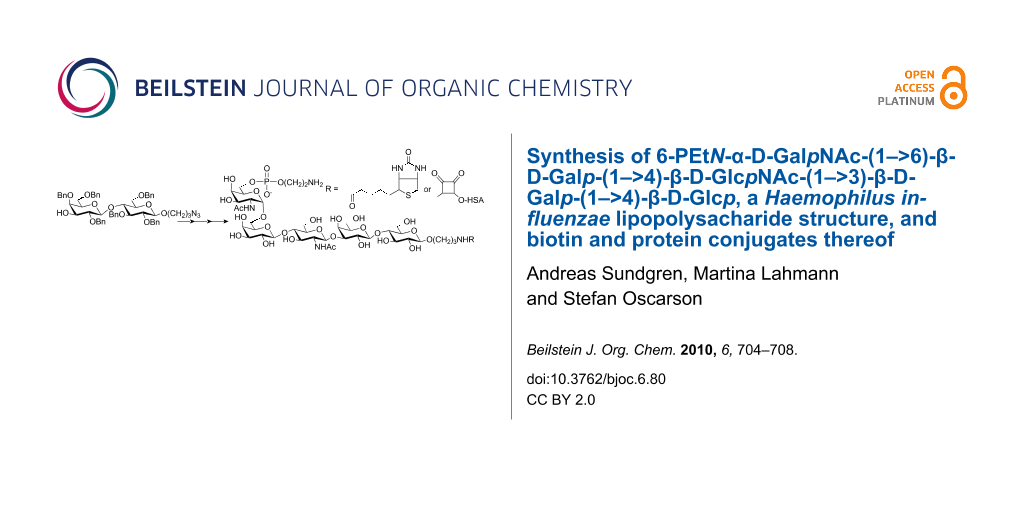
Abstract
Background: In bacteria with truncated lipopolysaccharide structures, i.e., lacking the O-antigen polysaccharide part, core structures are exposed to the immune system upon infection and thus their use as carbohydrate surface antigens in glycoconjugate vaccines can be considered and investigated. One such suggested structure from Haemophilus influenzae LPS is the phosphorylated pentasaccharide 6-PEtN-α-D-GalpNAc-(1→6)-β-D-Galp-(1→4)-β-D-GlcpNAc-(1→3)-β-D-Galp-(1→4)-β-D-Glcp.
Results: Starting from a spacer-containing lactose derivative a suitably protected lacto-N-neotetraose tetrasaccharide structure was constructed through subsequential couplings with two thioglycoside donors, a glucosamine residue followed by a galactose derivative, using NIS/AgOTf as promoter. Removal of a silyl protecting group at the primary position of the non-reducing end residue afforded an acceptor to which the terminal α-galactosamine moiety was introduced using a 2-azido bromo sugar and halide assisted coupling conditions. Global deprotection afforded the non-phosphorylated target pentasaccharide, whereas removal of a silyl group from the primary position of the non-reducing end residue produced a free hydroxy group which was phosphorylated using H-phosphonate chemistry to yield the phosphoethanolamine-containing protected pentasaccharide. Partial deprotection afforded the phosphorylated target pentasaccharide with a free spacer amino group but with a protected phosphoethanolamino group. Conjugation of the spacer amino group to biotin or dimethyl squarate followed by deprotection of the phosphoethanolamino group and, in the case of the squarate derivative, further reaction with a protein then afforded the title conjugates.
Conclusion: An effective synthesis of a biologically interesting pentasaccharide structure has been accomplished. The target pentasaccharide, an α-GalNAc substituted lacto-N-neotetraose structure, comprises a phosphoethanolamine motif and a spacer aglycon. Through the spacer, biotin and protein conjugates of the title compound have been constructed to allow further use in biological experiments.

