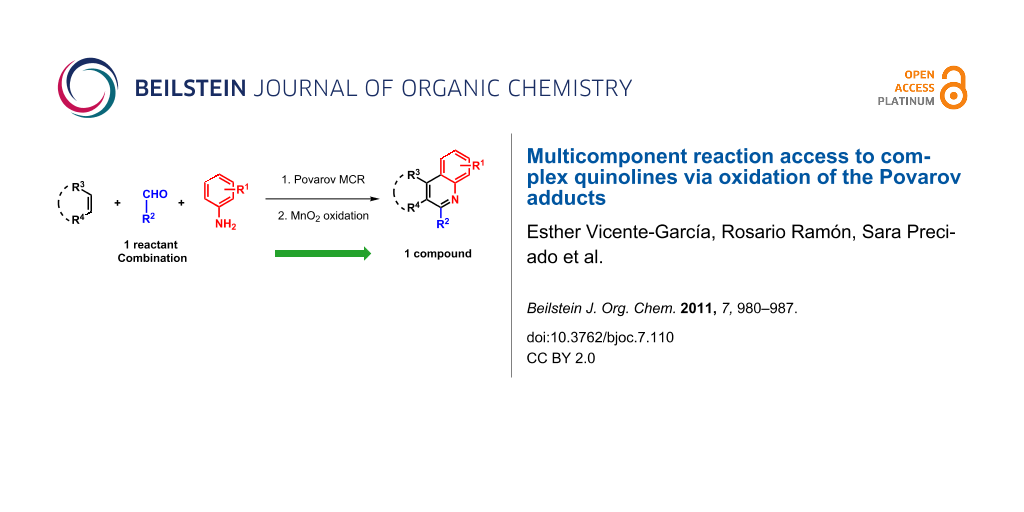
Abstract
The tetrahydroquinolines obtained through the Povarov multicomponent reaction have been oxidized to the corresponding quinoline, giving access to a single product through a two-step sequence. Several oxidizing agents were studied and manganese dioxide proved to be the reagent of choice, affording higher yields, cleaner reactions and practical protocols.

Graphical Abstract
Introduction
Heterocycles are ubiquitous scaffolds in pharmaceuticals, natural products and biologically active compounds. Quinoline systems in particular constitute a privileged substructure and are present in a large number of compounds with remarkable biological activity [1]. Although a variety of methods are used to prepare these heterocyclic compounds, the synthetic access to polysubstituted-polyfunctionalized derivatives remains a serious challenge [2]. Multistep sequences are widespread in the literature, but even in these cases the preparation of some substitution patterns and functional group combinations is particularly difficult. The recent introduction of multicomponent reactions (MCRs) into this field has brought interesting features typical of the ideal reaction, such as atom- and step economy, convergence, and exploratory power, together with new avenues in connectivity, leading to the straightforward synthesis of previously unobtainable scaffolds [3]. In this context, it is possible to obtain a wide variety of complex tetrahydroquinolines through the Povarov MCR (the interaction of anilines, aldehydes and activated olefin inputs under acid catalysis) [4-8]. Interestingly, this process allows cyclic enol ethers and enamines to be used as electron-rich alkenes, leading to heterocycle-fused tetrahydroquinolines, usually as a mixture of stereoisomers [9-13]. Unfortunately, no general methods for enantioselective Povarov reactions have been developed (for examples of catalytic enantioselective transformations operating in particular systems, see [14,15]), and this constitutes a serious drawback in the use of this reaction for library preparation, as one reaction affords several products, when ideally it should give only one. However, these adducts can be subjected to oxidation, which will lead to the corresponding quinolines, preserving the substituents and functionalization already introduced in the preceding MCR. Despite the loss of all stereochemical information, in this way it would be feasible to obtain a single product from a multicomponent process (Scheme 1).
![[1860-5397-7-110-i1]](/bjoc/content/inline/1860-5397-7-110-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Povarov oxidation access to substituted quinolines.
Scheme 1: Povarov oxidation access to substituted quinolines.
The oxidation step itself is challenging as it involves the formal removal of four hydrogens from a tetrahydropyridine moiety to reach the fully aromatic species. The literature contains scattered reports of the use of oxidants for this transformation: 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), ceric ammonium nitrate (CAN), nitrobenzene, elemental sulfur, palladium and manganese dioxide among others, all of them far from being ideally suited for these substrates.
