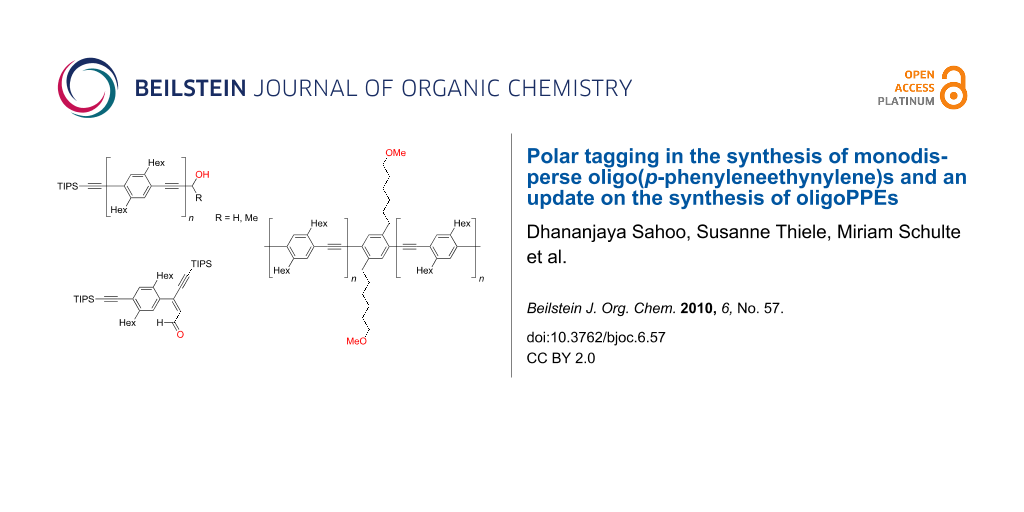
Abstract
One important access to monodisperse (functionalized) oligoPPEs is based on the orthogonality of the alkyne protecting groups triisopropylsilyl and hydroxymethyl (HOM) and on the polar tagging with the hydroxymethyl moiety for an easy chromatographic separation of the products. This paper provides an update of this synthetic route. For the deprotection of HOM protected alkynes, γ-MnO2 proved to be better than (highly) activated MnO2. The use of HOM as an alkyne protecting group is accompanied by carbometalation as a side reaction in the alkynyl–aryl coupling. The extent of carbometalation can be distinctly reduced through substitution of HOM for 1-hydroxyethyl. The strategy of polar tagging is extended by embedding ether linkages within the solubilising side chains. With building blocks such as 1,4-diiodo-2,5-bis(6-methoxyhexyl) less steps are needed to assemble oligoPPEs with functional end groups and the isolation of pure compounds becomes simple. For the preparation of 1,4-dialkyl-2,5-diiodobenzene a better procedure is presented together with the finding that 1,4-dialkyl-2,3-diiodobenzene, a constitutional isomer of 1,4-dialkyl-2,5-diiodobenzene, is one of the byproducts.

Graphical Abstract
Introduction
Oligo(p-phenyleneethynylene)s (oligoPPEs) have been frequently used as structural units for nanoscopic molecules because of their geometry and their electronic and photophysical properties [1-19]. For their preparation three widely used synthetic routes have emerged:
- The repeating unit by repeating unit approach (Scheme 1a) with desilylation and coupling of the (functionalized) arylethyne with 1-iodo-4-(2-trimethylsilylethynyl)benzene as the two repeating steps [6,9,20-24]. The oligomer grows slowly repeating unit by repeating unit. In the related bidirectional approach two repeating units are added in each coupling step (Scheme 1b) [25].
- The divergent-convergent Moore–Tour-route (Scheme 2a) [26-29] which employs the diethyltriazenyl group to mask an iodo substituent [30,31]. 1-(Diethyltriazenyl)-4-(2-trimethylsilylethynyl)benzene is the parent compound. Desilylation and exchange of the triazenyl substituent for an iodo substituent are the two divergent steps followed by the alkynyl–aryl coupling, the convergent step. The dialkyltriazenyl group decomposes during chromatography on silica gel [28].
- The divergent-convergent route which makes use of the orthogonality of the two alkyne protecting groups triisopropylsilyl (TIPS) and hydroxymethyl (HOM) (Scheme 2b) [32]. The reaction sequence starts with the HOM and TIPS protected 1,4-diethynylbenzene 1a1 from which the monoprotected 1,4-diethynylbenzenes 21 and 3a1 are derived by the removal of either the HOM or the TIPS group. The HOM protected 1,4-diethynylbenzene 3a1 is coupled with 1,4-diiodobenzene to obtain aryl iodide 4a2. This is coupled with the TIPS protected 1,4-diethynylbenzene 21 in the convergent step. It has been shown that HOM can be exchanged for 1-hydroxy-1-methylethyl (2-hydroxyprop-2-yl, HOP) [33-38].
![[1860-5397-6-57-i1]](/bjoc/content/inline/1860-5397-6-57-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of oligoPPEs by a unidirectional (a) or bidirectional (b) repeating unit by repeating unit approach. R denotes solubilising substituents.
Scheme 1: Synthesis of oligoPPEs by a unidirectional (a) or bidirectional (b) repeating unit by repeating uni...
A rather rarely utilized third divergent-convergent approach (Scheme 2c) [39-41] relies on the bromo iodo selectivity of the alkynyl–aryl coupling and bromo iodo exchange via halogen metal exchange.
The principles underlying these methods have been applied to building blocks with additional substituents including functional groups as well as to other aromatic building blocks, such as biphenyl [33], bipyridine [36,42], thiophene [36,43,44], fluorene [45], and triptycene [46], and other shapes, e.g. starlike compounds [2,7,12,34,37].
![[1860-5397-6-57-i2]](/bjoc/content/inline/1860-5397-6-57-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Three divergent-convergent routes to oligoPPEs. R denotes solubilising substituents such as hexyl.
Scheme 2: Three divergent-convergent routes to oligoPPEs. R denotes solubilising substituents such as hexyl.
The divergent-convergent route that employs the two orthogonal alkyne protecting groups TIPS and HOM (Scheme 2b) does not only allow the fast growth of oligomers – only four steps for doubling the number of repeating units with two of the four steps being experimentally extremely simple – but is especially satisfying because of a trouble-free separation of the desired alkynyl–aryl coupling product and the accompanying oxidative alkyne dimerization product (Glaser coupling product). In our experience, under the standard coupling conditions – i.e. Pd(PPh3)2Cl2, CuI, piperidine, THF, room temperature – Glaser coupling is much faster than the alkynyl–aryl coupling. Therefore, even traces of oxygen in the reaction vessel will lead to alkyne dimerization. Furthermore, most experimentalists prefer to work up the reaction mixtures under standard conditions which means exposing the reaction mixture to air. Opening the flask will immediately cause any unreacted terminal alkyne to undergo Glaser coupling. This is of no concern provided the alkyne dimer and the alkynyl–aryl coupling product can be easily separated, and this is what the HOM and related HOP group guarantee since they act as polar tags for the chromatographic separation. Polar tagging with HOM [47-49] or HOP [34,42,45,50-57] has been the key to the successful syntheses of a variety of aryleneethynylene building blocks and oligomers [42,45,47-52] and of oligoeneynes [53] including the natural marine compound callyberyne [54].
Since we disclosed this strategy several years have elapsed during which time we have gained more experience with it, became aware of some problems concerning it and improved it. Because the strategy has been adopted in whole or in part by other groups [33,34,47,49,58,59] and oligoPPEs are still a topic of great interest [1-19], we would like to share our results and present an update and an extension of our route in this paper. There are four issues that we want to address: (1) The type of MnO2 used for the removal of the HOM group, (2) carbometalation, a side reaction when using hydroxymethyl as an alkyne protecting group, (3) purity of 1,4-dihexyl-2,5-diiodobenzene, and (4) polar tags in the side chains of building blocks to reduce the number of steps in oligomer synthesis.
Results and Discussion
Type of MnO2 used for alkyne deprotection
The original paper on the oxidation-decarbonylation of HOM-protected alkynes [60,61] through treatment with MnO2 and powdered KOH does not contain any details about the type of MnO2. We applied this method to the synthesis of oligoPPEs, only exchanging benzene for diethyl ether, and obtained satisfactory results with commercially available active MnO2 (Aldrich) [32]. However, the deprotection of the HOM-protected arylalkynes 1an took several hours, especially when the reaction was run on larger scale, i.e. with 500 mg or more of starting material [62]. Even more annoying was that the required reaction time varied drastically from one experiment to another. The best procedure was to add portions of a mixture of MnO2 and KOH in intervals of 15 to 60 minutes until the reaction was complete. The reaction can be easily monitored by thin layer chromatography.
To improve the procedure, we tested the activated MnO2 (purchased; Aldrich), highly activated MnO2 (self-made) [63-65], BaMnO4 (purchased) [66-68], and γ-MnO2 (self-made) [63,64] on 3-(4-bromophenyl)prop-2-ynol in the presence of powdered KOH in diethyl ether at room temperature. The reaction with γ-MnO2 was the fastest. Even more important, γ-MnO2 when applied to the oligomers 1an proved to be highly reliable in its oxidizing power making it the reagent of choice for the removal of HOM groups. Some experimental results hint at a reduced activity of γ-MnO2 after it was stored for more than half a year in a closed jar under ambient conditions.