Graphical Abstract
Introduction
Haemophilus influenzae are Gram-negative bacteria divided into six serotypes, a–f, related to the structure of the capsular polysaccharide usually surrounding the bacterium [1]. Among these serotypes, type b is the cause of the most severe diseases, i.a. meningitis and pneumonia. However, there are now several commercial vaccines against this serotype that have proven to be highly effective [2]. These vaccines are glycoconjugate vaccines, based on capsular poly- or oligosaccharide structures, either native or synthetic [3,4], linked to a carrier protein. The lipopolysaccharide (LPS) of H. influenzae shows a huge structural variety and hence non-capsulated bacteria are referred to as non-typable H. influenzae (NTHi) [5,6]. This structural diversity is a good defence mechanism against the human acquired immune system, and NTHi often cause repetitive infections. Another way used by H. influenzae to avoid the human immune system seems to be molecular mimicry, i.e. the bacteria express common human carbohydrate structures on their surface that the host recognises as self-structures and do not produce antibodies against. Construction of a glycoconjugate vaccine against NTHi is thus quite complex. Carbohydrate structures that are both exposed on the surface of most bacteria and also immunogenic in humans must be found and produced. To tackle this problem we are presently pursuing two possible routes. A common conserved LPS inner-core pentasaccharide structure has been identified, and efforts to produce this structure, i.a. through synthesis, are on-going [7-9]. Furthermore, analysis of the LPS of NTHi strains to find frequent non-human outer-core structures is continuously performed [5,6]. One candidate recently suggested is a lacto-N-neotetraose structure substituted with a PEtN-GalNAc residue (Figure 1) [10]. Herein we describe the synthesis of this structure and its conjugation to biotin and a carrier protein to form glycoconjugates that can be used in biological experiments to evaluate the immunological properties of the title structure.
![[1860-5397-6-80-1]](/bjoc/content/figures/1860-5397-6-80-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The H. influenzae outer core target structure.
Figure 1: The H. influenzae outer core target structure.
Results and Discussion
The target structure is, as noted above, a substituted lacto-N-neotetraose structure that we have experience of from, i.a., former synthetic work related to Streptococcus pneumoniae type 14 CPS structures [11]. To allow for the introduction of the GalNAc residue at the 6IV-position and the subsequent phosphorylation of the 6V-hydroxy group, two new galactosyl donors were designed and synthesised (Scheme 1). Reductive ring opening, with BH3/Bu2BOTf [12], of the benzylidene acetal in the known ethyl thioglycoside 1 [13] gave the 6-hydroxy derivative 2 (85%), which was then silylated to afford donor 3 (90%). Regioselective silylation of the 2-azido-galactose ethyl thioglycoside 4 [14] yielded the 6-O-silylated compound 5 (92%), benzylation of which gave donor 6 (85%). A benzyl group in the 4-position was preferred to an ester group to avoid the risk of acyl migration during subsequent reactions; desilylation, glycosidation and phosphorylation.
![[1860-5397-6-80-i1]](/bjoc/content/inline/1860-5397-6-80-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: i. BH3, Bu2BOTf, THF/CH2Cl2, 85%; ii. TBDMSCl, pyridine, CH2Cl2, 90%; iii. TBDMSCl, pyridine, 92%; iv. BnBr, NaH, DMF, 85%.
Scheme 1: i. BH3, Bu2BOTf, THF/CH2Cl2, 85%; ii. TBDMSCl, pyridine, CH2Cl2, 90%; iii. TBDMSCl, pyridine, 92%; ...
3II,4II-Diols of lactose are often used as acceptors for regioselective glycosidations in the 3-position. However, the selectivity is dependent on the donor, promoter and conditions employed [15], e.g., it has been shown that donors containing a 4,6-O-benzylidene acetal can give a mixture of products [16]. Hence, the diol 7 [17] was transformed using benzaldehyde dimethyl acetal and camphorsulfonic acid stereo-selectively into the corresponding 3,4-endo-benzylidene derivative 8 which was in turn converted to the 3-hydroxy derivative 9 by NaBH3CN/HCl-mediated reductive opening of the acetal ring [18] (80% overall yield from 7, Scheme 2). NIS/AgOTf-promoted glycosidation of this acceptor with donor 10 [19] (1.4 equiv) then efficiently gave the β-linked trisaccharide 11 (83%). At this stage the phthalimido group was removed by aminolysis and the resulting amino compound acetylated to yield 12 (93%) with the target acetamido function in place. Once more reductive opening of a benzylidene acetal (NaBH3CN/HCl) gave a new mono-hydroxy compound, the acceptor 13 (81%).
