakai, R.; Satoh, T.; Kakuchi, T. Macromolecules 2009, 42, 3892–3897. doi:10.1021/ma900409h
Return to citation in text: [1] [2] -
Caltagirone, C.; Bates, G. W.; Gale, P. A.; Light, M. A. Chem. Commun. 2008, 61–63. doi:10.1039/b713431b
Return to citation in text: [1] [2] -
Berryman, O. B.; Johnson, C. A., II; Zakharov, L. N.; Haley, M. M.; Johnson, D. W. Angew. Chem., Int. Ed. 2008, 47, 117–120. doi:10.1002/anie.200703971
Return to citation in text: [1] [2] -
Shang, X. F.; Lin, H.; Lin, H. K. J. Fluorine Chem. 2007, 128, 530–534. doi:10.1016/j.jfluchem.2007.01.005
Return to citation in text: [1] [2] -
Wu, Y.; Peng, X.; Fan, J.; Gao, S.; Tian, M.; Zhao, J.; Sun, S. J. Org. Chem. 2007, 72, 62–70. doi:10.1021/jo061634c
Return to citation in text: [1] [2] -
Kavallieratos, K.; Sabucedo, A. J.; Pau, A. T.; Rodriguez, J. M. J. Am. Soc. Mass Spectrom. 2005, 16, 1377–1383. doi:10.1016/j.jasms.2005.04.008
Return to citation in text: [1] [2] -
Kondo, S.-I.; Suzuki, T.; Toyama, T.; Yano, Y. Bull. Chem. Soc. Jpn. 2005, 78, 1348–1350. doi:10.1246/bcsj.78.1348
Return to citation in text: [1] [2] -
Smith, B. D.; Lambert, T. N. Chem. Commun. 2003, 2261–2268. doi:10.1039/b303359g
Return to citation in text: [1] [2] -
Kondo, S.-I.; Suzuki, T.; Yano, Y. Tetrahedron Lett. 2002, 43, 7059–7061. doi:10.1016/S0040-4039(02)01543-5
Return to citation in text: [1] [2] -
Kavallieratos, K.; Moyer, B. A. Chem. Commun. 2001, 1620–1621. doi:10.1039/b102152b
Return to citation in text: [1] [2] -
Chrisstoffels, L. A. J.; de Jong, F.; Reinhoudt, D. N. Chem.–Eur. J. 2000, 6, 1376–1385. doi:10.1002/(SICI)1521-3765(20000417)6:8<1376::AID-CHEM1376>3.0.CO;2-I
Return to citation in text: [1] [2] -
Kavallieratos, K.; Bertao, C. M.; Crabtree, R. H. J. Org. Chem. 1999, 64, 1675–1683. doi:10.1021/jo982382l
Return to citation in text: [1] [2] [3] [4] -
Davis, A. P.; Perry, J. J.; Williams, R. P. J. Am. Chem. Soc. 1997, 119, 1793–1794. doi:10.1021/ja9629930
Return to citation in text: [1] [2] -
Morzherin, Y.; Rudkevich, D. M.; Verboom, W.; Reinhoudt, D. N. J. Org. Chem. 1993, 58, 7602–7605. doi:10.1021/jo00078a052
Return to citation in text: [1] [2] -
Li, J.; Wisner, J. A.; Jennings, M. C. Org. Lett. 2007, 9, 3267–3269. doi:10.1021/ol071171c
Return to citation in text: [1] -
Brooks, S. J.; Garcia-Garrido, S. E.; Light, M. E.; Cole, P. A.; Gale, P. A. Chem.–Eur. J. 2007, 13, 3320–3329. doi:10.1002/chem.200601647
Return to citation in text: [1] -
Schmuck, C.; Machon, U. Chem.–Eur. J. 2005, 11, 1109–1118. doi:10.1002/chem.200400652
Return to citation in text: [1] -
Hunter, C. A.; Purvis, D. A. Angew. Chem., Int. Ed. Engl. 1992, 31, 792. doi:10.1002/anie.199207921
Return to citation in text: [1] -
Wisner, J. A.; Li, J. unpublished results.
