Abstract
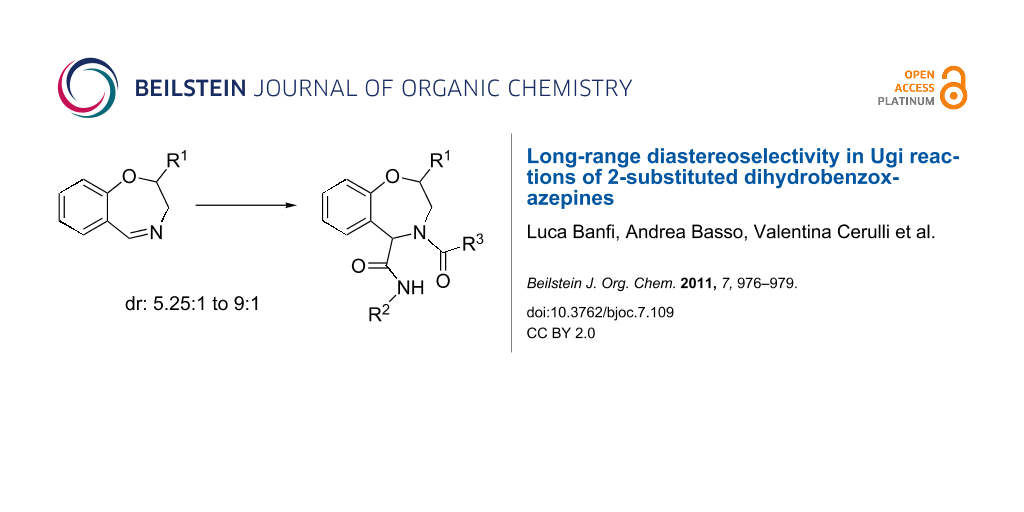
The Ugi reaction of 2-substituted dihydrobenzoxazepines was found to proceed with unexpectedly good diastereoselectivitiy (diastereoisomeric ratios up to 9:1), despite the large distance between the pre-existing stereogenic centre and the newly generated one. This result represents the first good 1,4 asymmetric induction in an Ugi reaction as well as the first example of diastereoselective Ugi reaction of seven membered cyclic imines. It allows the diversity-oriented synthesis of various tetrahydro[f][1,4]benzoxazepines.

Graphical Abstract
Introduction
The Ugi reaction is probably the most renowned and widely used multicomponent reaction. Its great utility in the highly convergent and diversity-oriented synthesis of libraries of heterocyclic compounds, stemming from the possibility to introduce up to four diversity inputs in a single step, has been fully demonstrated [1-5]. However, a main drawback of this venerable reaction is the poor diastereoselectivity typically experienced when using chiral inputs. It is well known that chiral isocyanides, aldehydes/ketones and carboxylic acids always bring about no or very little diastereoselectivity, whereas some relative asymmetric induction has been reported only with chiral amines as auxiliaries [6], or with chiral cyclic imines.
The use of cyclic imines (three-component Ugi–Joullié reaction) [7,8] is particularly useful, because the resulting Ugi products are necessarily nitrogen heterocycles. However, good diastereoselectivity has been obtained so far only with a few types of chiral substrates [9-15]. In most cases these are represented by five-membered imines (pyrrolines) with a stereogenic centre α to the imine carbon (1,2-induction), although this relative arrangement is not a guarantee of good stereoselectivity [8,14,16]. Examples of 1,3-induction on chiral imines with the stereocentre β to the carbon [14,16], or α to the nitrogen, of the C=N moiety [13,15,17] are rarer. More often, when the stereocentre is not in α, poor diastereoselectivity is observed [18,19]. This fact limits the diversity of heterocycles that can be accessed stereoselectively from the three-component Ugi–Joullié reaction of cyclic imines.
Results and Discussion
We report here some preliminary results disclosing a new family of chiral 7-membered cyclic imines that afford good levels of diastereoselectivity when submitted to an Ugi–Joullié reaction, despite the fact that the stereogenic centre is only γ to the imine carbon (1,4 relative induction). This is, to our knowledge, the first example of 1,4 asymmetric induction in an isocyanide based multicomponent reaction of chiral carbonyl compounds or imines, and the first example of diastereoselective Ugi reaction on chiral seven-membered imines [20,21].