![[1860-5397-6-80-i2]](/bjoc/content/inline/1860-5397-6-80-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: i. PhCH(OMe)2, CSA; ii. NaBH3CN, HCl/Et2O, THF, 80%; iii. NIS/AgOTf, CH2Cl2, 83%; iv. a) NaOMe, MeOH; b) NH2(CH2)2NH2, EtOH; c) Ac2O, pyridine, 93%; v. NaBH3CN, HCl/Et2O, THF, 81%.
Scheme 2: i. PhCH(OMe)2, CSA; ii. NaBH3CN, HCl/Et2O, THF, 80%; iii. NIS/AgOTf, CH2Cl2, 83%; iv. a) NaOMe, MeO...
The 4-hydroxy group in GlcNAc derivatives is known to be quite unreactive towards glycosylations, which, i.a., has led to development of new protecting group patterns to improve the reactivity [20,21]. Here, however, NIS/AgOTf-promoted glycosylation of acceptor 13 with donor 3 proceeded without difficulties to produce the tetrasaccharide 14 in high yield (77%, Scheme 3). Before the introduction of the azido-galactose residue, the spacer azido function was reduced to an amino group, which was protected as a benzyl carbamate (→ 15, 91%). Removal of the TBDMS group with TBAF then gave the tetrasaccharide acceptor 16 (89%). To obtain complete α-selectivity with 2-azido-galactose donors is not trivial. A 4,6-silyl acetal has been suggested as one way to improve the selectivity [22]. We have earlier tried halide-assisted conditions [23], which is not that common due to the incompatibility between the mild coupling conditions and the low reactivity of 2-azido donors, with good results when simple spacer alcohols were used as aglycons [24]. This approach also worked well here, although extended reaction time was necessary. A halide-assisted coupling between acceptor 16 and the bromosugar, obtained from thioglycoside 6, gave (after 11 days) the α-linked pentasaccharide 17 in 79% yield. Due to the excess of donor used and the long reaction time required the acetamido carbonyl oxygen also partly behaved as a nucleophile and gave a pseudohexasaccharide acetimidate side product according to MALDI-TOF and NMR. Similar side products have earlier been described [25-28]. Mild acid treatment of the glycosylation mixture prior to purification led to cleavage of the formed imidate and afforded the desired pentasaccharide in the above noted yield.
![[1860-5397-6-80-i3]](/bjoc/content/inline/1860-5397-6-80-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: i. NIS/AgOTf, CH2Cl2, 77%; ii. a) H2S, pyridine, Et3N; b) CbzCl, pyridine, CH2Cl2, 91%; iii. TBAF, THF, 89%; iv. Br2, CH2Cl2; v. Et4NBr, DMF, CH2Cl2, 79%; vi. a) H2S, pyridine, Et3N; b) Ac2O, pyridine, CH2Cl2, 91%; vii. TBAF, THF, 85%.
Scheme 3: i. NIS/AgOTf, CH2Cl2, 77%; ii. a) H2S, pyridine, Et3N; b) CbzCl, pyridine, CH2Cl2, 91%; iii. TBAF, ...
Reduction of the azido group in 17 and subsequent acetylation afforded compound 18 (91%). Removal of the TBDMS group, again using TBAF, gave derivative 19 (85%), ready for the introduction of the phosphoethanolamine. Compound 19 was also completely deprotected by sodium methoxide treatment followed by catalytic hydrogenolysis to give the non-phosphorylated target structure 20 (66%, Scheme 4), to be used as a reference in biological experiments. Earlier we used the Cbz-protected ethanolamine H-phosphonate monoester as a reagent in the formation of phosphoethanolamines [29]. Since the amino group in the spacer was already Cbz-protected and we wanted to be able to differentiate between the two amino groups during conjugation, a Boc-protected H-phosphonate monoester 21 was synthesised and used in the phosphorylation step. Activation of 21 with pivaloyl chloride in the presence of 19 afforded the H-phosphonate diester, which was oxidised with I2/pyridine in water to afford the phosphate diester 22 [30]. Deprotection with sodium methoxide followed by catalytic hydrogenolysis then afforded the still Boc-protected phosphoethanolamine pentasaccharide 23 (44% from 19) ready for conjugation via the free spacer amino group.