Carbometalation
When we published the synthesis of oligoPPEs via the divergent-convergent route which is based on the orthogonality of the alkyne protecting groups TIPS and HOM, we reported the carbometalation product 5a (Scheme 3) which formed as a side product in the coupling of iodo monomer 4a1 with TIPSethyne [32]. In the original procedure a reaction temperature of 50 °C was employed. Much later we found that the reaction goes to completion even at room temperature. Lowering the temperature reduced the amount of the carbometalation product 5a from 2–16% to 1–5%. Nevertheless, large scale preparative chromatographic separation on silica gel is tedious. Carbometalation product 5a and monomer 1a1 have very similar Rf-values and, unfortunately, the byproduct is eluted first.
![[1860-5397-6-57-i3]](/bjoc/content/inline/1860-5397-6-57-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the building blocks 11, 21, and 31. The depicted alkene configuration of 5 was chosen assuming a carbometalation process for the formation of 5 and thus a syn addition of the alkyne onto the hydroxypropyne moiety.
Scheme 3: Synthesis of the building blocks 11, 21, and 31. The depicted alkene configuration of 5 was chosen ...
Luckily, contamination of monomer 1a1 with carbometalation product 5a is of no concern if this material is used to prepare the TIPS protected 1,4-diethynylbenzene 21 (Scheme 3). Under standard reaction conditions – γ-MnO2, powdered KOH, Et2O, room temperature – the alcohol groups of both compounds 1a1 and 5a are oxidized. Whereas the oxidation product of 1a1, aldehyde 8a1, reacts with KOH to give the ethynyl anion and formic acid which immediately exchange a proton providing deprotected alkyne 21, the oxidation product of 5a, aldehyde 6a, is inert under the reaction conditions [69,70]. This finding is attributed to the higher energy content and therefore lower nucleofugicity of a vinyl anion as compared to an ethynyl anion. The products, alkyne 21 and the oxidized carbometalation product 6a, are easily separable by chromatography, which resembles a simple filtration through silica gel because 6a stays anchored on the solid phase through its polar carbonyl group. In this way pure alkyne 21 can be obtained even if carbometalation product 5a is present in the starting material.
The case is quite different when monomer 1a1 is used as the precursor for the HOM protected 1,4-diethynylbenzene 3a1. Treatment of a mixture of 1a1 and 5a with n-Bu4NF will not only remove the TIPS group of 1a1 but also the two TIPS groups of 5a (Scheme 3). The two products are as difficult to separate as the starting compounds. Furthermore, the ethynyl groups of both products are expected to have the same reactivity which can make the isolation of pure compounds of subsequent coupling reactions even more challenging. Finally, we found that carbometalation not only occurs during the preparation of monomer 1a1 and its trimethylsilyl (TMS)-analogue but also, even though extremely rarely, at later stages of the oligoPPE synthesis. For example, on one occasion we isolated compound 9a (2%, isolated yield) from the reaction between alkyne 21 and iodo monomer 4a1 which gave dimer 1a2 as the major product (79%, isolated yield) (Scheme 4).
![[1860-5397-6-57-i4]](/bjoc/content/inline/1860-5397-6-57-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Carbometalation, an occasionally detected side reaction. The depicted alkene configuration was chosen assuming a carbometalation process and thus a syn addition of the alkyne onto the hydroxypropyne moiety.
Scheme 4: Carbometalation, an occasionally detected side reaction. The depicted alkene configuration was chos...
In other reactions of this type, the carbometalation product may have remained undetected due to the limited sensitivity of 1H NMR spectroscopy, which we use as a routine method to assess the composition of the crude product and the chromatographic fractions, although the characteristic triplet at 6.38 ppm (J = 7 Hz) [71] arising from the vinyl proton of the carbometalation products is easily observed. If the 1H NMR spectrum displays the carbon satellites of an aromatic proton signal from the major product, the threshold for detection the carbometalation product is as low as 0.5%.
While compiling records on carbometalation of hydroxymethyl protected arylalkynes we never found any evidence for carbometalation of TMS or TIPS protected arylalkynes, even when conducting the aryl–alkyne coupling at 50 °C [72-75]. Possibly, the OH-group of the HOM group coordinates to the alkyne loaded Pd(II)-complex and thus acts as a directing and rate increasing group. It may as well be that the bulky trialkylsilyl substituents simply act as steric shields for the arylalkyne. If the latter is true, then the use of 1-hydroxyethyl (HOE) or HOP instead of HOM as polar protecting groups for alkynes could prevent carbometalation. Although HOP is a sterically more demanding group, we decided in favor of HOE because the removal of HOP requires refluxing in toluene for several hours in the presence of sodium hydride or potassium hydroxide [34,36-38,42,50-52,76] whereas we expected that HOE could be detached through treatment with γ-MnO2 and powdered KOH in diethyl ether at room temperature, i.e. under the same, comparatively mild reaction conditions that had been used for HOM protected alkynes.
For a comparison of the influence of HOM and HOE on the carbometalation, the iodo monomers 4a1 and 4b1 were coupled with TIPSethyne in THF and piperidine using Pd(PPh3)2Cl2 and CuI as the catalysts. A reaction temperature of 40 °C was chosen in order to boost carbometalation. These two experiments were carried out at the same time, thus providing the best basis for a comparison. After an aqueous workup, the reaction products were analyzed by 1H NMR spectroscopy. In both cases the conversion of 41 was complete and the main component of the crude product was the coupling product 11. The spectra gave no indication of the presence of carbometalation product 5b whereas carbometalation product 5a (ca. 3%) had formed. However, when another member of our group performed the coupling of TIPSethyne with HOE protected iodo monomer 4b1, he found a trace of carbometalation product 5b (1%; as determined by 1H NMR spectroscopy; the characteristic signal is the doublet at 6.15 ppm with J = 9 Hz in CDCl3 which is assigned to the vinyl proton) in his crude product, although the reaction had been performed at room temperature. The same co-worker generally obtains a comparatively large amount of carbometalation product. Thus, the extent to which carbometalation occurs varies with the operator. So far we have no clue what the relevant factor is. The conclusion is that the alkyne protecting group HOE reduces the amount of accompanying carbometalation product when compared with HOM, however it does not inhibit it completely.
As expected, the HOE group is as smoothly removed as the HOM group through treatment of the protected alkynes 1bn with γ-MnO2 and powdered KOH in diethyl ether at room temperature. This is illustrated for the conversion of hexamer 1b6 and heptamer 1b7 into the alkynes 26 (98% isolated yield) and 27 (90% isolated yield), respectively.
Iodination of 1,4-dihexylbenzene
The preparation of 1,4-dihexyl-2,5-diiodobenzene (10a) by the iodination of 1,4-dihexylbenzene with a mixture of iodine and potassium iodate in HOAc, H2SO4, and water at 70 °C [32,77,78] gave variable yields and on occasions failed. We obtained 10a much more reliably via iodination with iodine and periodic acid in HOAc, H2SO4, water, and dichloromethane at 70 °C [79,80]. Dichloromethane probably acts as a phase compatibiliser. Nevertheless, the reaction never went to completion: In all cases monoiodination product 11a was found (Scheme 5). Additionally, irrespective of which procedure was followed, the crude product always contained 1,4-dihexyl-2,3-diiodobenzene (12a) (ca. 3%), a constitutional isomer of 10a. The structure elucidation of these byproducts based on 1H NMR spectra is outlined in Supporting Information File 1.
![[1860-5397-6-57-i5]](/bjoc/content/inline/1860-5397-6-57-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Iodination of 1,4-dihexylbenzene.
Scheme 5: Iodination of 1,4-dihexylbenzene.
At least twofold recrystallization is needed to obtain material which contains less than 0.5% of these byproducts (as determined by 1H NMR spectroscopy. The 13C-satellites of the signal of the aromatic protons of 10a were used as the reference). Whereas monoiodination product 11a is of minor concern because it is monofunctional, the constitutional isomer 12a is a severe threat to the structural purity of oligoPPEs and especially polyPPEs. To illustrate this point let us assume that 10a and thereof derived 1,4-diethynyl-2,5-dihexylbenzene, both contaminated with only 0.1% of the respective constitutional isomers, are polymerized to give a polymer batch with an average polymerisation degree of 1000. The consequence is that on average each of the polymer chains in this sample will have a kink, i.e. a severe structural defect.
Polar tags in the side chain
In spite of its efficiency, the divergent-convergent synthesis of oligoPPEs involves considerable effort, especially as a result of the chromatography which is required after each alkynyl–aryl coupling. In the case of the synthesis of oligoPPEs with terminal functional groups, it is tempting to reduce the number of steps through the coupling of a diiodo compound with oligoPPEs which carry one functional group and have about half of the number of repeating units of the target compound. To give one concrete example (Scheme 6): Starting from 1a3 the synthesis of 14a through the coupling of 13 with diiodobenzene 10a (Scheme 6, route A) requires only two alkynyl–aryl couplings (four steps overall), whereas the alternative (Scheme 6, route B) via heptamer 1a7 would take three or four cross coupling reactions (seven or eight steps overall) [81,82].
![[1860-5397-6-57-i6]](/bjoc/content/inline/1860-5397-6-57-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Different routes to compound 14, a representative of the large group of functionalized oligoPPEs.
Scheme 6: Different routes to compound 14, a representative of the large group of functionalized oligoPPEs.