The two imines 5a,b (Scheme 1) have been convergently synthesized in three steps from Weinreb hydroxamate 1, in turn prepared in one step from salicylic acid (Supporting Information File 1). The key step of the synthesis is the intramolecular condensation of 1 with racemic alcohols 2a,b through a Mitsunobu reaction. The moderate yields are due to the consumption of alcohols 2, which undergo side-reactions, resulting in incomplete transformation of 1, even when using 1.3–1.5 equiv of 2. The use of a larger excess of 2 would probably increase the yields, but this is not particularly convenient (especially if one plans to use enantiomerically pure 2). In any case, unreacted 1 may be recovered. Alcohol 2b behaves somewhat better than 2a in this reaction. The other two steps proceeded with no problems to give imines 5a,b in high yield. It is worth noting that the Mitsunobu reaction is not effective on unprotected salicylaldehyde. 2,3-Dihydrobenzo[f][1,4]oxazepines similar to 5a,b have been previously prepared, but through less general routes [22-24].
![[1860-5397-7-109-i1]](/bjoc/content/inline/1860-5397-7-109-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: a) TBAD [(t-BuO2C−N=)2], PPh3, THF, −15 °C → rt, 49% (3a), 62% (3b); b) LiAlH4, Et2O–THF, 0 °C, 90% (4a), 88% (4b); c) HCl, CH2Cl2–H2O, rt; d) R2NC, R3CO2H, MeOH, rt.
Scheme 1: a) TBAD [(t-BuO2C−N=)2], PPh3, THF, −15 °C → rt, 49% (3a), 62% (3b); b) LiAlH4, Et2O–THF, 0 °C, 90%...
Compounds 5a,b were reacted with a series of isocyanides and carboxylic acids to give, in good yields, nine different tetrahydro[f][1.4]benzoxazepines 6, equipped with three diversity points.
As shown in Table 1, all the tested Ugi reactions proceeded with remarkably high diastereoselectivity, if one considers that the R1 substituent is quite far away from the imine carbon. This long range diastereoselectivity (from 5.25:1 up to 9:1) is completely unprecedented for an isocyanide based multicomponent reaction.
Table 1: Results of Ugi reactions of imines 5a,b.
| Product | R1 | R2 | R3 | Yielda | drb |
|---|---|---|---|---|---|
| 6a | Me | Cy | Et | 70% | 85:15 |
| 6b | Me | t-Bu | MeOCH2 | 77% | 87:13 |
| 6c | Me | Bn | BocNHCH2 | 71% | 84:16 |
| 6d | iBu | 4-BnOC6H4CH2CH2 | MeOCH2 | 56% | 90:10 |
| 6e | iBu | Cy | Et | 59% | 86:14 |
| 6f | iBu | t-Bu | Bn | 64% | 88:12 |
| 6g | iBu | n-Bu | 3-BrC6H4 | 57% | 88:12 |
| 6h | iBu | t-Bu | 5-Cl-2-thienyl | 78% | 89:11 |
| 6i | iBu | n-Bu | Z-NH-CH2CH2 | 70% | 88:12 |
aIsolated yields (after chromatography) from aldehydes 4a,b. bDetermined by HPLC or by 1H NMR (for 6f, 6h, 6i only by NMR).
A slight increase in the dr was observed on passing from R1 = Me to bulkier R1 = iBu. On the other hand the structure and the nature of both isocyanides and carboxylic acids seem to have little influence on the diastereoselectivity. NMR characterization of the products is reported in Supporting Information File 2. Minimization of the cyclic imine 5a using CSC Chem3D (v10) indicates that there are only two significant conformations, and that the one with the substituent at C-2 in the equatorial position is strongly favored. In this situation (Figure 1), the substituent at C-2 should be quite far away from the site of isocyanide attack, being unable to discriminate the two diastereotopic faces.
![[1860-5397-7-109-1]](/bjoc/content/figures/1860-5397-7-109-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Model of the expected preferred conformation of imine 5a, as minimized using CSC Chem3D (MOPAC-PM3).
Figure 1: Model of the expected preferred conformation of imine 5a, as minimized using CSC Chem3D (MOPAC-PM3)....