One of the most commonly used is DDQ, which affords quinolines in acceptable yields. The main advantages of this oxidizing agent lie in its chemoselectivity and a requirement for relatively mild conditions, allowing it to be used in the presence of a wide range of substituents of the starting tetrahydroquinoline, such as O-, N- and C-linked residues (Scheme 2) [8,9,12,13,16-18]. Unfortunately, the alternative oxidation–elimination products (5 and 8) are often observed, therefore suggesting an acid catalyzed process. This would account for the elimination of alcohol and amine moieties, leading to dihydroquinoline intermediates that, after spontaneous oxidation in air, provide the final fragmented quinolines. The ability of DDQ to act as a Lewis acid and promote this alternative pathway has some precedent in the literature [19]. Furthermore, TFA treatment of Povarov adducts in oxygenated atmospheres also affords the oxidation–elimination products 5 and 8 (Scheme 2) [8,12,20].
![[1860-5397-7-110-i2]](/bjoc/content/inline/1860-5397-7-110-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The alternative oxidation–elimination pathway is predominant in some CAN-promoted oxidations of different Povarov adducts 3. Incidentally, this reagent is also used as a catalyst in the Povarov MCR without oxidative interference [18]. The same trend (oxidation–fragmentation) can be observed using nitrobenzene [21] as the oxidant. Analogously, elemental sulfur and palladium, although requiring drastic conditions, also lead to the fragmented quinolines when the substrates bear O- and N-substituents [22-25] (for related isoquinoline oxidations, see [26,27]).
Related oxidative processes involve, for instance, a cascade Povarov–hydrogen transfer reaction using Tf2NH as a catalyst and the imine as an oxidant, as recently described [28]. In addition, Povarov adducts resulting from the reaction between 3-aminocoumarin, aldehydes and cyclic enol ethers have been oxidized with different types of reagents, such as bromide, palladium, DDQ, sodium periodate, manganese dioxide or CAN, but in all cases the main product was the elimination–oxidation compound [29].
Finally, chemical manganese dioxide (CMD) has been widely used in this type of transformations, and already in 1982 the oxidation of tetrahydroisoquinoline (11, Scheme 2) was reported to yield the corresponding isoquinoline 12, the intermediate dihydroisoquinoline 13 being obtained as a by-product [26]. Later, Thompson et al. described the oxidation of fused pyrrolohydroquinolines (type 6) using MnO2 obtained from batteries. A kinetic competition between two processes was observed, and the desired double oxidation to the corresponding fused quinoline 7 took place, along with the oxidation–elimination sequence leading to 8. A large excess of oxidant was required in order to obtain the desired quinoline 7 as the major product (Scheme 2) [30,31].
Results and Discussion
Experiments were performed with the goal of developing a general and practical protocol for the oxidation of Povarov adducts to furnish the corresponding fused quinolines, avoiding elimination by-products. After unsuccessful attempts using palladium on carbon (decomposition), CuCl (partial oxidative elimination), Fremy’s salt (unreactive) and IBX (a complex reaction leading to unknown compounds), we focused our attention on MnO2 as the oxidant of choice. A literature search revealed different reactivity patterns depending on the type and origin of the reagent, with the commercial source being particularly important [32-36]. A systematic study was therefore conducted to determine the influence of different reaction conditions, commercial reagents and additives on the oxidation of an elimination-prone Povarov tetrahydroquinoline substrate.
In this way, tetrahydroquinolines 17,17' were synthesized as a mixture of isomers from the enol ether 14, p-bromoaniline (15) and p-chlorobenzaldehyde (16) under Sc(OTf)3 catalysis using standard reaction conditions (Scheme 3) [9]. Subsequently, these adducts 17,17' were oxidized with DDQ by the standard protocol [9], to isolate the desired quinoline 18 and its fragmented derivative 19, and they could also be subjected to an acid treatment to obtain selectively the latter product [8]. All compounds were purified and unequivocally characterized by NMR and HPLC methods.
![[1860-5397-7-110-i3]](/bjoc/content/inline/1860-5397-7-110-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the Povarov adducts and their oxidation products.
Scheme 3: Synthesis of the Povarov adducts and their oxidation products.
Taking into account that the oxidation of thiazolidines to thiazoles with MnO2 (25 equiv) in toluene (55 °C) in the presence of pyridine (1.25 equiv) is a clean and efficient method [35], a first experiment was set up to test these conditions with an old (≈40 years) MnO2 sample of unknown origin (particle size 11.46 µm, see below). A promising result was obtained, achieving a 39% conversion to the desired product 18, albeit with a high ratio of the elimination–oxidation compound 19. Next, the equivalents of oxidant and pyridine were increased to 100 and 6, respectively, and under these optimized conditions, a 72% isolated yield of quinoline 18 was obtained, and no starting material or elimination–oxidation compound was detected.
Unfortunately, we were not able to reproduce the above results when using brand new samples of MnO2. It was decided to test different commercially available MnO2 sources (Aldrich, Acros and Wako) of distinct activation degrees (particle size, powder or activated reagent, Table 1) in order to find a suitable reagent leading to comparable results.