![[1860-5397-6-80-i4]](/bjoc/content/inline/1860-5397-6-80-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: i. NaOMe, MeOH; ii. H2, Pd/C, MeOH/H2O; iii. 21, PivCl, pyridine, MeCN; iv. I2, H2O, pyridine; v. Dimethyl squarate or NHS-biotin, buffer pH 7; vi. TFA (10% aqueous).
Scheme 4: i. NaOMe, MeOH; ii. H2, Pd/C, MeOH/H2O; iii. 21, PivCl, pyridine, MeCN; iv. I2, H2O, pyridine; v. D...
The protein conjugations were carried out using squarate ester methodology [31,32]. Reaction of compound 23 with dimethyl squarate at neutral pH afforded the monomethyl ester squarate amide of 23, from which the Boc-group was removed by acid hydrolysis to afford derivative 24. Protein conjugations were performed by reaction of 24 (20 equiv) with human serum albumin (HSA). Compound 20 was similarly activated with dimethyl squarate and conjugated to HSA. MALDI-TOF MS of the HSA-conjugates of compounds 20 and 24 showed a loading of 16 oligosaccharides/protein molecule and 7 oligosaccharides/protein molecule, respectively. Since derivative 24 contains a free amino group there is a possibility of oligomerisation in addition to protein conjugation. To what extent this happened was not examined. Compound 23 was also biotinylated for use in ELISA-screening of antibodies raised against native LPS. Reaction with the commercial NHS-activated ester of biotin followed by TFA-treatment to remove the Boc-group afforded derivative 25.
Supporting Information
| Supporting Information File 1: Experimental Section | ||
| Format: PDF | Size: 106.4 KB | Download |
References
-
Kenne, L.; Lindberg, B. In The Polysaccharides; Aspinall, G. O., Ed.; Academic Press: New York, 1995; Vol. 2, pp 287–363.
Return to citation in text: [1] -
Ravenscroft, N.; Jones, C. Curr. Opin. Drug Discovery Dev. 2000, 3, 222–231.
Return to citation in text: [1] -
Lindberg, A. A. Vaccine 1999, 17, S28–S36. doi:10.1016/S0264-410X(99)00232-7
Return to citation in text: [1] -
Verez-Bencomo, V.; Fernández-Santana, V.; Hardy, E.; Toledo, M. E.; Rodríguez, M. C.; Heynngnezz, L.; Rodriguez, A.; Baly, A.; Herrera, L.; Izquierdo, M.; Villar, A.; Valdés, Y.; Cosme, K.; Deler, M. L.; Montane, M.; Garcia, E.; Ramos, A.; Aguilar, A.; Medina, E.; Toraño, G.; Sosa, I.; Hernandez, I.; Martínez, R.; Muzachio, A.; Carmenates, A.; Costa, L.; Cardoso, F.; Campa, C.; Diaz, M.; Roy, R. Science 2004, 305, 522–525. doi:10.1126/science.1095209
Return to citation in text: [1] -
Richards, J. C.; Cox, A. D.; Schweda, E. K. H.; Martin, A.; Hood, D. W.; Moxon, E. R. Adv. Exp. Med. Biol. 2001, 491, 515–524.
and references cited therein.
Return to citation in text: [1] [2] -
Yildirim, H. H.; Hood, D. W.; Moxon, E. R.; Schweda, E. K. H. Eur. J. Biochem. 2003, 270, 3153–3167. doi:10.1046/j.1432-1033.2003.03693.x
and references cited therein.