Thus, rationalization of the observed stereoselectivity is not trivial, also because we have not yet proved unambiguously the relative configuration of the major adducts. Some authors have suggested, for six membered rings, a preferential axial attack of the isocyanide [14,16], since it relieves unfavorable steric strain in the forming tetrahedric adduct (Scheme 2). In our case, equatorial attack, leading to intermediate 9, would experience steric strain with the peri H-7. Therefore, if the preferred conformation of the imine is the one depicted in Figure 1, with R1 equatorial, axial attack would give the cis adducts. The importance of the unfavorable peri interaction is confirmed by the fact that the isocyanide derived substituent prefers an axial position in both stereoisomers, as demonstrated by NOE experiments carried out on 6e (Supporting Information File 2). Thus, after attack, the trans initial adduct 10 undergoes a conformational change to 11. The two vicinal J2-3 (i.e., 2.1 and 9.3 Hz for 6h) in the major diastereomers are in agreement with the chair-like conformation 8 of the cis adduct, whereas the same coupling constants in the minor diastereoisomer (i.e., 3.6 and 8.7 Hz. for 6h) fit the boat-like conformation 11 of the trans adduct. However, the difference between these coupling constants for the two stereoisomers is not large enough to guarantee the undisputable assignment of the cis relative configuration to the major adduct. In the presented hypothesis, the function of the substituent at C-2 would therefore not be to shield one of the two diastereotopic faces, but only to fix the conformation by favoring an equatorial disposition of R1. We are planning to prove the relative configuration of the major adducts and to prepare analogues with further substituents in order to get more clues on this unusual diastereoselectivity and, hopefully, to further improve stereoselectivity.
![[1860-5397-7-109-i2]](/bjoc/content/inline/1860-5397-7-109-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Possible explanation of diastereoselectivity in Ugi reactions of imines 5.
Scheme 2: Possible explanation of diastereoselectivity in Ugi reactions of imines 5.
In conclusion, the methodology presented herein appears particularly well suited for the stereoselective preparation of libraries of peptidomimetics based on the tetrahydrobenzoxazepine ring. Although structures of general formula 6 are unprecedented, other tetrahydrobenzoxazepines have shown interesting pharmacological properties [25,26]. The possibility to introduce up to 4 points of diversity (including also substituents on the aromatic ring), and to obtain enantiomerically pure compounds, starting from enantiomerically pure alcohols 2a,b, will be explored, too.
References
-
Akritopoulou-Zanze, I.; Djuric, S. W. Heterocycles 2007, 73, 125–147. doi:10.3987/REV-07-SR(U)3
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Riva, R. In Synthesis of Heterocycles via Multicomponent Reactions I; Orru, R. V. A.; Ruijter, E., Eds.; Springer: Berlin/Heidelberg, 2010; Vol. 23, pp 1–39.
Return to citation in text: [1] -