Table 1: Survey of different MnO2 reagents.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-110-i5.svg?max-width=637&scale=1.0)
|
||||
| entry | MnO2 trademark, characteristics (reagent code) | particle size (median diameter, d50, µm)a | reaction conditions | product ratios (17,17')/18/19 |
|---|---|---|---|---|
| 1 |
Aldrich, reagent grade
(310700) |
4.3 | 25 equiv of oxidant | 54/3/43 |
| 2 |
Aldrich, reagent grade
(310700) |
4.3 | pyridine (50 equiv) | 48/8/44 |
| 3 |
Aldrich, reagent grade
(310700) |
4.3 | 25 equiv of oxidant K2CO3 (6 equiv) | 37/6/57 |
| 4 |
Aldrich, reagent grade
(310700) |
4.3 | 55 °C for 14 h | 37/13/50 |
| 5 |
Aldrich, reagent grade
(310700) |
4.3 | rt for 48 h | 51/0/49 |
| 6 |
Aldrich, reagent plus
(243442) |
138.4 | general conditionsb | 100/0/0 |
| 7 |
Aldrich, reagent plus
(243442) |
138.4 | 110 °C for 14 h | 61/0/39 |
| 8 |
Aldrich, activated
(217646) |
4.2 | general conditionsb | 8/14/78 |
| 9 |
Acros, powder
(213490010) |
7.6 | general conditionsb | 75/0/25 |
| 10 |
Wako, 1st grade powder
(138-09675) |
25.7 | general conditionsb | 0/100/0 |
aAll manganese dioxide samples were analyzed with a LSTM 13 320 series Laser diffraction particle size analyzer. For more details, see Supporting Information File 1. bUnless otherwise stated, the reactions were performed in toluene as the solvent, using 100 equiv of oxidant, 6 equiv of pyridine at 55 °C for 2 h.
Aldrich MnO2 (reagent grade) did not afford the desired quinoline 18 (entry 1, Table 1), the main products being the fragmented quinoline 19 and starting material. Modifications including the use of a greater excess of pyridine, the addition of K2CO3 as a heterogeneous base (entries 2 and 3), and adjustment of the reaction time or temperature (entries 4 and 5) did not substantially change the outcome. MnO2 (Aldrich, reagent plus) was completely inefficient at 55 °C (entry 6), and on heating to 110 °C for 14 h it promoted a 39% conversion but led exclusively to the elimination product (entry 7). On the other hand, using activated MnO2 (Aldrich), some oxidized quinoline 18 was observed, although again the predominant product was the fragmentation compound 19 (entry 8). Next, the reagents from Acros (entry 9) and Wako (entry 10) were tested, the latter being selective in the formation of the desired oxidation product, completely avoiding the elimination pathway. The results were reproducible, allowing the isolation of quinoline 18 in 66% yield in gram scale quantities.
In an attempt to improve the reaction conditions, Et3N was tested as a base, and molecular sieves (4 Å) and MgSO4 were introduced as dehydrating agents, but no meaningful changes were observed in any case. As the elimination–oxidation product 19 is thought to be generated by the acid characteristics of the oxidation reagents, an activated MnO2 sample was treated with an aqueous basic (NaCO3) solution, in an attempt to neutralize the acidic impurities, but the ratio of the elimination–oxidation product did not decrease. We then analysed the particle size of all samples using a laser diffraction technique (see Supporting Information File 1). Although a straightforward conclusion is not evident, it seems that all samples with a small (around 4 µm) or large particle size (138 µm) were inefficient in promoting the desired oxidation. On the other hand, medium size samples (Wako and the old sample of unknown origin) were the most selective oxidants (see Supporting Information File 1).
Using this reliable reagent, different reaction conditions were tested in order to optimize the process, especially regarding reagent consumption (Figure 1). The effects of varying the amounts of Wako MnO2 (from 10 to 100 equiv) and pyridine (from 2 to 20 equiv) in the standard solvent (toluene), reaction time and temperature (2 h, 55 °C) were studied. The gradual increment in the amount of oxidant resulted in a progressive increase in the yield of compound 18 and the simultaneous decrease of the elimination quinoline 19. No productive transformation to quinoline 18 was observed using 10 equiv of oxidant, the fragmented compound 19 being the predominant species. It is worth noting that the conversion of the starting material was only complete when at least 80 equiv of MnO2 were used, but even in these conditions the elimination pathway could not be completely avoided, despite the huge excess of pyridine (up to 20 equiv). As such large amounts of pyridine were not beneficial, the use of 6 equiv of this reagent was a practical compromize, leading to the same essential outcome. In an attempt to disaggregate the Wako MnO2 powder, and in this way reduce the amount of reagent, the reaction was performed in an ultrasonic bath under the general conditions, but no improvement was observed in the reaction profile.
![[1860-5397-7-110-1]](/bjoc/content/figures/1860-5397-7-110-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Optimization of the reaction conditions for the preparation of quinoline 18.