Return to citation in text: [1] [2] -
Segerstedt, E.; Mannerstedt, K.; Johansson, M.; Oscarson, S. J. Carbohydr. Chem. 2004, 23, 443–452. doi:10.1081/CAR-200044580
Return to citation in text: [1] -
Bernlind, C.; Oscarson, S. J. Org. Chem. 1998, 63, 7780–7788. doi:10.1021/jo9808573
Return to citation in text: [1] -
Mannerstedt, K.; Segerstedt, E.; Olsson, J.; Oscarson, S. Org. Biomol. Chem. 2008, 6, 1087–1091. doi:10.1039/b717564g
Return to citation in text: [1] -
Schweda, E. K. H.; Richards, J. C.
Personal communication.
Return to citation in text: [1] -
Sundgren, A.; Lahmann, M.; Oscarson, S. J. Carbohydr. Chem. 2005, 24, 379–391. doi:10.1081/CAR-200066935
Return to citation in text: [1] -
Jiang, L.; Chan, T.-H. Tetrahedron Lett. 1998, 39, 355–358. doi:10.1016/S0040-4039(97)10599-8
Return to citation in text: [1] -
Yashunsky, D. V.; Higson, A. P.; Ross, A. J.; Nikolaev, A. V. Carbohydr. Res. 2001, 336, 243–248. doi:10.1016/S0008-6215(01)00274-9
Return to citation in text: [1] -
Paulsen, H.; Rauwald, W.; Weichert, U. Liebigs Ann. Chem. 1988, 75–86. doi:10.1002/jlac.198819880114
Return to citation in text: [1] -
Paulsen, H.; Steiger, K. M. Carbohydr. Res. 1987, 169, 105–125. doi:10.1016/0008-6215(87)80245-8
Return to citation in text: [1] -
Lahmann, M.; Bülow, L.; Teodorovic, P.; Gybäck, H.; Oscarson, S. Glycoconjugate J. 2004, 21, 251–256. doi:10.1023/B:GLYC.0000045097.19353.73
Return to citation in text: [1] -
Demchenko, A. V.; Boons, G.-J. J. Org. Chem. 2001, 66, 2547–2554. doi:10.1021/jo001477w
Return to citation in text: [1] -
Garegg, P. J.; Hultberg, H.; Wallin, S. Carbohydr. Res. 1982, 108, 97–101. doi:10.1016/S0008-6215(00)81894-7
Return to citation in text: [1] -
Kihlberg, J. O.; Leigh, D. A.; Bundle, D. R. J. Org. Chem. 1990, 55, 2860–2863. doi:10.1021/jo00296a055
Return to citation in text: [1] -
Crich, D.; Vinod, A. U. Org. Lett. 2003, 5, 1297–1300. doi:10.1021/ol0342305
Return to citation in text: [1] -
Crich, D.; Vinod, A. U. J. Org. Chem. 2005, 70, 1291–1296. doi:10.1021/jo0482559
Return to citation in text: [1] -
Imamura, A.; Ando, H.; Korogi, S.; Tanabe, G.; Muraoka, O.; Ishida, H.; Kiso, M. Tetrahedron Lett. 2003, 44, 6725–6728. doi:10.1016/S0040-4039(03)01647-2
Return to citation in text: [1] -
Lemieux, R. U.; Hendriks, K. B.; Stick, R. V.; James, K. J. Am. Chem. Soc. 1975, 97, 4056–4062. doi:10.1021/ja00847a032
Return to citation in text: [1] -
Hansson, J.; Garegg, P. J.; Oscarson, S. J. Org. Chem. 2001, 66, 6234–6243. doi:10.1021/jo001302m
Return to citation in text: [1] -
Haraldsson, M. Chem. Commun. (Stockholm Univ.) 1987, No. 4.
Return to citation in text: [1] -
Arnarp, J.; Haraldsson, M.; Lönngren, J. J. Chem. Soc., Perkin Trans. 1 1985, 535–539. doi:10.1039/P19850000535
Return to citation in text: [1] -
Pougny, R. J.; Nasser, M.; Naulet, N.; Sinaÿ, P. Nouv. J. Chim. 1978, 2, 389–395.