Banfi, L.; Riva, R.; Basso, A. Synlett 2010, 23–41. doi:10.1055/s-0029-1218527
Return to citation in text: [1] -
Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728
Return to citation in text: [1] -
Hulme, C.; Dietrich, J. Mol. Div. 2009, 13, 195–207. doi:10.1007/s11030-009-9111-6
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Guanti, R.; Riva, R. Asymmetric Isocyanide-Based MCRs. In Multicomponent Reactions; Zhu, J. P.; Bienaymé, H., Eds.; Wiley: Weinheim, 2005; pp 1–32. doi:10.1002/3527605118.ch1
Return to citation in text: [1] -
Nutt, R. F.; Joullié, M. M. J. Am. Chem. Soc. 1982, 104, 5852–5853. doi:10.1021/ja00385a080
Return to citation in text: [1] -
Bowers, M. M.; Carroll, P.; Joullié, M. M. J. Chem. Soc., Perkin Trans. 1 1989, 857–865. doi:10.1039/P19890000857
Return to citation in text: [1] [2] -
Znabet, A.; Ruijter, E.; de Kanter, F. J. J.; Köhler, V.; Helliwell, M.; Turner, N. J.; Orru, R. V. A. Angew. Chem., Int. Ed. 2010, 49, 5289–5292. doi:10.1002/anie.201001592
Return to citation in text: [1] -
Bonger, K. M.; Wennekes, T.; Filippov, D. V.; Lodder, G.; van der Marel, G. A.; Overkleeft, H. S. Eur. J. Org. Chem. 2008, 3678–3688. doi:10.1002/ejoc.200800340
Return to citation in text: [1] -
Chapman, T. M.; Davies, I. G.; Gu, B.; Block, T. M.; Scopes, D. I. C.; Hay, P. A.; Courtney, S. M.; McNeill, L. A.; Schofield, C. J.; Davis, B. G. J. Am. Chem. Soc. 2005, 127, 506–507. doi:10.1021/ja043924l
Return to citation in text: [1] -
Timmer, M. S. M.; Risseeuw, M. D. P.; Verdoes, M.; Filippov, D. V.; Plaisier, J. R.; van der Marel, G. A.; Overkleeft, H. S.; van Boom, J. H. Tetrahedron: Asymmetry 2005, 16, 177–185. doi:10.1016/j.tetasy.2004.11.079
Return to citation in text: [1] -
Maison, W.; Lützen, A.; Kosten, M.; Schlemminger, I.; Westerhoff, O.; Saak, W.; Martens, J. J. Chem. Soc., Perkin Trans. 1 2000, 1867–1871. doi:10.1039/B002258F
Return to citation in text: [1] [2] -
Sperger, C. A.; Mayer, P.; Wanner, K. T. Tetrahedron 2009, 65, 10463–10469. doi:10.1016/j.tet.2009.10.020
Return to citation in text: [1] [2] [3] [4] -
Zhu, D.; Chen, R.; Liang, H.; Li, S.; Pan, L.; Chen, X. Synlett 2010, 897–900. doi:10.1055/s-0029-1219533
Return to citation in text: [1] [2] -
El Kaïm, L.; Grimaud, L.; Oble, J.; Wagschal, S. Tetrahedron Lett. 2009, 50, 1741–1743. doi:10.1016/j.tetlet.2009.01.145
Return to citation in text: [1] [2] [3] -
Gröger, H.; Hatam, M.; Martens, J. Tetrahedron 1995, 51, 7173–7180. doi:10.1016/0040-4020(95)00365-F
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Guanti, G.; Merlo, S.; Repetto, C.; Riva, R. Tetrahedron 2008, 64, 1114–1134. doi:10.1016/j.tet.2007.10.058
Return to citation in text: [1] -
Iizuka, T.; Takiguchi, S.; Kumakura, Y.; Tsukioka, N.; Higuchi, K.; Kawasaki, T. Tetrahedron Lett. 2010, 51, 6003–6005. doi:10.1016/j.tetlet.2010.09.026
Return to citation in text: [1] -
Gulevich, A. V.; Shevchenko, N. F.; Balenkova, E. S.; Roeschenthaler, G. V.; Nenajdenko, V. G. Synlett 2009, 403–406. doi:10.1055/s-0028-1087529
Return to citation in text: [1] -