However, all of these routes will be plagued by the difficulty in separating 14a from the accompanying alkyne dimer (Glaser coupling product). These two products differ only in the number of repeating units. In our experience with functionalized oligoPPEs their chromatographic properties on silica gel are very weakly influenced by the number of the non polar repeating unit, 2,5-dihexyl-1,4-phenyleneethynylene, but dominated by the polar functional groups. Therefore, if the two products differ in the number of polar groups, chromatographic separation can become easy. This idea was put to the test for the shortest route, route A, by employing methoxyhexyl substituted diiodobenzene 10b instead of hexyl substituted diiodobenzene 10a. The two methoxy groups influence the chromatographic behaviour distinctly (Rf(14b) = 0.29, Rf(15) = 0.71; silica gel, CH2Cl2/n-pentane 6:4). The oxygen atoms are intentionally inserted remote from the polyconjugated backbone in order not to change the optical properties of the oligomers.
Polar tagging with e.g. the rather inert ether moiety within the side chains at a site distant from the backbone appears to us a generally useful concept for the synthesis of mesoscopic molecules which very often have unbranched or slightly branched alkyl substituents present for solubility reasons.
Supporting Information
Supporting information features the syntheses of compounds used for the discussed experiments, the detailed experimental procedures, and the structure elucidation of the products from the iodination of 1,4-dihexylbenzene.
| Supporting Information File 1: | ||
| Format: PDF | Size: 189.0 KB | Download |
References
-
Daniell, H. W.; Klotz, E. J. F.; Odell, B.; Claridge, T. D. W.; Anderson, H. L. Angew. Chem. 2007, 119, 6969–6972. doi:10.1002/ange.200702349
Return to citation in text: [1] [2] -
Tour, J. M. J. Org. Chem. 2007, 72, 7477–7496. doi:10.1021/jo070543s
Return to citation in text: [1] [2] [3] -
Hu, W.; Zhu, N.; Tang, W.; Zhao, D. Org. Lett. 2008, 10, 2669–2672. doi:10.1021/ol800753z
Return to citation in text: [1] [2] -
Blakskjær, P.; Gothelf, K. V. Org. Biomol. Chem. 2006, 4, 3442–3447. doi:10.1039/b605844b
Return to citation in text: [1] [2] -
Andersen, C. S.; Gothelf, K. V. Org. Biomol. Chem. 2009, 7, 58–60. doi:10.1039/b815099k
Return to citation in text: [1] [2] -
Ljungdahl, T.; Pettersson, K.; Albinsson, B.; Mårtensson, J. Eur. J. Org. Chem. 2006, 3087–3096. doi:10.1002/ejoc.200600240
Return to citation in text: [1] [2] [3] -
Zhao, Y.; Shirai, Y.; Slepkov, A. D.; Cheng, L.; Alemany, L. B.; Sasaki, T.; Hegmann, F. A.; Tour, J. M. Chem.–Eur. J. 2005, 11, 3643–3658. doi:10.1002/chem.200401198
Return to citation in text: [1] [2] [3] -
Huber, R.; Gonzáles, M. T.; Wu, S.; Langer, M.; Grunder, S.; Horhoiu, V.; Mayor, M.; Bryce, M. R.; Wang, C.; Jitchati, R.; Schönenberger, C.; Calame, M. J. Am. Chem. Soc. 2008, 130, 1080–1084. doi:10.1021/ja0767940
Return to citation in text: [1] [2] -
Mayr, A.; Srisailas, M.; Zhao, Q.; Gao, Y.; Hsieh, H.; Hoshmand-Kochi, M.; St. Fleur, N. Tetrahedron 2007, 63, 8206–8217. doi:10.1016/j.tet.2007.05.116
Return to citation in text: [1] [2] [3] -
Guo, X.; Whalley, A.; Klare, J. E.; Huang, L.; O’Brien, S.; Steigerwald, M.; Nuckolls, C. Nano Lett. 2007, 7, 1119–1122. doi:10.1021/nl070245a
Return to citation in text: [1] [2] -
Lu, Q.; Liu, K.; Zhang, H.; Du, Z.; Wang, X.; Wang, F. ACS Nano 2009, 3, 3861–3868. doi:10.1021/nn9012687
Return to citation in text: [1] [2] -
Jeschke, G.; Sajid, M.; Schulte, M.; Godt, A. Phys. Chem. Chem. Phys. 2009, 11, 6580–6591. doi:10.1039/b905724b
Return to citation in text: [1] [2] [3] -
Polyhach, Y.; Godt, A.; Bauer, C.; Jeschke, G. J. Magn. Reson. 2007, 185, 118–129. doi:10.1016/j.jmr.2006.11.012
Return to citation in text: [1] [2] -
Godt, A.; Schulte, M.; Zimmermann, H.; Jeschke, G. Angew. Chem. 2006, 118, 7722–7726. doi:10.1002/ange.200602807
Return to citation in text: [1] [2] -
Babgi, B.; Rigamonti, L.; Cifuentes, M. P.; Corkery, T. C.; Randles, M. D.; Schwich, T.; Petrie, S.; Stranger, R.; Teshome, A.; Asselberghs, I.; Clays, K.; Samoc, M.; Humphrey, M. G. J. Am. Chem. Soc. 2009, 131, 10293–10307. doi:10.1021/ja902793z
Return to citation in text: [1] [2] -
Ochi, T.; Yamaguchi, Y.; Wakamiya, T.; Matsubara, Y.; Yoshida, Z. Org. Biomol. Chem. 2008, 6, 1222–1231. doi:10.1039/b717832h
Return to citation in text: [1] [2] -
Albinsson, B.; Mårtensson, J. J. Photochem. Photobiol., C: Photochem. Rev. 2008, 9, 138–155. doi:10.1016/j.jphotochemrev.2008.01.002
Return to citation in text: [1] [2] -
Myahkostupov, M.; Piotrowiak, P.; Wang, D.; Galoppini, E. J. Phys. Chem. C 2007, 111, 2827–2829. doi:10.1021/jp0679257
Return to citation in text: [1] [2] -
Yatabe, T.; Suzuki, Y.; Kawanishi, Y. J. Mater. Chem. 2008, 18, 4468–4477. doi:10.1039/b808036d
Return to citation in text: [1] [2] -
Lavastre, O.; Ollivier, L.; Dixneuf, P. H.; Sibandhit, S. Tetrahedron 1996, 52, 5495–5504. doi:10.1016/0040-4020(96)00240-2
Return to citation in text: [1] -
Aujard, I.; Baltaze, J.-P.; Baudin, J.-B.; Cogné, E.; Ferrage, F.; Jullien, L.; Perez, E.; Prévost, V.; Qian, L. M.; Ruel, O. J. Am. Chem. Soc. 2001, 123, 8177–8188. doi:10.1021/ja010019h
Return to citation in text: [1] -
Hwang, J.-J.; Tour, J. M. Tetrahedron 2002, 58, 10387–10405. doi:10.1016/S0040-4020(02)01409-6
Return to citation in text: [1] -
Zhi, Y.-G.; Lai, S.-W.; Chan, Q. K.-W.; Law, Y.-C.; Tong, G. S.-M.; Che, C.-M. Eur. J. Org. Chem. 2006, 3125–3139. doi:10.1002/ejoc.200600103
Return to citation in text: [1] -
Nierengarten, J.-F.; Gu, T.; Hadziioannou, G.; Tsamouras, D.; Krasnikov, V. Helv. Chim. Acta 2004, 87, 2948–2966. doi:10.1002/hlca.200490266
Another stepwise approach consisting of coupling with 4-iodobenzaldehyde and conversion of the aldehyde group into an ethyne moiety via the Corey-Fuchs reaction.
Return to citation in text: [1] -
Huang, S.; Tour, J. M. Tetrahedron Lett. 1999, 40, 3347–3350. doi:10.1016/S0040-4039(99)00463-3
Return to citation in text: [1] -
Zhang, J.; Pesak, D. J.; Ludwick, J. L.; Moore, J. S. J. Am. Chem. Soc. 1994, 116, 4227–4239. doi:10.1021/ja00089a012
Return to citation in text: [1] -
Jones, L., II; Schumm, J. S.; Tour, J. M. J. Org. Chem. 1997, 62, 1388–1410. doi:10.1021/jo962336q
Return to citation in text: [1] -
Li, G.; Wang, X.; Wang, F. Tetrahedron Lett. 2005, 46, 8971–8973. doi:10.1016/j.tetlet.2005.10.113
Return to citation in text: [1] [2] -
Hortholary, C.; Coudret, C. J. Org. Chem. 2003, 68, 2167–2174. doi:10.1021/jo026735z
Return to citation in text: [1] -
The use of the triflate group for the alkynyl–aryl coupling and its masking as the precursory OH group offers an alternative that was applied to the synthesis of phenyleneethynylene dendrimers [31], however, not (yet) to the synthesis of oligoPPEs.
Return to citation in text: [1] -
Pan, Y.; Peng, Z.; Melinger, J. S. Tetrahedron 2003, 59, 5495–5506. doi:10.1016/S0040-4020(03)00827-5
Return to citation in text: [1] [2] -
Kukula, H.; Veit, S.; Godt, A. Eur. J. Org. Chem. 1999, 277–286. doi:10.1002/(SICI)1099-0690(199901)1999:1<277::AID-EJOC277>3.0.CO;2-R
Return to citation in text: [1] [2] [3] [4] -
Wang, C.; Batsanov, A. S.; Bryce, M. R. J. Org. Chem. 2006, 71, 108–116. doi:10.1021/jo051711o
Return to citation in text: [1] [2] [3] -
Chandra, K. L.; Zhang, S.; Gorman, C. B. Tetrahedron 2007, 63, 7120–7132. doi:10.1016/j.tet.2007.05.006
Return to citation in text: [1] [2] [3] [4] [5] -
We like to call attention to the recent reports that trimethylsilyl and HOP are orthogonal alkyne protecting groups which make HOP a very interesting protecting group [36,37]. The same is true for tert-butyldimethylsilyl and HOP [38].