Return to citation in text: [1] -
Liao, L.; Auzanneau, F. I. Org. Lett. 2003, 5, 2607–2610. doi:10.1021/ol034669x
Return to citation in text: [1] -
Stewart, A.; Bernlind, C.; Martin, A.; Oscarson, S.; Richards, J. C.; Schweda, E. K. H. Carbohydr. Res. 1998, 313, 193–202. doi:10.1016/S0008-6215(98)00271-7
Return to citation in text: [1] -
Garegg, P. J.; Regberg, T.; Stawinski, J.; Strömberg, R. Chem. Scr. 1986, 26, 59–62.
Return to citation in text: [1] -
Tietze, L. F.; Schröter, C.; Gabius, S.; Brink, U.; Goerlach-Graw, A.; Gabius, H.-J. Bioconjugate Chem. 1991, 2, 148–153. doi:10.1021/bc00009a003
Return to citation in text: [1] -
Tietze, L. F.; Arlt, M.; Beller, M.; Gluesenkamp, K.-H.; Jähde, E.; Rajewsky, M. F. Chem. Ber. 1991, 124, 1215–1221. doi:10.1002/cber.19911240539
Return to citation in text: [1]
| 23. | Lemieux, R. U.; Hendriks, K. B.; Stick, R. V.; James, K. J. Am. Chem. Soc. 1975, 97, 4056–4062. doi:10.1021/ja00847a032 |
| 20. | Crich, D.; Vinod, A. U. Org. Lett. 2003, 5, 1297–1300. doi:10.1021/ol0342305 |
| 21. | Crich, D.; Vinod, A. U. J. Org. Chem. 2005, 70, 1291–1296. doi:10.1021/jo0482559 |
| 22. | Imamura, A.; Ando, H.; Korogi, S.; Tanabe, G.; Muraoka, O.; Ishida, H.; Kiso, M. Tetrahedron Lett. 2003, 44, 6725–6728. doi:10.1016/S0040-4039(03)01647-2 |
| 1. | Kenne, L.; Lindberg, B. In The Polysaccharides; Aspinall, G. O., Ed.; Academic Press: New York, 1995; Vol. 2, pp 287–363. |
| 7. | Segerstedt, E.; Mannerstedt, K.; Johansson, M.; Oscarson, S. J. Carbohydr. Chem. 2004, 23, 443–452. doi:10.1081/CAR-200044580 |
| 8. | Bernlind, C.; Oscarson, S. J. Org. Chem. 1998, 63, 7780–7788. doi:10.1021/jo9808573 |
| 9. | Mannerstedt, K.; Segerstedt, E.; Olsson, J.; Oscarson, S. Org. Biomol. Chem. 2008, 6, 1087–1091. doi:10.1039/b717564g |
| 18. | Garegg, P. J.; Hultberg, H.; Wallin, S. Carbohydr. Res. 1982, 108, 97–101. doi:10.1016/S0008-6215(00)81894-7 |
| 5. |
Richards, J. C.; Cox, A. D.; Schweda, E. K. H.; Martin, A.; Hood, D. W.; Moxon, E. R. Adv. Exp. Med. Biol. 2001, 491, 515–524.