Recently, a very high diastereoselectivity leading to seven-membered ring was obtained in an intramolecular Ugi reaction of an oxoacid endowed with axial chirality: Mehta, V. P.; Modha, S. G.; Ruijter, E.; Van Hecke, K.; Van Meervelt, L.; Pannecouque, C.; Balzarini, J.; Orru, R. V. A.; Van der Eycken, E. J. Org. Chem. 2011, 76, 2828–2839. doi:10.1021/jo200251q
Return to citation in text: [1] -
Del Buttero, P.; Molteni, G.; Papagni, A.; Miozzo, L. Tetrahedron: Asymmetry 2004, 15, 2555–2559. doi:10.1016/j.tetasy.2004.06.037
Return to citation in text: [1] -
Prabhu, K. R.; Sivanand, P. S.; Chandrsekaran, S. Synlett 1998, 1998, 47–48. doi:10.1055/s-1998-1057884
Return to citation in text: [1] -
Takács, M.; Vámos, J.; Tóth, G.; Mikó-Hideg, Z. Arch. Pharm. 2000, 333, 48–52. doi:10.1002/(SICI)1521-4184(200002)333:2/3<48::AID-ARDP48>3.0.CO;2-N
Return to citation in text: [1] -
Díaz-Gavilán, M.; Rodríguez-Serrano, F.; Gómez-Vidal, J. A.; Marchal, J. A.; Aránega, A.; Gallo, M. Á.; Espinosa, A.; Campos, J. M. Tetrahedron 2004, 60, 11547–11557. doi:10.1016/j.tet.2004.09.072
Return to citation in text: [1] -
Mishra, J. K.; Samanta, K.; Jain, M.; Dikshit, M.; Panda, G. Bioorg. Med. Chem. Lett. 2010, 20, 244–247. doi:10.1016/j.bmcl.2009.10.126
Return to citation in text: [1]
| 1. | Akritopoulou-Zanze, I.; Djuric, S. W. Heterocycles 2007, 73, 125–147. doi:10.3987/REV-07-SR(U)3 |
| 2. | Banfi, L.; Basso, A.; Riva, R. In Synthesis of Heterocycles via Multicomponent Reactions I; Orru, R. V. A.; Ruijter, E., Eds.; Springer: Berlin/Heidelberg, 2010; Vol. 23, pp 1–39. |
| 3. | Banfi, L.; Riva, R.; Basso, A. Synlett 2010, 23–41. doi:10.1055/s-0029-1218527 |
| 4. | Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728 |
| 5. | Hulme, C.; Dietrich, J. Mol. Div. 2009, 13, 195–207. doi:10.1007/s11030-009-9111-6 |
| 8. | Bowers, M. M.; Carroll, P.; Joullié, M. M. J. Chem. Soc., Perkin Trans. 1 1989, 857–865. doi:10.1039/P19890000857 |
| 14. | Sperger, C. A.; Mayer, P.; Wanner, K. T. Tetrahedron 2009, 65, 10463–10469. doi:10.1016/j.tet.2009.10.020 |
| 16. | El Kaïm, L.; Grimaud, L.; Oble, J.; Wagschal, S. Tetrahedron Lett. 2009, 50, 1741–1743. doi:10.1016/j.tetlet.2009.01.145 |
| 9. | Znabet, A.; Ruijter, E.; de Kanter, F. J. J.; Köhler, V.; Helliwell, M.; Turner, N. J.; Orru, R. V. A. Angew. Chem., Int. Ed. 2010, 49, 5289–5292. doi:10.1002/anie.201001592 |
| 10. | Bonger, K. M.; Wennekes, T.; Filippov, D. V.; Lodder, G.; van der Marel, G. A.; Overkleeft, H. S. Eur. J. Org. Chem. 2008, 3678–3688. doi:10.1002/ejoc.200800340 |
| 11. | Chapman, T. M.; Davies, I. G.; Gu, B.; Block, T. M.; Scopes, D. I. C.; Hay, P. A.; Courtney, S. M.; McNeill, L. A.; Schofield, C. J.; Davis, B. G. J. Am. Chem. Soc. 2005, 127, 506–507. doi:10.1021/ja043924l |
| 12. | Timmer, M. S. M.; Risseeuw, M. D. P.; Verdoes, M.; Filippov, D. V.; Plaisier, J. R.; van der Marel, G. A.; Overkleeft, H. S.; van Boom, J. H. Tetrahedron: Asymmetry 2005, 16, 177–185. doi:10.1016/j.tetasy.2004.11.079 |
| 13. | Maison, W.; Lützen, A.; Kosten, M.; Schlemminger, I.; Westerhoff, O.; Saak, W.; Martens, J. J. Chem. Soc., Perkin Trans. 1 2000, 1867–1871. doi:10.1039/B002258F |
| 14. | Sperger, C. A.; Mayer, P.; Wanner, K. T. Tetrahedron 2009, 65, 10463–10469. doi:10.1016/j.tet.2009.10.020 |