Return to citation in text: [1] -
Goeb, S.; De Nicola, A.; Ziessel, R. J. Org. Chem. 2005, 70, 1518–1529. doi:10.1021/jo048435i
Return to citation in text: [1] [2] [3] [4] [5] -
Rodríguez, J. G.; Esquivias, J.; Lafuente, A.; Díaz, C. J. Org. Chem. 2003, 68, 8120–8128. doi:10.1021/jo034972b
Return to citation in text: [1] [2] [3] [4] -
Shimizu, H.; Fujimoto, K.; Furusyo, M.; Maeda, H.; Nanai, Y.; Mizuno, K.; Inouye, M. J. Org. Chem. 2007, 72, 1530–1533. doi:10.1021/jo061959t
Return to citation in text: [1] [2] [3] -
Ziener, U.; Godt, A. J. Org. Chem. 1997, 62, 6137–6143. doi:10.1021/jo970548x
Return to citation in text: [1] -
Acharya, J. R.; Zhang, H.; Li, X.; Nesterov, E. E. J. Am. Chem. Soc. 2009, 131, 880–881. doi:10.1021/ja807621z
Return to citation in text: [1] -
Hsung, R. P.; Chidsey, C. E. D.; Sita, L. R. Organometallics 1995, 14, 4808–4815. doi:10.1021/om00010a049
Return to citation in text: [1] -
Ley, K. D.; Li, Y.; Johnson, J. V.; Powell, D. H.; Schanze, K. S. Chem. Commun. 1999, 1749–1750. doi:10.1039/a903476e
Return to citation in text: [1] [2] [3] [4] -
Li, G.; Wang, X.; Li, J.; Zhao, X.; Wang, F. Tetrahedron 2006, 62, 2576–2582. doi:10.1016/j.tet.2005.12.043
Return to citation in text: [1] -
Pearson, D. L.; Tour, J. M. J. Org. Chem. 1997, 62, 1376–1387. doi:10.1021/jo962335y
Return to citation in text: [1] -
Zeng, X.; Wang, C.; Bryce, M. R.; Batsanov, A. S.; Sirichantaropass, S.; García-Suárez, V. M.; Lambert, C. J.; Sage, I. Eur. J. Org. Chem. 2007, 5244–5249. doi:10.1002/ejoc.200700507
Return to citation in text: [1] [2] [3] -
Maag, D.; Kottke, T.; Schulte, M.; Godt, A. J. Org. Chem. 2009, 74, 7733–7742. doi:10.1021/jo9009744
Return to citation in text: [1] -
Yang, J.; Ng, M.-K. Synthesis 2006, 3075–3079. doi:10.1055/s-2006-942529
Return to citation in text: [1] [2] [3] -
Robinson, J. M. A.; Kariuki, B. M.; Harris, K. D. M.; Philp, D. J. Chem. Soc., Perkin Trans. 2 1998, 2459–2469. doi:10.1039/a804676j
Return to citation in text: [1] [2] -
Harriman, A.; Mallon, L.; Ziessel, R. Chem.–Eur. J. 2008, 14, 11461–11473. doi:10.1002/chem.200801384
Return to citation in text: [1] [2] [3] -
Zhao, Z.; Yu, S.; Xu, L.; Wang, H.; Lu, P. Tetrahedron 2007, 63, 7809–7815. doi:10.1016/j.tet.2007.05.095
Return to citation in text: [1] [2] [3] -
Rodríguez, J. G.; Tejedor, J. L. J. Org. Chem. 2002, 67, 7631–7640. doi:10.1021/jo0203589
Return to citation in text: [1] [2] [3] -
Kaneko, T.; Horie, T.; Matsumoto, S.; Teraguchi, M.; Aoki, T. Macromol. Chem. Phys. 2009, 210, 22–36. doi:10.1002/macp.200800429
Return to citation in text: [1] [2] [3] -
Takayama, Y.; Delas, C.; Muraoka, K.; Uemura, M.; Sato, F. J. Am. Chem. Soc. 2003, 125, 14163–14167. doi:10.1021/ja037975e
Return to citation in text: [1] [2] -
López, S.; Fernández-Trillo, F.; Midón, P.; Castedo, L.; Saá, C. J. Org. Chem. 2006, 71, 2802–2810. doi:10.1021/jo052609u
Return to citation in text: [1] [2] -
Silicon based alkyne protecting groups with a cyano group as the polar tag [54,56,57].
Return to citation in text: [1] -
Höger, S.; Bonrad, K. J. Org. Chem. 2000, 65, 2243–2245. doi:10.1021/jo991746m
Return to citation in text: [1] [2] -
Gaefke, G.; Höger, S. Synthesis 2008, 2155–2157. doi:10.1055/s-2008-1067141
Return to citation in text: [1] [2] -
Keller, J. M.; Schanze, K. S. Organometallics 2009, 28, 4210–4216. doi:10.1021/om900195p
Return to citation in text: [1] -
Weibel, N.; Mishchenko, A.; Wandlowski, T.; Neuburger, M.; Leroux, Y.; Mayor, M. Eur. J. Org. Chem. 2009, 6140–6150. doi:10.1002/ejoc.200900751
Return to citation in text: [1] -
Bumagin, N. A.; Ponomaryov, A. B.; Beletskaya, I. P. Synthesis 1984, 728–729. doi:10.1055/s-1984-30947
Return to citation in text: [1] -
Atkinson, R. E.; Curtis, R. F.; Jones, D. M.; Taylor, J. A. J. Chem. Soc. C 1969, 2173–2176. doi:10.1039/J39690002173
The suggestion to use the HOM group as a protecting group that can be removed via oxidation–decarbonylation was made much earlier here.
Return to citation in text: [1] -
The largely different reaction times given in the experimental part of [48] probably indicate that the experimenters of that reference experienced the same difficulties.
Return to citation in text: [1] -
Burke, S. D.; Danheiser, R. L., Eds. Handbook of Reagents for Organic Synthesis, Oxidizing and Reducing Agents; Wiley: Chichester, U.K., 2004; p 232.
Return to citation in text: [1] [2] -
Fatiadi, A. J. Synthesis 1976, 65–104. doi:10.1055/s-1976-23961
Return to citation in text: [1] [2] -
For the preparation of highly activated MnO2 we followed the procedure given in [63]. In this procedure less MnCl2 • 4 H2O (200 g vs. 220 g) but the same amount of KMnO4 (160 g) and of solvent was used in comparison to the procedure given in [64].
Return to citation in text: [1] -
As purchased; 80% technical grade.
Return to citation in text: [1] -
Fatiadi, A. J. Synthesis 1987, 85–127. doi:10.1055/s-1987-27859
Return to citation in text: [1] -
Firouzabadi, H.; Ghaderi, E. Tetrahedron Lett. 1978, 19, 839–840. doi:10.1016/S0040-4039(01)85412-5
Return to citation in text: [1] -
Experimental proof: Carbometalation product 5a was treated with γ-MnO2 and powdered KOH in diethylether at room temperature. The 1H NMR spectrum of the crude product shows unambigously the signals of the expected aldehyde 6a. There are no signals that fit to the characteristic signals of the unsymmetrically 1,1-disubstituted alkene 7, the product in case 6a had lost the formyl group.
The two sets of 1H NMR signals in an intensity ratio of 22:1 for the aryl protons, the aldehyde proton, and the vinyl proton indicate a mixture of E- and Z-alkene. Alkene isomerization upon oxidation with MnO2 has been reported [70]. The alkene isomerization can also be explained by a reversible addition of hydroxide to the electron acceptor substituted alkene of aldehyde 6a. Characteristic signals of the major isomer: δ = 9.38 (d, J = 8.2 Hz, 1 H, CHO), 7.32 and 7.00 (2 s, 1 H each, ArH), 6.47 (d, J = 8.2 Hz, 1 H, C=CH); Characteristic signals of the minor isomer: δ = 10.28 (d, J = 8.2 Hz, 1 H, CHO), 7.30 and 7.09 (2 s, 1 H each, ArH), 6.31 (d, J = 8.2 Hz, 1 H, C=CH).
Return to citation in text: [1] -
Burrell, J. W. K.; Garwood, R. F.; Jackman, L. M.; Oskay, E.; Weedon, B. C. L. J. Chem. Soc. C 1966, 2144–2154. doi:10.1039/J39660002144
Return to citation in text: [1] [2] -
Characteristic 1H NMR signals of the carbometalation product 9a in CDCl3: δ = 7.37, 7.31, 7.29, 7.22, 7.17, 6.98 (6 s, 1 H each, ArH), 6.38 (t, J = 7 Hz, 1 H, C=CHCH2OH), 4.06 (t-shaped signal, slightly broadened, J = 6 Hz, 2 H, CH2OH).