Figure 1: Optimization of the reaction conditions for the preparation of quinoline 18.
The optimized oxidation conditions were applied to another class of tetrahydroquinolines, which contain a fused lactam ring (20,20', Scheme 4) [12]. These new substrates were prepared through the Povarov MCR from the corresponding unsaturated lactam, aldehyde and aniline. The oxidation and elimination products (21 and 22, respectively) were independently prepared with DDQ under acid catalysis in an oxygenated atmosphere (O2-TFA), and characterized by NMR and HPLC methods. The optimized conditions with the Wako reagent were productive and selectively afforded the corresponding quinolines 21 in high yields, and the elimination product 22 was not detected. The processes were slower (5–8 h) than those involving the pyran-fused substrates 17,17' (Scheme 3). Interestingly, although DDQ is also capable of promoting these transformations, it is not as selective as Wako MnO2, and apart from yielding the fragmented quinolines 22, it also oxidizes the benzylic hydrogens (a series, R2 = Me) leading to the corresponding aldehyde derivative 21c [12]. Studies are ongoing to expand this set of transformations to fused oxygenated and nitrogenated 5-membered ring systems.
![[1860-5397-7-110-i4]](/bjoc/content/inline/1860-5397-7-110-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Oxidation of lactam-fused tetrahydroquinolines 20,20'.
Scheme 4: Oxidation of lactam-fused tetrahydroquinolines 20,20'.
Conclusion
In conclusion, we have described a fast, practical and reliable methodology to oxidize complex polysubstituted tetrahydroquinolines, arising from Povarov MCRs, to the corresponding quinolines, using MnO2. The influence of the reagent source, stoichiometry, additives and reaction conditions has been determined. Wako CMD is the oxidant of choice and the presence of pyridine is critical to avoid the fragmentation pathway, a side reaction often found in this type of transformation. This process enables the selective preparation of heterocycle-fused quinolines arising from a single combination of aldehydes, anilines and activated alkenes in a short sequence, involving Povarov MCR and oxidation steps.
Experimental
General
1H and 13C NMR spectra were recorded on a Varian Mercury 400 spectrometer. Unless otherwise stated, NMR spectra were recorded in CDCl3 solution with TMS as an internal reference. Data for 1H NMR spectra are reported as follows: Chemical shift (δ ppm), multiplicity, integration and coupling constants (Hz). Data for 13C NMR spectra are reported in terms of chemical shift (δ ppm). Signals were assigned by means of two-dimensional NMR spectroscopy: 1H,1H-COSY, 1H,13C-COSY (HSQC: heteronuclear single quantum coherence) and long-range 1H,13C-COSY (HMBC: heteronuclear multiple bond connectivity). IR spectra were recorded using a Thermo Nicolet Nexus spectrometer and are reported in wavenumbers (cm−1). High resolution mass spectrometry was performed by the University of Barcelona Mass Spectrometry Service.
General procedure A [9,12]
To a solution of compound 17,17' or 20,20' (1 mmol) in 15 mL of CHCl3, DDQ (2 mmol) was added and the mixture was stirred for 24 h in an open vessel at room temperature. An aqueous saturated NaHCO3 solution (10 mL) was added, and the resulting mixture was extracted with CHCl3 (3 × 10 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The reaction mixture was purified by flash chromatography (hexane–EtOAc) to afford the desired product.
General procedure B [9,12]
To a solution of compound 17,17' or 20,20' (1 mmol) in CH3CN/H2O or CHCl3/H2O (1:1, 6 mL), TFA (2 mmol) was added. The reaction mixture was stirred for 24 h at room temperature, quenched with an aqueous saturated NaHCO3 solution (10 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo to give a residue which was purified by flash chromatography (hexane–ethyl acetate) to afford the desired product.
General procedure C
To a solution of compound 17,17' or 20,20' (1 mmol) in 50 mL of toluene, pyridine (6 mmol) and MnO2 Wako (100 mmol) were added and the mixture was stirred in an open vessel at 55 °C. The progress of the reaction was controlled by TLC or HPLC, until the starting material completely disappeared or no evolution was observed. The crude mixture was filtered through Celite, and the filtrate was concentrated in vacuo. The reaction mixture was purified by flash chromatography (hexane–EtOAc) to afford the desired product.