Return to citation in text: [1] -
Hynes, M. J. J. Chem. Soc., Dalton Trans. 1993, 311. doi:10.1039/DT9930000311
Return to citation in text: [1] -
Eagle, C. T.; Kavallieratos, K.; Bryan, J. C. J. Chem. Crystallogr. 2002, 32, 165. doi:10.1023/A:1020283701925
Return to citation in text: [1] -
Brownstein, S.; Stillman, A. E. J. Phys. Chem. 1959, 63, 2061–2062. doi:10.1021/j150582a022
Return to citation in text: [1]
| 1. | Sessler, J. L.; Gale, P. A.; Cho, W.-S. Anion Receptor Chemistry; Royal Society of Chemistry: Cambridge, U.K., 2006. |
| 2. | Katayev, E. A.; Kolesnikov, G. V.; Sessler, J. L. Chem. Soc. Rev. 2009, 38, 1572–1586. doi:10.1039/b806468g |
| 8. | López, M. V.; Bermejo, M. R.; Vásquez, M. E.; Taglietti, A.; Zaragoza, G.; Pedrido, R.; Martínez-Calvo, M. Org. Biomol. Chem. 2010, 8, 357–362. doi:10.1039/b916040j |
| 9. | Warr, R. J.; Westra, A. N.; Bell, K. J.; Chartres, J.; Ellis, R.; Tong, C.; Simmance, T. G.; Gadzhieva, A.; Blake, A. J.; Tasker, P. A.; Schröder, M. Chem.–Eur. J. 2009, 15, 4836–4850. doi:10.1002/chem.200802377 |
| 10. | Kakuchi, R.; Kodama, T.; Shimada, R.; Tago, Y.; Sakai, R.; Satoh, T.; Kakuchi, T. Macromolecules 2009, 42, 3892–3897. doi:10.1021/ma900409h |
| 11. | Caltagirone, C.; Bates, G. W.; Gale, P. A.; Light, M. A. Chem. Commun. 2008, 61–63. doi:10.1039/b713431b |
| 12. | Berryman, O. B.; Johnson, C. A., II; Zakharov, L. N.; Haley, M. M.; Johnson, D. W. Angew. Chem., Int. Ed. 2008, 47, 117–120. doi:10.1002/anie.200703971 |
| 13. | Shang, X. F.; Lin, H.; Lin, H. K. J. Fluorine Chem. 2007, 128, 530–534. doi:10.1016/j.jfluchem.2007.01.005 |
| 14. | Wu, Y.; Peng, X.; Fan, J.; Gao, S.; Tian, M.; Zhao, J.; Sun, S. J. Org. Chem. 2007, 72, 62–70. doi:10.1021/jo061634c |
| 15. | Kavallieratos, K.; Sabucedo, A. J.; Pau, A. T.; Rodriguez, J. M. J. Am. Soc. Mass Spectrom. 2005, 16, 1377–1383. doi:10.1016/j.jasms.2005.04.008 |
| 16. | Kondo, S.-I.; Suzuki, T.; Toyama, T.; Yano, Y. Bull. Chem. Soc. Jpn. 2005, 78, 1348–1350. doi:10.1246/bcsj.78.1348 |
| 17. | Smith, B. D.; Lambert, T. N. Chem. Commun. 2003, 2261–2268. doi:10.1039/b303359g |
| 18. | Kondo, S.-I.; Suzuki, T.; Yano, Y. Tetrahedron Lett. 2002, 43, 7059–7061. doi:10.1016/S0040-4039(02)01543-5 |
| 19. | Kavallieratos, K.; Moyer, B. A. Chem. Commun. 2001, 1620–1621. doi:10.1039/b102152b |
| 20. | Chrisstoffels, L. A. J.; de Jong, F.; Reinhoudt, D. N. Chem.–Eur. J. 2000, 6, 1376–1385. doi:10.1002/(SICI)1521-3765(20000417)6:8<1376::AID-CHEM1376>3.0.CO;2-I |
| 21. | Kavallieratos, K.; Bertao, C. M.; Crabtree, R. H. J. Org. Chem. 1999, 64, 1675–1683. doi:10.1021/jo982382l |
| 22. | Davis, A. P.; Perry, J. J.; Williams, R. P. J. Am. Chem. Soc. 1997, 119, 1793–1794. doi:10.1021/ja9629930 |
| 23. | Morzherin, Y.; Rudkevich, D. M.; Verboom, W.; Reinhoudt, D. N. J. Org. Chem. 1993, 58, 7602–7605. doi:10.1021/jo00078a052 |
| 31. | Brownstein, S.; Stillman, A. E. J. Phys. Chem. 1959, 63, 2061–2062. doi:10.1021/j150582a022 |
| 6. | Cho, W.-S.; Sessler, J. L. Artificial pyrrole-based anion receptors. In Functional Synthetic Receptors; Schrader, T.; Hamilton, A. D., Eds.; Wiley-VCH: Weinheim, Germany, 2005; pp 165–256. doi:10.1002/352760572X.ch4 |
| 7. | Gale, P. A. Chem. Commun. 2008, 4525–4540. doi:10.1039/b809508f |
| 5. | Zhang, Z.; Schreiner, P. R. Chem. Soc. Rev. 2009, 38, 1187–1198. doi:10.1039/b801793j |