Return to citation in text: [1] -
We know of only two publications that report on carbometalation of 1-aryl-2-trialkylsilylethynes [73,74]. The carbometalation reported in [73] is possibly induced by the hydroxy group of the hydroxymethyl substituent in ortho-position to the 2-(trimethylsilyl)ethynyl group. Note also the related Pd-catalyzed addition of TMSethyne onto 3-(trimethylsilyl)prop-2-ynol giving Z-2-(2-trimethylsilylethynyl)-3-trimethylsilylprop-2-enol in [75].
Return to citation in text: [1] -
Stará, I. G.; Starý, I.; Kollárovič, A.; Teplý, F.; Šaman, D.; Fiedler, P. Tetrahedron 1998, 54, 11209–11234. doi:10.1016/S0040-4020(98)00655-3
Return to citation in text: [1] [2] [3] -
Batenburg-Nguyen, B.; Ung, A. T.; Pyne, S. G. Tetrahedron 2009, 65, 318–327. doi:10.1016/j.tet.2008.10.052
Return to citation in text: [1] [2] -
Trost, B. M.; McIntosh, M. C. Tetrahedron Lett. 1997, 38, 3207–3210. doi:10.1016/S0040-4039(97)00614-X
Return to citation in text: [1] [2] -
Mal’kina, A. G.; Brandsma, L.; Vasilevsky, S. F.; Trofimov, B. A. Synthesis 1996, 589–590. doi:10.1055/s-1996-4265
Return to citation in text: [1] -
McQuade, D. T.; Kim, J.; Swager, T. M. J. Am. Chem. Soc. 2000, 122, 5885–5886. doi:10.1021/ja000553+
Return to citation in text: [1] -
Werz, D. B.; Fischer, F. R.; Kornmayer, S. C.; Rominger, F.; Gleiter, R. J. Org. Chem. 2008, 73, 8021–8029. doi:10.1021/jo801378p
Return to citation in text: [1] -
The use of CCl4 instead of CH2Cl2 as described in [80] for iodination of 1,4-dialkoxybenzene resulted in an even higher conversion. However, the aromatic substitution reaction was no longer the only occuring reaction as especially the signals around 6 ppm in the 1H NMR spectrum of the crude product reveal.
Return to citation in text: [1] -
Bao, Z.; Chen, Y.; Cai, R.; Yu, L. Macromolecules 1993, 26, 5281–5286. doi:10.1021/ma00072a002
Return to citation in text: [1] [2] -
The eight step synthesis is outlined in analogy to the synthesis of the corresponding oligoPPE diesters in [82]. The eight step route is to be preferred over the seven step route because of the instability of oligoPPEs with two unprotected terminal ethyne moieties.
Return to citation in text: [1] -
Hensel, V.; Godt, A.; Popovitz-Biro, R.; Cohen, H.; Jensen, T. R.; Kjaer, K.; Weissbuch, I.; Lifshitz, E.; Lahav, M. Chem.–Eur. J. 2002, 8, 1413–1423. doi:10.1002/1521-3765(20020315)8:6<1413::AID-CHEM1413>3.0.CO;2-6
Return to citation in text: [1] [2]
| 63. | Burke, S. D.; Danheiser, R. L., Eds. Handbook of Reagents for Organic Synthesis, Oxidizing and Reducing Agents; Wiley: Chichester, U.K., 2004; p 232. |
| 64. | Fatiadi, A. J. Synthesis 1976, 65–104. doi:10.1055/s-1976-23961 |
| 32. | Kukula, H.; Veit, S.; Godt, A. Eur. J. Org. Chem. 1999, 277–286. doi:10.1002/(SICI)1099-0690(199901)1999:1<277::AID-EJOC277>3.0.CO;2-R |
| 69. |
Experimental proof: Carbometalation product 5a was treated with γ-MnO2 and powdered KOH in diethylether at room temperature. The 1H NMR spectrum of the crude product shows unambigously the signals of the expected aldehyde 6a. There are no signals that fit to the characteristic signals of the unsymmetrically 1,1-disubstituted alkene 7, the product in case 6a had lost the formyl group.
The two sets of 1H NMR signals in an intensity ratio of 22:1 for the aryl protons, the aldehyde proton, and the vinyl proton indicate a mixture of E- and Z-alkene. Alkene isomerization upon oxidation with MnO2 has been reported [70]. The alkene isomerization can also be explained by a reversible addition of hydroxide to the electron acceptor substituted alkene of aldehyde 6a. Characteristic signals of the major isomer: δ = 9.38 (d, J = 8.2 Hz, 1 H, CHO), 7.32 and 7.00 (2 s, 1 H each, ArH), 6.47 (d, J = 8.2 Hz, 1 H, C=CH); Characteristic signals of the minor isomer: δ = 10.28 (d, J = 8.2 Hz, 1 H, CHO), 7.30 and 7.09 (2 s, 1 H each, ArH), 6.31 (d, J = 8.2 Hz, 1 H, C=CH). |
| 70. | Burrell, J. W. K.; Garwood, R. F.; Jackman, L. M.; Oskay, E.; Weedon, B. C. L. J. Chem. Soc. C 1966, 2144–2154. doi:10.1039/J39660002144 |
| 31. | Pan, Y.; Peng, Z.; Melinger, J. S. Tetrahedron 2003, 59, 5495–5506. doi:10.1016/S0040-4020(03)00827-5 |
| 36. | Goeb, S.; De Nicola, A.; Ziessel, R. J. Org. Chem. 2005, 70, 1518–1529. doi:10.1021/jo048435i |
| 37. | Rodríguez, J. G.; Esquivias, J.; Lafuente, A.; Díaz, C. J. Org. Chem. 2003, 68, 8120–8128. doi:10.1021/jo034972b |
| 79. | The use of CCl4 instead of CH2Cl2 as described in [80] for iodination of 1,4-dialkoxybenzene resulted in an even higher conversion. However, the aromatic substitution reaction was no longer the only occuring reaction as especially the signals around 6 ppm in the 1H NMR spectrum of the crude product reveal. |
| 80. | Bao, Z.; Chen, Y.; Cai, R.; Yu, L. Macromolecules 1993, 26, 5281–5286. doi:10.1021/ma00072a002 |
| 81. | The eight step synthesis is outlined in analogy to the synthesis of the corresponding oligoPPE diesters in [82]. The eight step route is to be preferred over the seven step route because of the instability of oligoPPEs with two unprotected terminal ethyne moieties. |
| 82. | Hensel, V.; Godt, A.; Popovitz-Biro, R.; Cohen, H.; Jensen, T. R.; Kjaer, K.; Weissbuch, I.; Lifshitz, E.; Lahav, M. Chem.–Eur. J. 2002, 8, 1413–1423. doi:10.1002/1521-3765(20020315)8:6<1413::AID-CHEM1413>3.0.CO;2-6 |
| 34. | Chandra, K. L.; Zhang, S.; Gorman, C. B. Tetrahedron 2007, 63, 7120–7132. doi:10.1016/j.tet.2007.05.006 |
| 36. | Goeb, S.; De Nicola, A.; Ziessel, R. J. Org. Chem. 2005, 70, 1518–1529. doi:10.1021/jo048435i |
| 37. | Rodríguez, J. G.; Esquivias, J.; Lafuente, A.; Díaz, C. J. Org. Chem. 2003, 68, 8120–8128. doi:10.1021/jo034972b |
| 38. | Shimizu, H.; Fujimoto, K.; Furusyo, M.; Maeda, H.; Nanai, Y.; Mizuno, K.; Inouye, M. J. Org. Chem. 2007, 72, 1530–1533. doi:10.1021/jo061959t |
| 42. | Ley, K. D.; Li, Y.; Johnson, J. V.; Powell, D. H.; Schanze, K. S. Chem. Commun. 1999, 1749–1750. doi:10.1039/a903476e |
| 50. | Zhao, Z.; Yu, S.; Xu, L.; Wang, H.; Lu, P. Tetrahedron 2007, 63, 7809–7815. doi:10.1016/j.tet.2007.05.095 |
| 51. | Rodríguez, J. G.; Tejedor, J. L. J. Org. Chem. 2002, 67, 7631–7640. doi:10.1021/jo0203589 |
| 52. | Kaneko, T.; Horie, T.; Matsumoto, S.; Teraguchi, M.; Aoki, T. Macromol. Chem. Phys. 2009, 210, 22–36. doi:10.1002/macp.200800429 |