9-bromo-5-(4-chlorophenyl)-3,4-dihydro-2H-pyrano[3,2-c]quinoline (18)
Following the general procedure A, the oxidation of 17,17' afforded compound 18 as a white solid (68%). Following the general procedure C for 2 h with Wako MnO2, the oxidation of 17,17' afforded compound 18 as a white solid (66%). 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 2.2 Hz, 1H), 7.85 (d, J = 8.9 Hz, 1H), 7.70 (dd, J = 2.3, 8.9 Hz, 1H), 7.53–7.48 (m, 2H), 7.45–7.40 (m, 2H), 4.46–4.39 (m, 2H), 2.72 (t, J = 6.3 Hz, 2H), 2.03–1.97 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 159.91, 156.71, 145.86, 138.58, 134.58, 132.71, 130.76, 130.24, 128.54, 123.91, 121.18, 119.46, 111.35, 67.21, 23.80, 21.75; IR (film): 3319, 3058, 2987, 2949, 2917, 2859, 1905, 1585, 1476, 1392, 1348, 1322, 1162, 1123, 1085, 989 cm−1; HRMS (ESI+, m/z): [M + H]+ calcd for C18H14BrClNO, 373.9942; found, 373.9933.
3-(6-bromo-2-(4-chlorophenyl)quinolin-3-yl)propan-1-ol (19)
Following the general procedure B, the oxidation of 17,17' afforded compound 19 as a white solid (60%). 1H NMR (400 MHz, CDCl3) δ 7.92–7.87 (m, 3H), 7.67 (dd, J = 2.2, 8.9 Hz, 1H), 7.44–7.37 (m, 4H), 3.51 (t, J = 6.2 Hz, 2H), 2.85–2.77 (m, 2H), 1.75–1.65 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 159.9, 145.2, 139.0, 135.3, 134.8, 134.3, 132.8, 131.2, 130.3, 129.2, 128.9, 128.9, 120.8, 62.0, 33.3, 29.4; IR (film): 3353, 2924, 2847, 1783, 1732, 1598, 1476, 1431, 1393, 1258, 1188, 1085, 1059, 1009, 919, 823 cm−1; HRMS (ESI+, m/z): [M + H]+ calcd for C18H16BrClNO, 376.0098; found, 376.0090.
Supporting Information
Supporting information features the characterization data of compounds 18, 19, 21 and 22, copies of their 1H NMR and 13C NMR spectra, and the particle size analyses of MnO2 samples.
| Supporting Information File 1: Experimental details. | ||
| Format: PDF | Size: 1.1 MB | Download |
Acknowledgements
This work was supported by DGICYT – Spain (project BQUCTQ2009-07758), Generalitat de Catalunya (project 2009SGR 1024) and Barcelona Science Park. Grupo Ferrer (Barcelona, Spain) is thanked for financial support. We thank Dr. Pedro Grima (Grupo Ferrer, R+D center, Barcelona) for useful comments.
References
-
Eicher, T.; Hauptmann, S. The Chemistry of Heterocycles, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 316–336. doi:10.1002/352760183X
Return to citation in text: [1] -
Larsen, L. D.; Cai, D. Quinolines. In Six-Membered Hetarenes with One Nitrogen or Phosphorus Atom; Black, D., Ed.; Science of Synthesis, Vol. 15; Georg Thieme Verlag: Stuttgart, Germany, 2005; pp 551–660.
Return to citation in text: [1] -
Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Povarov, L. S. Russ. Chem. Rev. 1967, 36, 656–670. doi:10.1070/RC1967v036n09ABEH001680
Return to citation in text: [1] -
Kouznetsov, V. V. Tetrahedron 2009, 65, 2721–2750. doi:10.1016/j.tet.2008.12.059
Return to citation in text: [1] -
Glushkov, V. A.; Tolstikov, A. G. Russ. Chem. Rev. 2008, 77, 137–159. doi:10.1070/RC2008v077n02ABEH003749
Return to citation in text: [1] -
Bello, D.; Ramón, R.; Lavilla, R. Curr. Org. Chem. 2010, 14, 332–356. doi:10.2174/138527210790231883
Return to citation in text: [1] -
Smith, C. D.; Gavrilyuk, J. I.; Lough, A. J.; Batey, R. A. J. Org. Chem. 2010, 75, 702–715. doi:10.1021/jo9021106
Return to citation in text: [1] [2] [3] [4] -
Jiménez, O.; de la Rosa, G.; Lavilla, R. Angew. Chem., Int. Ed. 2005, 44, 6521–6525. doi:10.1002/anie.200501548
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Camps, P.; Formosa, X.; Galdeano, C.; Muñoz-Torrero, D.; Ramírez, L.; Gómez, E.; Isambert, N.; Lavilla, R.; Badia, A.; Clos, M. V.; Bartolini, M.; Mancini, F.; Andrisano, V.; Arce, M. P.; Rodríguez-Franco, M. I.; Huertas, O.; Dafni, T.; Luque, F. J. J. Med. Chem. 2009, 52, 5365–5379. doi:10.1021/jm900859q
Return to citation in text: [1] -
Powell, D. A.; Batey, R. A. Org. Lett. 2002, 4, 2913–2916. doi:10.1021/ol026293d
Return to citation in text: [1] -
Vicente-García, E.; Catti, F.; Ramón, R.; Lavilla, R. Org. Lett. 2010, 12, 860–863. doi:10.1021/ol902913j
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Stevenson, P. J.; Graham, I. ARKIVOC 2003, vii, 139–144.