| 15. | Zhu, D.; Chen, R.; Liang, H.; Li, S.; Pan, L.; Chen, X. Synlett 2010, 897–900. doi:10.1055/s-0029-1219533 |
| 7. | Nutt, R. F.; Joullié, M. M. J. Am. Chem. Soc. 1982, 104, 5852–5853. doi:10.1021/ja00385a080 |
| 8. | Bowers, M. M.; Carroll, P.; Joullié, M. M. J. Chem. Soc., Perkin Trans. 1 1989, 857–865. doi:10.1039/P19890000857 |
| 6. | Banfi, L.; Basso, A.; Guanti, R.; Riva, R. Asymmetric Isocyanide-Based MCRs. In Multicomponent Reactions; Zhu, J. P.; Bienaymé, H., Eds.; Wiley: Weinheim, 2005; pp 1–32. doi:10.1002/3527605118.ch1 |
| 20. | Gulevich, A. V.; Shevchenko, N. F.; Balenkova, E. S.; Roeschenthaler, G. V.; Nenajdenko, V. G. Synlett 2009, 403–406. doi:10.1055/s-0028-1087529 |
| 21. | Recently, a very high diastereoselectivity leading to seven-membered ring was obtained in an intramolecular Ugi reaction of an oxoacid endowed with axial chirality: Mehta, V. P.; Modha, S. G.; Ruijter, E.; Van Hecke, K.; Van Meervelt, L.; Pannecouque, C.; Balzarini, J.; Orru, R. V. A.; Van der Eycken, E. J. Org. Chem. 2011, 76, 2828–2839. doi:10.1021/jo200251q |
| 14. | Sperger, C. A.; Mayer, P.; Wanner, K. T. Tetrahedron 2009, 65, 10463–10469. doi:10.1016/j.tet.2009.10.020 |
| 16. | El Kaïm, L.; Grimaud, L.; Oble, J.; Wagschal, S. Tetrahedron Lett. 2009, 50, 1741–1743. doi:10.1016/j.tetlet.2009.01.145 |
| 18. | Banfi, L.; Basso, A.; Guanti, G.; Merlo, S.; Repetto, C.; Riva, R. Tetrahedron 2008, 64, 1114–1134. doi:10.1016/j.tet.2007.10.058 |
| 19. | Iizuka, T.; Takiguchi, S.; Kumakura, Y.; Tsukioka, N.; Higuchi, K.; Kawasaki, T. Tetrahedron Lett. 2010, 51, 6003–6005. doi:10.1016/j.tetlet.2010.09.026 |
| 25. | Díaz-Gavilán, M.; Rodríguez-Serrano, F.; Gómez-Vidal, J. A.; Marchal, J. A.; Aránega, A.; Gallo, M. Á.; Espinosa, A.; Campos, J. M. Tetrahedron 2004, 60, 11547–11557. doi:10.1016/j.tet.2004.09.072 |
| 26. | Mishra, J. K.; Samanta, K.; Jain, M.; Dikshit, M.; Panda, G. Bioorg. Med. Chem. Lett. 2010, 20, 244–247. doi:10.1016/j.bmcl.2009.10.126 |
| 13. | Maison, W.; Lützen, A.; Kosten, M.; Schlemminger, I.; Westerhoff, O.; Saak, W.; Martens, J. J. Chem. Soc., Perkin Trans. 1 2000, 1867–1871. doi:10.1039/B002258F |
| 15. | Zhu, D.; Chen, R.; Liang, H.; Li, S.; Pan, L.; Chen, X. Synlett 2010, 897–900. doi:10.1055/s-0029-1219533 |
| 17. | Gröger, H.; Hatam, M.; Martens, J. Tetrahedron 1995, 51, 7173–7180. doi:10.1016/0040-4020(95)00365-F |
| 14. | Sperger, C. A.; Mayer, P.; Wanner, K. T. Tetrahedron 2009, 65, 10463–10469. doi:10.1016/j.tet.2009.10.020 |
| 16. | El Kaïm, L.; Grimaud, L.; Oble, J.; Wagschal, S. Tetrahedron Lett. 2009, 50, 1741–1743. doi:10.1016/j.tetlet.2009.01.145 |
| 22. | Del Buttero, P.; Molteni, G.; Papagni, A.; Miozzo, L. Tetrahedron: Asymmetry 2004, 15, 2555–2559. doi:10.1016/j.tetasy.2004.06.037 |
| 23. | Prabhu, K. R.; Sivanand, P. S.; Chandrsekaran, S. Synlett 1998, 1998, 47–48. doi:10.1055/s-1998-1057884 |
| 24. | Takács, M.; Vámos, J.; Tóth, G.; Mikó-Hideg, Z. Arch. Pharm. 2000, 333, 48–52. doi:10.1002/(SICI)1521-4184(200002)333:2/3<48::AID-ARDP48>3.0.CO;2-N |
© 2011 Banfi et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)