| 76. | Mal’kina, A. G.; Brandsma, L.; Vasilevsky, S. F.; Trofimov, B. A. Synthesis 1996, 589–590. doi:10.1055/s-1996-4265 |
| 32. | Kukula, H.; Veit, S.; Godt, A. Eur. J. Org. Chem. 1999, 277–286. doi:10.1002/(SICI)1099-0690(199901)1999:1<277::AID-EJOC277>3.0.CO;2-R |
| 77. | McQuade, D. T.; Kim, J.; Swager, T. M. J. Am. Chem. Soc. 2000, 122, 5885–5886. doi:10.1021/ja000553+ |
| 78. | Werz, D. B.; Fischer, F. R.; Kornmayer, S. C.; Rominger, F.; Gleiter, R. J. Org. Chem. 2008, 73, 8021–8029. doi:10.1021/jo801378p |
| 71. | Characteristic 1H NMR signals of the carbometalation product 9a in CDCl3: δ = 7.37, 7.31, 7.29, 7.22, 7.17, 6.98 (6 s, 1 H each, ArH), 6.38 (t, J = 7 Hz, 1 H, C=CHCH2OH), 4.06 (t-shaped signal, slightly broadened, J = 6 Hz, 2 H, CH2OH). |
| 72. | We know of only two publications that report on carbometalation of 1-aryl-2-trialkylsilylethynes [73,74]. The carbometalation reported in [73] is possibly induced by the hydroxy group of the hydroxymethyl substituent in ortho-position to the 2-(trimethylsilyl)ethynyl group. Note also the related Pd-catalyzed addition of TMSethyne onto 3-(trimethylsilyl)prop-2-ynol giving Z-2-(2-trimethylsilylethynyl)-3-trimethylsilylprop-2-enol in [75]. |
| 73. | Stará, I. G.; Starý, I.; Kollárovič, A.; Teplý, F.; Šaman, D.; Fiedler, P. Tetrahedron 1998, 54, 11209–11234. doi:10.1016/S0040-4020(98)00655-3 |
| 74. | Batenburg-Nguyen, B.; Ung, A. T.; Pyne, S. G. Tetrahedron 2009, 65, 318–327. doi:10.1016/j.tet.2008.10.052 |
| 75. | Trost, B. M.; McIntosh, M. C. Tetrahedron Lett. 1997, 38, 3207–3210. doi:10.1016/S0040-4039(97)00614-X |
| 38. | Shimizu, H.; Fujimoto, K.; Furusyo, M.; Maeda, H.; Nanai, Y.; Mizuno, K.; Inouye, M. J. Org. Chem. 2007, 72, 1530–1533. doi:10.1021/jo061959t |
| 54. | López, S.; Fernández-Trillo, F.; Midón, P.; Castedo, L.; Saá, C. J. Org. Chem. 2006, 71, 2802–2810. doi:10.1021/jo052609u |
| 56. | Höger, S.; Bonrad, K. J. Org. Chem. 2000, 65, 2243–2245. doi:10.1021/jo991746m |
| 57. | Gaefke, G.; Höger, S. Synthesis 2008, 2155–2157. doi:10.1055/s-2008-1067141 |
| 48. | Robinson, J. M. A.; Kariuki, B. M.; Harris, K. D. M.; Philp, D. J. Chem. Soc., Perkin Trans. 2 1998, 2459–2469. doi:10.1039/a804676j |
| 80. | Bao, Z.; Chen, Y.; Cai, R.; Yu, L. Macromolecules 1993, 26, 5281–5286. doi:10.1021/ma00072a002 |
| 82. | Hensel, V.; Godt, A.; Popovitz-Biro, R.; Cohen, H.; Jensen, T. R.; Kjaer, K.; Weissbuch, I.; Lifshitz, E.; Lahav, M. Chem.–Eur. J. 2002, 8, 1413–1423. doi:10.1002/1521-3765(20020315)8:6<1413::AID-CHEM1413>3.0.CO;2-6 |
| 73. | Stará, I. G.; Starý, I.; Kollárovič, A.; Teplý, F.; Šaman, D.; Fiedler, P. Tetrahedron 1998, 54, 11209–11234. doi:10.1016/S0040-4020(98)00655-3 |
| 75. | Trost, B. M.; McIntosh, M. C. Tetrahedron Lett. 1997, 38, 3207–3210. doi:10.1016/S0040-4039(97)00614-X |
| 70. | Burrell, J. W. K.; Garwood, R. F.; Jackman, L. M.; Oskay, E.; Weedon, B. C. L. J. Chem. Soc. C 1966, 2144–2154. doi:10.1039/J39660002144 |
| 73. | Stará, I. G.; Starý, I.; Kollárovič, A.; Teplý, F.; Šaman, D.; Fiedler, P. Tetrahedron 1998, 54, 11209–11234. doi:10.1016/S0040-4020(98)00655-3 |
| 74. | Batenburg-Nguyen, B.; Ung, A. T.; Pyne, S. G. Tetrahedron 2009, 65, 318–327. doi:10.1016/j.tet.2008.10.052 |
| 63. | Burke, S. D.; Danheiser, R. L., Eds. Handbook of Reagents for Organic Synthesis, Oxidizing and Reducing Agents; Wiley: Chichester, U.K., 2004; p 232. |
| 1. | Daniell, H. W.; Klotz, E. J. F.; Odell, B.; Claridge, T. D. W.; Anderson, H. L. Angew. Chem. 2007, 119, 6969–6972. doi:10.1002/ange.200702349 |
| 2. | Tour, J. M. J. Org. Chem. 2007, 72, 7477–7496. doi:10.1021/jo070543s |
| 3. | Hu, W.; Zhu, N.; Tang, W.; Zhao, D. Org. Lett. 2008, 10, 2669–2672. doi:10.1021/ol800753z |
| 4. | Blakskjær, P.; Gothelf, K. V. Org. Biomol. Chem. 2006, 4, 3442–3447. doi:10.1039/b605844b |
| 5. | Andersen, C. S.; Gothelf, K. V. Org. Biomol. Chem. 2009, 7, 58–60. doi:10.1039/b815099k |
| 6. | Ljungdahl, T.; Pettersson, K.; Albinsson, B.; Mårtensson, J. Eur. J. Org. Chem. 2006, 3087–3096. doi:10.1002/ejoc.200600240 |
| 7. | Zhao, Y.; Shirai, Y.; Slepkov, A. D.; Cheng, L.; Alemany, L. B.; Sasaki, T.; Hegmann, F. A.; Tour, J. M. Chem.–Eur. J. 2005, 11, 3643–3658. doi:10.1002/chem.200401198 |
| 8. | Huber, R.; Gonzáles, M. T.; Wu, S.; Langer, M.; Grunder, S.; Horhoiu, V.; Mayor, M.; Bryce, M. R.; Wang, C.; Jitchati, R.; Schönenberger, C.; Calame, M. J. Am. Chem. Soc. 2008, 130, 1080–1084. doi:10.1021/ja0767940 |
| 9. | Mayr, A.; Srisailas, M.; Zhao, Q.; Gao, Y.; Hsieh, H.; Hoshmand-Kochi, M.; St. Fleur, N. Tetrahedron 2007, 63, 8206–8217. doi:10.1016/j.tet.2007.05.116 |
| 10. | Guo, X.; Whalley, A.; Klare, J. E.; Huang, L.; O’Brien, S.; Steigerwald, M.; Nuckolls, C. Nano Lett. 2007, 7, 1119–1122. doi:10.1021/nl070245a |
| 11. | Lu, Q.; Liu, K.; Zhang, H.; Du, Z.; Wang, X.; Wang, F. ACS Nano 2009, 3, 3861–3868. doi:10.1021/nn9012687 |
| 12. | Jeschke, G.; Sajid, M.; Schulte, M.; Godt, A. Phys. Chem. Chem. Phys. 2009, 11, 6580–6591. doi:10.1039/b905724b |
| 13. | Polyhach, Y.; Godt, A.; Bauer, C.; Jeschke, G. J. Magn. Reson. 2007, 185, 118–129. doi:10.1016/j.jmr.2006.11.012 |
| 14. | Godt, A.; Schulte, M.; Zimmermann, H.; Jeschke, G. Angew. Chem. 2006, 118, 7722–7726. doi:10.1002/ange.200602807 |
| 15. | Babgi, B.; Rigamonti, L.; Cifuentes, M. P.; Corkery, T. C.; Randles, M. D.; Schwich, T.; Petrie, S.; Stranger, R.; Teshome, A.; Asselberghs, I.; Clays, K.; Samoc, M.; Humphrey, M. G. J. Am. Chem. Soc. 2009, 131, 10293–10307. doi:10.1021/ja902793z |
| 16. | Ochi, T.; Yamaguchi, Y.; Wakamiya, T.; Matsubara, Y.; Yoshida, Z. Org. Biomol. Chem. 2008, 6, 1222–1231. doi:10.1039/b717832h |
| 17. | Albinsson, B.; Mårtensson, J. J. Photochem. Photobiol., C: Photochem. Rev. 2008, 9, 138–155. doi:10.1016/j.jphotochemrev.2008.01.002 |
| 18. | Myahkostupov, M.; Piotrowiak, P.; Wang, D.; Galoppini, E. J. Phys. Chem. C 2007, 111, 2827–2829. doi:10.1021/jp0679257 |
| 19. | Yatabe, T.; Suzuki, Y.; Kawanishi, Y. J. Mater. Chem. 2008, 18, 4468–4477. doi:10.1039/b808036d |
| 30. | The use of the triflate group for the alkynyl–aryl coupling and its masking as the precursory OH group offers an alternative that was applied to the synthesis of phenyleneethynylene dendrimers [31], however, not (yet) to the synthesis of oligoPPEs. |