| 21. | Kavallieratos, K.; Bertao, C. M.; Crabtree, R. H. J. Org. Chem. 1999, 64, 1675–1683. doi:10.1021/jo982382l |
| 3. | Kubik, S. Chem. Soc. Rev. 2009, 38, 585–605. doi:10.1039/b810531f |
| 4. | Kang, S. O.; Begum, R. A.; Bowman-James, K. Angew. Chem., Int. Ed. 2006, 45, 7882–7894. doi:10.1002/anie.200602006 |
| 30. | Eagle, C. T.; Kavallieratos, K.; Bryan, J. C. J. Chem. Crystallogr. 2002, 32, 165. doi:10.1023/A:1020283701925 |
| 25. | Brooks, S. J.; Garcia-Garrido, S. E.; Light, M. E.; Cole, P. A.; Gale, P. A. Chem.–Eur. J. 2007, 13, 3320–3329. doi:10.1002/chem.200601647 |
| 26. | Schmuck, C.; Machon, U. Chem.–Eur. J. 2005, 11, 1109–1118. doi:10.1002/chem.200400652 |
| 21. | Kavallieratos, K.; Bertao, C. M.; Crabtree, R. H. J. Org. Chem. 1999, 64, 1675–1683. doi:10.1021/jo982382l |
| 29. | Hynes, M. J. J. Chem. Soc., Dalton Trans. 1993, 311. doi:10.1039/DT9930000311 |
| 24. | Li, J.; Wisner, J. A.; Jennings, M. C. Org. Lett. 2007, 9, 3267–3269. doi:10.1021/ol071171c |
| 8. | López, M. V.; Bermejo, M. R.; Vásquez, M. E.; Taglietti, A.; Zaragoza, G.; Pedrido, R.; Martínez-Calvo, M. Org. Biomol. Chem. 2010, 8, 357–362. doi:10.1039/b916040j |
| 9. | Warr, R. J.; Westra, A. N.; Bell, K. J.; Chartres, J.; Ellis, R.; Tong, C.; Simmance, T. G.; Gadzhieva, A.; Blake, A. J.; Tasker, P. A.; Schröder, M. Chem.–Eur. J. 2009, 15, 4836–4850. doi:10.1002/chem.200802377 |
| 10. | Kakuchi, R.; Kodama, T.; Shimada, R.; Tago, Y.; Sakai, R.; Satoh, T.; Kakuchi, T. Macromolecules 2009, 42, 3892–3897. doi:10.1021/ma900409h |
| 11. | Caltagirone, C.; Bates, G. W.; Gale, P. A.; Light, M. A. Chem. Commun. 2008, 61–63. doi:10.1039/b713431b |
| 12. | Berryman, O. B.; Johnson, C. A., II; Zakharov, L. N.; Haley, M. M.; Johnson, D. W. Angew. Chem., Int. Ed. 2008, 47, 117–120. doi:10.1002/anie.200703971 |
| 13. | Shang, X. F.; Lin, H.; Lin, H. K. J. Fluorine Chem. 2007, 128, 530–534. doi:10.1016/j.jfluchem.2007.01.005 |
| 14. | Wu, Y.; Peng, X.; Fan, J.; Gao, S.; Tian, M.; Zhao, J.; Sun, S. J. Org. Chem. 2007, 72, 62–70. doi:10.1021/jo061634c |
| 15. | Kavallieratos, K.; Sabucedo, A. J.; Pau, A. T.; Rodriguez, J. M. J. Am. Soc. Mass Spectrom. 2005, 16, 1377–1383. doi:10.1016/j.jasms.2005.04.008 |
| 16. | Kondo, S.-I.; Suzuki, T.; Toyama, T.; Yano, Y. Bull. Chem. Soc. Jpn. 2005, 78, 1348–1350. doi:10.1246/bcsj.78.1348 |
| 17. | Smith, B. D.; Lambert, T. N. Chem. Commun. 2003, 2261–2268. doi:10.1039/b303359g |
| 18. | Kondo, S.-I.; Suzuki, T.; Yano, Y. Tetrahedron Lett. 2002, 43, 7059–7061. doi:10.1016/S0040-4039(02)01543-5 |
| 19. | Kavallieratos, K.; Moyer, B. A. Chem. Commun. 2001, 1620–1621. doi:10.1039/b102152b |
| 20. | Chrisstoffels, L. A. J.; de Jong, F.; Reinhoudt, D. N. Chem.–Eur. J. 2000, 6, 1376–1385. doi:10.1002/(SICI)1521-3765(20000417)6:8<1376::AID-CHEM1376>3.0.CO;2-I |
| 21. | Kavallieratos, K.; Bertao, C. M.; Crabtree, R. H. J. Org. Chem. 1999, 64, 1675–1683. doi:10.1021/jo982382l |
| 22. | Davis, A. P.; Perry, J. J.; Williams, R. P. J. Am. Chem. Soc. 1997, 119, 1793–1794. doi:10.1021/ja9629930 |
| 23. | Morzherin, Y.; Rudkevich, D. M.; Verboom, W.; Reinhoudt, D. N. J. Org. Chem. 1993, 58, 7602–7605. doi:10.1021/jo00078a052 |
| 27. | Hunter, C. A.; Purvis, D. A. Angew. Chem., Int. Ed. Engl. 1992, 31, 792. doi:10.1002/anie.199207921 |
© 2010 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)