and references cited therein. |
| 6. |
Yildirim, H. H.; Hood, D. W.; Moxon, E. R.; Schweda, E. K. H. Eur. J. Biochem. 2003, 270, 3153–3167. doi:10.1046/j.1432-1033.2003.03693.x
and references cited therein. |
| 19. | Kihlberg, J. O.; Leigh, D. A.; Bundle, D. R. J. Org. Chem. 1990, 55, 2860–2863. doi:10.1021/jo00296a055 |
| 3. | Lindberg, A. A. Vaccine 1999, 17, S28–S36. doi:10.1016/S0264-410X(99)00232-7 |
| 4. | Verez-Bencomo, V.; Fernández-Santana, V.; Hardy, E.; Toledo, M. E.; Rodríguez, M. C.; Heynngnezz, L.; Rodriguez, A.; Baly, A.; Herrera, L.; Izquierdo, M.; Villar, A.; Valdés, Y.; Cosme, K.; Deler, M. L.; Montane, M.; Garcia, E.; Ramos, A.; Aguilar, A.; Medina, E.; Toraño, G.; Sosa, I.; Hernandez, I.; Martínez, R.; Muzachio, A.; Carmenates, A.; Costa, L.; Cardoso, F.; Campa, C.; Diaz, M.; Roy, R. Science 2004, 305, 522–525. doi:10.1126/science.1095209 |
| 16. | Lahmann, M.; Bülow, L.; Teodorovic, P.; Gybäck, H.; Oscarson, S. Glycoconjugate J. 2004, 21, 251–256. doi:10.1023/B:GLYC.0000045097.19353.73 |
| 31. | Tietze, L. F.; Schröter, C.; Gabius, S.; Brink, U.; Goerlach-Graw, A.; Gabius, H.-J. Bioconjugate Chem. 1991, 2, 148–153. doi:10.1021/bc00009a003 |
| 32. | Tietze, L. F.; Arlt, M.; Beller, M.; Gluesenkamp, K.-H.; Jähde, E.; Rajewsky, M. F. Chem. Ber. 1991, 124, 1215–1221. doi:10.1002/cber.19911240539 |
| 17. | Demchenko, A. V.; Boons, G.-J. J. Org. Chem. 2001, 66, 2547–2554. doi:10.1021/jo001477w |
| 12. | Jiang, L.; Chan, T.-H. Tetrahedron Lett. 1998, 39, 355–358. doi:10.1016/S0040-4039(97)10599-8 |
| 14. | Paulsen, H.; Rauwald, W.; Weichert, U. Liebigs Ann. Chem. 1988, 75–86. doi:10.1002/jlac.198819880114 |
| 29. | Stewart, A.; Bernlind, C.; Martin, A.; Oscarson, S.; Richards, J. C.; Schweda, E. K. H. Carbohydr. Res. 1998, 313, 193–202. doi:10.1016/S0008-6215(98)00271-7 |
| 11. | Sundgren, A.; Lahmann, M.; Oscarson, S. J. Carbohydr. Chem. 2005, 24, 379–391. doi:10.1081/CAR-200066935 |
| 15. | Paulsen, H.; Steiger, K. M. Carbohydr. Res. 1987, 169, 105–125. doi:10.1016/0008-6215(87)80245-8 |
| 30. | Garegg, P. J.; Regberg, T.; Stawinski, J.; Strömberg, R. Chem. Scr. 1986, 26, 59–62. |
| 24. | Hansson, J.; Garegg, P. J.; Oscarson, S. J. Org. Chem. 2001, 66, 6234–6243. doi:10.1021/jo001302m |
| 5. |
Richards, J. C.; Cox, A. D.; Schweda, E. K. H.; Martin, A.; Hood, D. W.; Moxon, E. R. Adv. Exp. Med. Biol. 2001, 491, 515–524.
and references cited therein. |
| 6. |
Yildirim, H. H.; Hood, D. W.; Moxon, E. R.; Schweda, E. K. H. Eur. J. Biochem. 2003, 270, 3153–3167. doi:10.1046/j.1432-1033.2003.03693.x
and references cited therein. |
| 13. | Yashunsky, D. V.; Higson, A. P.; Ross, A. J.; Nikolaev, A. V. Carbohydr. Res. 2001, 336, 243–248. doi:10.1016/S0008-6215(01)00274-9 |
| 25. | Haraldsson, M. Chem. Commun. (Stockholm Univ.) 1987, No. 4. |
| 26. | Arnarp, J.; Haraldsson, M.; Lönngren, J. J. Chem. Soc., Perkin Trans. 1 1985, 535–539. doi:10.1039/P19850000535 |
| 27. | Pougny, R. J.; Nasser, M.; Naulet, N.; Sinaÿ, P. Nouv. J. Chim. 1978, 2, 389–395. |
| 28. | Liao, L.; Auzanneau, F. I. Org. Lett. 2003, 5, 2607–2610. doi:10.1021/ol034669x |
© 2010 Sundgren et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)