| 31. | Pan, Y.; Peng, Z.; Melinger, J. S. Tetrahedron 2003, 59, 5495–5506. doi:10.1016/S0040-4020(03)00827-5 |
| 2. | Tour, J. M. J. Org. Chem. 2007, 72, 7477–7496. doi:10.1021/jo070543s |
| 7. | Zhao, Y.; Shirai, Y.; Slepkov, A. D.; Cheng, L.; Alemany, L. B.; Sasaki, T.; Hegmann, F. A.; Tour, J. M. Chem.–Eur. J. 2005, 11, 3643–3658. doi:10.1002/chem.200401198 |
| 12. | Jeschke, G.; Sajid, M.; Schulte, M.; Godt, A. Phys. Chem. Chem. Phys. 2009, 11, 6580–6591. doi:10.1039/b905724b |
| 34. | Chandra, K. L.; Zhang, S.; Gorman, C. B. Tetrahedron 2007, 63, 7120–7132. doi:10.1016/j.tet.2007.05.006 |
| 37. | Rodríguez, J. G.; Esquivias, J.; Lafuente, A.; Díaz, C. J. Org. Chem. 2003, 68, 8120–8128. doi:10.1021/jo034972b |
| 26. | Zhang, J.; Pesak, D. J.; Ludwick, J. L.; Moore, J. S. J. Am. Chem. Soc. 1994, 116, 4227–4239. doi:10.1021/ja00089a012 |
| 27. | Jones, L., II; Schumm, J. S.; Tour, J. M. J. Org. Chem. 1997, 62, 1388–1410. doi:10.1021/jo962336q |
| 28. | Li, G.; Wang, X.; Wang, F. Tetrahedron Lett. 2005, 46, 8971–8973. doi:10.1016/j.tetlet.2005.10.113 |
| 29. | Hortholary, C.; Coudret, C. J. Org. Chem. 2003, 68, 2167–2174. doi:10.1021/jo026735z |
| 47. | Yang, J.; Ng, M.-K. Synthesis 2006, 3075–3079. doi:10.1055/s-2006-942529 |
| 48. | Robinson, J. M. A.; Kariuki, B. M.; Harris, K. D. M.; Philp, D. J. Chem. Soc., Perkin Trans. 2 1998, 2459–2469. doi:10.1039/a804676j |
| 49. | Harriman, A.; Mallon, L.; Ziessel, R. Chem.–Eur. J. 2008, 14, 11461–11473. doi:10.1002/chem.200801384 |
| 25. | Huang, S.; Tour, J. M. Tetrahedron Lett. 1999, 40, 3347–3350. doi:10.1016/S0040-4039(99)00463-3 |
| 45. | Zeng, X.; Wang, C.; Bryce, M. R.; Batsanov, A. S.; Sirichantaropass, S.; García-Suárez, V. M.; Lambert, C. J.; Sage, I. Eur. J. Org. Chem. 2007, 5244–5249. doi:10.1002/ejoc.200700507 |
| 6. | Ljungdahl, T.; Pettersson, K.; Albinsson, B.; Mårtensson, J. Eur. J. Org. Chem. 2006, 3087–3096. doi:10.1002/ejoc.200600240 |
| 9. | Mayr, A.; Srisailas, M.; Zhao, Q.; Gao, Y.; Hsieh, H.; Hoshmand-Kochi, M.; St. Fleur, N. Tetrahedron 2007, 63, 8206–8217. doi:10.1016/j.tet.2007.05.116 |
| 20. | Lavastre, O.; Ollivier, L.; Dixneuf, P. H.; Sibandhit, S. Tetrahedron 1996, 52, 5495–5504. doi:10.1016/0040-4020(96)00240-2 |
| 21. | Aujard, I.; Baltaze, J.-P.; Baudin, J.-B.; Cogné, E.; Ferrage, F.; Jullien, L.; Perez, E.; Prévost, V.; Qian, L. M.; Ruel, O. J. Am. Chem. Soc. 2001, 123, 8177–8188. doi:10.1021/ja010019h |
| 22. | Hwang, J.-J.; Tour, J. M. Tetrahedron 2002, 58, 10387–10405. doi:10.1016/S0040-4020(02)01409-6 |
| 23. | Zhi, Y.-G.; Lai, S.-W.; Chan, Q. K.-W.; Law, Y.-C.; Tong, G. S.-M.; Che, C.-M. Eur. J. Org. Chem. 2006, 3125–3139. doi:10.1002/ejoc.200600103 |
| 24. |
Nierengarten, J.-F.; Gu, T.; Hadziioannou, G.; Tsamouras, D.; Krasnikov, V. Helv. Chim. Acta 2004, 87, 2948–2966. doi:10.1002/hlca.200490266
Another stepwise approach consisting of coupling with 4-iodobenzaldehyde and conversion of the aldehyde group into an ethyne moiety via the Corey-Fuchs reaction. |
| 46. | Maag, D.; Kottke, T.; Schulte, M.; Godt, A. J. Org. Chem. 2009, 74, 7733–7742. doi:10.1021/jo9009744 |
| 39. | Ziener, U.; Godt, A. J. Org. Chem. 1997, 62, 6137–6143. doi:10.1021/jo970548x |
| 40. | Acharya, J. R.; Zhang, H.; Li, X.; Nesterov, E. E. J. Am. Chem. Soc. 2009, 131, 880–881. doi:10.1021/ja807621z |
| 41. | Hsung, R. P.; Chidsey, C. E. D.; Sita, L. R. Organometallics 1995, 14, 4808–4815. doi:10.1021/om00010a049 |
| 36. | Goeb, S.; De Nicola, A.; Ziessel, R. J. Org. Chem. 2005, 70, 1518–1529. doi:10.1021/jo048435i |
| 42. | Ley, K. D.; Li, Y.; Johnson, J. V.; Powell, D. H.; Schanze, K. S. Chem. Commun. 1999, 1749–1750. doi:10.1039/a903476e |
| 33. | Wang, C.; Batsanov, A. S.; Bryce, M. R. J. Org. Chem. 2006, 71, 108–116. doi:10.1021/jo051711o |
| 34. | Chandra, K. L.; Zhang, S.; Gorman, C. B. Tetrahedron 2007, 63, 7120–7132. doi:10.1016/j.tet.2007.05.006 |
| 35. | We like to call attention to the recent reports that trimethylsilyl and HOP are orthogonal alkyne protecting groups which make HOP a very interesting protecting group [36,37]. The same is true for tert-butyldimethylsilyl and HOP [38]. |
| 36. | Goeb, S.; De Nicola, A.; Ziessel, R. J. Org. Chem. 2005, 70, 1518–1529. doi:10.1021/jo048435i |
| 37. | Rodríguez, J. G.; Esquivias, J.; Lafuente, A.; Díaz, C. J. Org. Chem. 2003, 68, 8120–8128. doi:10.1021/jo034972b |
| 38. | Shimizu, H.; Fujimoto, K.; Furusyo, M.; Maeda, H.; Nanai, Y.; Mizuno, K.; Inouye, M. J. Org. Chem. 2007, 72, 1530–1533. doi:10.1021/jo061959t |
| 36. | Goeb, S.; De Nicola, A.; Ziessel, R. J. Org. Chem. 2005, 70, 1518–1529. doi:10.1021/jo048435i |
| 43. | Li, G.; Wang, X.; Li, J.; Zhao, X.; Wang, F. Tetrahedron 2006, 62, 2576–2582. doi:10.1016/j.tet.2005.12.043 |
| 44. | Pearson, D. L.; Tour, J. M. J. Org. Chem. 1997, 62, 1376–1387. doi:10.1021/jo962335y |
| 32. | Kukula, H.; Veit, S.; Godt, A. Eur. J. Org. Chem. 1999, 277–286. doi:10.1002/(SICI)1099-0690(199901)1999:1<277::AID-EJOC277>3.0.CO;2-R |
| 28. | Li, G.; Wang, X.; Wang, F. Tetrahedron Lett. 2005, 46, 8971–8973. doi:10.1016/j.tetlet.2005.10.113 |
| 33. | Wang, C.; Batsanov, A. S.; Bryce, M. R. J. Org. Chem. 2006, 71, 108–116. doi:10.1021/jo051711o |
| 53. | Takayama, Y.; Delas, C.; Muraoka, K.; Uemura, M.; Sato, F. J. Am. Chem. Soc. 2003, 125, 14163–14167. doi:10.1021/ja037975e |
| 34. | Chandra, K. L.; Zhang, S.; Gorman, C. B. Tetrahedron 2007, 63, 7120–7132. doi:10.1016/j.tet.2007.05.006 |
| 42. | Ley, K. D.; Li, Y.; Johnson, J. V.; Powell, D. H.; Schanze, K. S. Chem. Commun. 1999, 1749–1750. doi:10.1039/a903476e |
| 45. | Zeng, X.; Wang, C.; Bryce, M. R.; Batsanov, A. S.; Sirichantaropass, S.; García-Suárez, V. M.; Lambert, C. J.; Sage, I. Eur. J. Org. Chem. 2007, 5244–5249. doi:10.1002/ejoc.200700507 |
| 50. | Zhao, Z.; Yu, S.; Xu, L.; Wang, H.; Lu, P. Tetrahedron 2007, 63, 7809–7815. doi:10.1016/j.tet.2007.05.095 |
| 51. | Rodríguez, J. G.; Tejedor, J. L. J. Org. Chem. 2002, 67, 7631–7640. doi:10.1021/jo0203589 |
| 52. | Kaneko, T.; Horie, T.; Matsumoto, S.; Teraguchi, M.; Aoki, T. Macromol. Chem. Phys. 2009, 210, 22–36. doi:10.1002/macp.200800429 |
| 53. | Takayama, Y.; Delas, C.; Muraoka, K.; Uemura, M.; Sato, F. J. Am. Chem. Soc. 2003, 125, 14163–14167. doi:10.1021/ja037975e |