Return to citation in text: [1] [2] -
Liu, H.; Dagousset, G.; Masson, G.; Retailleau, P.; Zhu, J. J. Am. Chem. Soc. 2009, 131, 4598–4599. doi:10.1021/ja900806q
Return to citation in text: [1] -
Ishitani, H.; Kobayashi, S. Tetrahedron Lett. 1996, 37, 7357–7360. doi:10.1016/0040-4039(96)01655-3
Return to citation in text: [1] -
Rano, T. A.; Sieber-McMaster, E.; Pelton, P. D.; Yang, M.; Demarest, K. T.; Kuo, G.-H. Bioorg. Med. Chem. Lett. 2009, 19, 2456–2460. doi:10.1016/j.bmcl.2009.03.051
Return to citation in text: [1] -
Spaller, M. R.; Thielemann, W. T.; Brennan, P. E.; Bartlett, P. A. J. Comb. Chem. 2002, 4, 516–522. doi:10.1021/cc020027+
Return to citation in text: [1] -
Sridharan, V.; Avendaño, C.; Menéndez, J. C. Tetrahedron 2009, 65, 2087–2096. doi:10.1016/j.tet.2008.12.077
Return to citation in text: [1] [2] -
Oku, A.; Kinugasa, M.; Kamada, T. Chem. Lett. 1993, 22, 165–168. doi:10.1246/cl.1993.165
Return to citation in text: [1] -
Borrione, E.; Prato, M.; Scorrano, G.; Stivanello, M.; Lucchini, V. J. Heterocycl. Chem. 1988, 25, 1831–1835. doi:10.1002/jhet.5570250644
Return to citation in text: [1] -
Zaitsev, V. P.; Mikhailova, N. M.; Orlova, D. N.; Nikitina, E. V.; Boltukhina, E. V.; Zubkov, F. I. Chem. Heterocycl. Compd. 2009, 45, 308–316. doi:10.1007/s10593-009-0271-1
Return to citation in text: [1] -
Kouznetsov, V. V.; Bohórquez, A. R. R.; Saavedra, L. A. Synthesis 2009, 4219–4225. doi:10.1055/s-0029-1217049
Return to citation in text: [1] -
Fadel, F.; Titouani, S. L.; Soufiaoui, M.; Ajamay, H.; Mazzah, A. Tetrahedron Lett. 2004, 45, 5905–5908. doi:10.1016/j.tetlet.2004.05.127
Return to citation in text: [1] -
Saavedra, L. A.; Vallejos C., G.; Kouznetsov, V. V.; Gutierrez C., M.; Meléndez Gómez, C. M.; Vargas Méndez, L. Y.; Bermúdez Jaimes, J. H. Synthesis 2010, 593–600. doi:10.1055/s-0029-1218613
Return to citation in text: [1] -
Kouznetsov, V. V.; Meléndez Gomez, C. M.; Bermúdez Jaimes, J. H. J. Heterocycl. Chem. 2010, 47, 1148–1152. doi:10.1002/jhet.441
Return to citation in text: [1] -
Kashdan, D. S.; Schwartz, J. A.; Rapoport, H. J. Org. Chem. 1982, 47, 2638–2643. doi:10.1021/jo00134a025
Return to citation in text: [1] [2] -
Teske, J. A.; Deiters, A. J. Org. Chem. 2008, 73, 342–345. doi:10.1021/jo7020955
Return to citation in text: [1] -
Shindoh, N.; Tokuyama, H.; Takemoto, Y.; Takasu, K. J. Org. Chem. 2008, 73, 7451–7456. doi:10.1021/jo8009243
Return to citation in text: [1] -
Kudale, A. A.; Kendell, J.; Miller, D. O.; Collins, J. L.; Bodwell, G. J. J. Org. Chem. 2008, 73, 8437–8447. doi:10.1021/jo801411p
Return to citation in text: [1] -
Hadden, M.; Nieuwenhuyzen, M.; Potts, D.; Stevenson, P. J.; Thompson, N. Tetrahedron 2001, 57, 5615–5624. doi:10.1016/S0040-4020(01)00466-5
Return to citation in text: [1] -
Kajino, M.; Hird, N. W.; Tarui, N.; Banno, H.; Kawano, Y.; Inatomi, N. Fused quinoline derivativeand use thereof. PCT WO2005/105802 A1, Nov 10, 2005.
Chem. Abstr. 2005, 143, 440393.