| 54. | López, S.; Fernández-Trillo, F.; Midón, P.; Castedo, L.; Saá, C. J. Org. Chem. 2006, 71, 2802–2810. doi:10.1021/jo052609u |
| 55. | Silicon based alkyne protecting groups with a cyano group as the polar tag [54,56,57]. |
| 56. | Höger, S.; Bonrad, K. J. Org. Chem. 2000, 65, 2243–2245. doi:10.1021/jo991746m |
| 57. | Gaefke, G.; Höger, S. Synthesis 2008, 2155–2157. doi:10.1055/s-2008-1067141 |
| 42. | Ley, K. D.; Li, Y.; Johnson, J. V.; Powell, D. H.; Schanze, K. S. Chem. Commun. 1999, 1749–1750. doi:10.1039/a903476e |
| 45. | Zeng, X.; Wang, C.; Bryce, M. R.; Batsanov, A. S.; Sirichantaropass, S.; García-Suárez, V. M.; Lambert, C. J.; Sage, I. Eur. J. Org. Chem. 2007, 5244–5249. doi:10.1002/ejoc.200700507 |
| 47. | Yang, J.; Ng, M.-K. Synthesis 2006, 3075–3079. doi:10.1055/s-2006-942529 |
| 48. | Robinson, J. M. A.; Kariuki, B. M.; Harris, K. D. M.; Philp, D. J. Chem. Soc., Perkin Trans. 2 1998, 2459–2469. doi:10.1039/a804676j |
| 49. | Harriman, A.; Mallon, L.; Ziessel, R. Chem.–Eur. J. 2008, 14, 11461–11473. doi:10.1002/chem.200801384 |
| 50. | Zhao, Z.; Yu, S.; Xu, L.; Wang, H.; Lu, P. Tetrahedron 2007, 63, 7809–7815. doi:10.1016/j.tet.2007.05.095 |
| 51. | Rodríguez, J. G.; Tejedor, J. L. J. Org. Chem. 2002, 67, 7631–7640. doi:10.1021/jo0203589 |
| 52. | Kaneko, T.; Horie, T.; Matsumoto, S.; Teraguchi, M.; Aoki, T. Macromol. Chem. Phys. 2009, 210, 22–36. doi:10.1002/macp.200800429 |
| 63. | Burke, S. D.; Danheiser, R. L., Eds. Handbook of Reagents for Organic Synthesis, Oxidizing and Reducing Agents; Wiley: Chichester, U.K., 2004; p 232. |
| 64. | Fatiadi, A. J. Synthesis 1976, 65–104. doi:10.1055/s-1976-23961 |
| 65. | For the preparation of highly activated MnO2 we followed the procedure given in [63]. In this procedure less MnCl2 • 4 H2O (200 g vs. 220 g) but the same amount of KMnO4 (160 g) and of solvent was used in comparison to the procedure given in [64]. |
| 66. | As purchased; 80% technical grade. |
| 67. | Fatiadi, A. J. Synthesis 1987, 85–127. doi:10.1055/s-1987-27859 |
| 68. | Firouzabadi, H.; Ghaderi, E. Tetrahedron Lett. 1978, 19, 839–840. doi:10.1016/S0040-4039(01)85412-5 |
| 32. | Kukula, H.; Veit, S.; Godt, A. Eur. J. Org. Chem. 1999, 277–286. doi:10.1002/(SICI)1099-0690(199901)1999:1<277::AID-EJOC277>3.0.CO;2-R |
| 62. | The largely different reaction times given in the experimental part of [48] probably indicate that the experimenters of that reference experienced the same difficulties. |
| 1. | Daniell, H. W.; Klotz, E. J. F.; Odell, B.; Claridge, T. D. W.; Anderson, H. L. Angew. Chem. 2007, 119, 6969–6972. doi:10.1002/ange.200702349 |
| 2. | Tour, J. M. J. Org. Chem. 2007, 72, 7477–7496. doi:10.1021/jo070543s |
| 3. | Hu, W.; Zhu, N.; Tang, W.; Zhao, D. Org. Lett. 2008, 10, 2669–2672. doi:10.1021/ol800753z |
| 4. | Blakskjær, P.; Gothelf, K. V. Org. Biomol. Chem. 2006, 4, 3442–3447. doi:10.1039/b605844b |
| 5. | Andersen, C. S.; Gothelf, K. V. Org. Biomol. Chem. 2009, 7, 58–60. doi:10.1039/b815099k |
| 6. | Ljungdahl, T.; Pettersson, K.; Albinsson, B.; Mårtensson, J. Eur. J. Org. Chem. 2006, 3087–3096. doi:10.1002/ejoc.200600240 |
| 7. | Zhao, Y.; Shirai, Y.; Slepkov, A. D.; Cheng, L.; Alemany, L. B.; Sasaki, T.; Hegmann, F. A.; Tour, J. M. Chem.–Eur. J. 2005, 11, 3643–3658. doi:10.1002/chem.200401198 |
| 8. | Huber, R.; Gonzáles, M. T.; Wu, S.; Langer, M.; Grunder, S.; Horhoiu, V.; Mayor, M.; Bryce, M. R.; Wang, C.; Jitchati, R.; Schönenberger, C.; Calame, M. J. Am. Chem. Soc. 2008, 130, 1080–1084. doi:10.1021/ja0767940 |
| 9. | Mayr, A.; Srisailas, M.; Zhao, Q.; Gao, Y.; Hsieh, H.; Hoshmand-Kochi, M.; St. Fleur, N. Tetrahedron 2007, 63, 8206–8217. doi:10.1016/j.tet.2007.05.116 |
| 10. | Guo, X.; Whalley, A.; Klare, J. E.; Huang, L.; O’Brien, S.; Steigerwald, M.; Nuckolls, C. Nano Lett. 2007, 7, 1119–1122. doi:10.1021/nl070245a |
| 11. | Lu, Q.; Liu, K.; Zhang, H.; Du, Z.; Wang, X.; Wang, F. ACS Nano 2009, 3, 3861–3868. doi:10.1021/nn9012687 |
| 12. | Jeschke, G.; Sajid, M.; Schulte, M.; Godt, A. Phys. Chem. Chem. Phys. 2009, 11, 6580–6591. doi:10.1039/b905724b |
| 13. | Polyhach, Y.; Godt, A.; Bauer, C.; Jeschke, G. J. Magn. Reson. 2007, 185, 118–129. doi:10.1016/j.jmr.2006.11.012 |
| 14. | Godt, A.; Schulte, M.; Zimmermann, H.; Jeschke, G. Angew. Chem. 2006, 118, 7722–7726. doi:10.1002/ange.200602807 |
| 15. | Babgi, B.; Rigamonti, L.; Cifuentes, M. P.; Corkery, T. C.; Randles, M. D.; Schwich, T.; Petrie, S.; Stranger, R.; Teshome, A.; Asselberghs, I.; Clays, K.; Samoc, M.; Humphrey, M. G. J. Am. Chem. Soc. 2009, 131, 10293–10307. doi:10.1021/ja902793z |
| 16. | Ochi, T.; Yamaguchi, Y.; Wakamiya, T.; Matsubara, Y.; Yoshida, Z. Org. Biomol. Chem. 2008, 6, 1222–1231. doi:10.1039/b717832h |
| 17. | Albinsson, B.; Mårtensson, J. J. Photochem. Photobiol., C: Photochem. Rev. 2008, 9, 138–155. doi:10.1016/j.jphotochemrev.2008.01.002 |
| 18. | Myahkostupov, M.; Piotrowiak, P.; Wang, D.; Galoppini, E. J. Phys. Chem. C 2007, 111, 2827–2829. doi:10.1021/jp0679257 |
| 19. | Yatabe, T.; Suzuki, Y.; Kawanishi, Y. J. Mater. Chem. 2008, 18, 4468–4477. doi:10.1039/b808036d |
| 60. | Bumagin, N. A.; Ponomaryov, A. B.; Beletskaya, I. P. Synthesis 1984, 728–729. doi:10.1055/s-1984-30947 |
| 61. |
Atkinson, R. E.; Curtis, R. F.; Jones, D. M.; Taylor, J. A. J. Chem. Soc. C 1969, 2173–2176. doi:10.1039/J39690002173
The suggestion to use the HOM group as a protecting group that can be removed via oxidation–decarbonylation was made much earlier here. |
| 54. | López, S.; Fernández-Trillo, F.; Midón, P.; Castedo, L.; Saá, C. J. Org. Chem. 2006, 71, 2802–2810. doi:10.1021/jo052609u |
| 33. | Wang, C.; Batsanov, A. S.; Bryce, M. R. J. Org. Chem. 2006, 71, 108–116. doi:10.1021/jo051711o |
| 34. | Chandra, K. L.; Zhang, S.; Gorman, C. B. Tetrahedron 2007, 63, 7120–7132. doi:10.1016/j.tet.2007.05.006 |
| 47. | Yang, J.; Ng, M.-K. Synthesis 2006, 3075–3079. doi:10.1055/s-2006-942529 |
| 49. | Harriman, A.; Mallon, L.; Ziessel, R. Chem.–Eur. J. 2008, 14, 11461–11473. doi:10.1002/chem.200801384 |
| 58. | Keller, J. M.; Schanze, K. S. Organometallics 2009, 28, 4210–4216. doi:10.1021/om900195p |
| 59. | Weibel, N.; Mishchenko, A.; Wandlowski, T.; Neuburger, M.; Leroux, Y.; Mayor, M. Eur. J. Org. Chem. 2009, 6140–6150. doi:10.1002/ejoc.200900751 |
© 2010 Sahoo et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)