Return to citation in text: [1] -
Lulinski, P.; Krassowska-Swiebocka, B.; Skulski, L. Molecules 2004, 9, 595–601. doi:10.3390/90700595
Return to citation in text: [1] -
Aoyama, T.; Sonoda, N.; Yamauchi, M.; Toriyama, K.; Anzai, M.; Ando, A.; Shioiri, T. Synlett 1998, 35–36. doi:10.1055/s-1998-1583
Return to citation in text: [1] -
Bhatnagar, I.; George, M. V. J. Org. Chem. 1968, 33, 2407–2411. doi:10.1021/jo01270a052
Return to citation in text: [1] -
Hamada, Y.; Shibata, M.; Sugiura, T.; Kato, S.; Shioiri, T. J. Org. Chem. 1987, 52, 1252–1255. doi:10.1021/jo00383a014
Return to citation in text: [1] [2] -
Sekine, Y. Atmos. Environ. 2002, 36, 5543–5547. doi:10.1016/S1352-2310(02)00670-2
Return to citation in text: [1]
| 1. | Eicher, T.; Hauptmann, S. The Chemistry of Heterocycles, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 316–336. doi:10.1002/352760183X |
| 9. | Jiménez, O.; de la Rosa, G.; Lavilla, R. Angew. Chem., Int. Ed. 2005, 44, 6521–6525. doi:10.1002/anie.200501548 |
| 10. | Camps, P.; Formosa, X.; Galdeano, C.; Muñoz-Torrero, D.; Ramírez, L.; Gómez, E.; Isambert, N.; Lavilla, R.; Badia, A.; Clos, M. V.; Bartolini, M.; Mancini, F.; Andrisano, V.; Arce, M. P.; Rodríguez-Franco, M. I.; Huertas, O.; Dafni, T.; Luque, F. J. J. Med. Chem. 2009, 52, 5365–5379. doi:10.1021/jm900859q |
| 11. | Powell, D. A.; Batey, R. A. Org. Lett. 2002, 4, 2913–2916. doi:10.1021/ol026293d |
| 12. | Vicente-García, E.; Catti, F.; Ramón, R.; Lavilla, R. Org. Lett. 2010, 12, 860–863. doi:10.1021/ol902913j |
| 13. | Stevenson, P. J.; Graham, I. ARKIVOC 2003, vii, 139–144. |
| 29. | Kudale, A. A.; Kendell, J.; Miller, D. O.; Collins, J. L.; Bodwell, G. J. J. Org. Chem. 2008, 73, 8437–8447. doi:10.1021/jo801411p |
| 4. | Povarov, L. S. Russ. Chem. Rev. 1967, 36, 656–670. doi:10.1070/RC1967v036n09ABEH001680 |
| 5. | Kouznetsov, V. V. Tetrahedron 2009, 65, 2721–2750. doi:10.1016/j.tet.2008.12.059 |
| 6. | Glushkov, V. A.; Tolstikov, A. G. Russ. Chem. Rev. 2008, 77, 137–159. doi:10.1070/RC2008v077n02ABEH003749 |
| 7. | Bello, D.; Ramón, R.; Lavilla, R. Curr. Org. Chem. 2010, 14, 332–356. doi:10.2174/138527210790231883 |
| 8. | Smith, C. D.; Gavrilyuk, J. I.; Lough, A. J.; Batey, R. A. J. Org. Chem. 2010, 75, 702–715. doi:10.1021/jo9021106 |
| 26. | Kashdan, D. S.; Schwartz, J. A.; Rapoport, H. J. Org. Chem. 1982, 47, 2638–2643. doi:10.1021/jo00134a025 |
| 3. | Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. |
| 26. | Kashdan, D. S.; Schwartz, J. A.; Rapoport, H. J. Org. Chem. 1982, 47, 2638–2643. doi:10.1021/jo00134a025 |
| 27. | Teske, J. A.; Deiters, A. J. Org. Chem. 2008, 73, 342–345. doi:10.1021/jo7020955 |
| 2. | Larsen, L. D.; Cai, D. Quinolines. In Six-Membered Hetarenes with One Nitrogen or Phosphorus Atom; Black, D., Ed.; Science of Synthesis, Vol. 15; Georg Thieme Verlag: Stuttgart, Germany, 2005; pp 551–660. |
| 28. | Shindoh, N.; Tokuyama, H.; Takemoto, Y.; Takasu, K. J. Org. Chem. 2008, 73, 7451–7456. doi:10.1021/jo8009243 |
| 8. | Smith, C. D.; Gavrilyuk, J. I.; Lough, A. J.; Batey, R. A. J. Org. Chem. 2010, 75, 702–715. doi:10.1021/jo9021106 |
| 12. | Vicente-García, E.; Catti, F.; Ramón, R.; Lavilla, R. Org. Lett. 2010, 12, 860–863. doi:10.1021/ol902913j |
| 20. | Borrione, E.; Prato, M.; Scorrano, G.; Stivanello, M.; Lucchini, V. J. Heterocycl. Chem. 1988, 25, 1831–1835. doi:10.1002/jhet.5570250644 |
| 21. | Zaitsev, V. P.; Mikhailova, N. M.; Orlova, D. N.; Nikitina, E. V.; Boltukhina, E. V.; Zubkov, F. I. Chem. Heterocycl. Compd. 2009, 45, 308–316. doi:10.1007/s10593-009-0271-1 |
| 19. | Oku, A.; Kinugasa, M.; Kamada, T. Chem. Lett. 1993, 22, 165–168. doi:10.1246/cl.1993.165 |
| 22. | Kouznetsov, V. V.; Bohórquez, A. R. R.; Saavedra, L. A. Synthesis 2009, 4219–4225. doi:10.1055/s-0029-1217049 |
| 23. | Fadel, F.; Titouani, S. L.; Soufiaoui, M.; Ajamay, H.; Mazzah, A. Tetrahedron Lett. 2004, 45, 5905–5908. doi:10.1016/j.tetlet.2004.05.127 |
| 24. | Saavedra, L. A.; Vallejos C., G.; Kouznetsov, V. V.; Gutierrez C., M.; Meléndez Gómez, C. M.; Vargas Méndez, L. Y.; Bermúdez Jaimes, J. H. Synthesis 2010, 593–600. doi:10.1055/s-0029-1218613 |
| 25. | Kouznetsov, V. V.; Meléndez Gomez, C. M.; Bermúdez Jaimes, J. H. J. Heterocycl. Chem. 2010, 47, 1148–1152. doi:10.1002/jhet.441 |
| 8. | Smith, C. D.; Gavrilyuk, J. I.; Lough, A. J.; Batey, R. A. J. Org. Chem. 2010, 75, 702–715. doi:10.1021/jo9021106 |
| 9. | Jiménez, O.; de la Rosa, G.; Lavilla, R. Angew. Chem., Int. Ed. 2005, 44, 6521–6525. doi:10.1002/anie.200501548 |
| 12. | Vicente-García, E.; Catti, F.; Ramón, R.; Lavilla, R. Org. Lett. 2010, 12, 860–863. doi:10.1021/ol902913j |
| 13. | Stevenson, P. J.; Graham, I. ARKIVOC 2003, vii, 139–144. |
| 16. | Rano, T. A.; Sieber-McMaster, E.; Pelton, P. D.; Yang, M.; Demarest, K. T.; Kuo, G.-H. Bioorg. Med. Chem. Lett. 2009, 19, 2456–2460. doi:10.1016/j.bmcl.2009.03.051 |
| 17. | Spaller, M. R.; Thielemann, W. T.; Brennan, P. E.; Bartlett, P. A. J. Comb. Chem. 2002, 4, 516–522. doi:10.1021/cc020027+ |
| 18. | Sridharan, V.; Avendaño, C.; Menéndez, J. C. Tetrahedron 2009, 65, 2087–2096. doi:10.1016/j.tet.2008.12.077 |
| 14. | Liu, H.; Dagousset, G.; Masson, G.; Retailleau, P.; Zhu, J. J. Am. Chem. Soc. 2009, 131, 4598–4599. doi:10.1021/ja900806q |
| 15. | Ishitani, H.; Kobayashi, S. Tetrahedron Lett. 1996, 37, 7357–7360. doi:10.1016/0040-4039(96)01655-3 |
| 18. | Sridharan, V.; Avendaño, C.; Menéndez, J. C. Tetrahedron 2009, 65, 2087–2096. doi:10.1016/j.tet.2008.12.077 |
| 9. | Jiménez, O.; de la Rosa, G.; Lavilla, R. Angew. Chem., Int. Ed. 2005, 44, 6521–6525. doi:10.1002/anie.200501548 |
| 30. | Hadden, M.; Nieuwenhuyzen, M.; Potts, D.; Stevenson, P. J.; Thompson, N. Tetrahedron 2001, 57, 5615–5624. doi:10.1016/S0040-4020(01)00466-5 |
| 31. |
Kajino, M.; Hird, N. W.; Tarui, N.; Banno, H.; Kawano, Y.; Inatomi, N. Fused quinoline derivativeand use thereof. PCT WO2005/105802 A1, Nov 10, 2005.
Chem. Abstr. 2005, 143, 440393. |
| 32. | Lulinski, P.; Krassowska-Swiebocka, B.; Skulski, L. Molecules 2004, 9, 595–601. doi:10.3390/90700595 |
| 33. | Aoyama, T.; Sonoda, N.; Yamauchi, M.; Toriyama, K.; Anzai, M.; Ando, A.; Shioiri, T. Synlett 1998, 35–36. doi:10.1055/s-1998-1583 |
| 34. | Bhatnagar, I.; George, M. V. J. Org. Chem. 1968, 33, 2407–2411. doi:10.1021/jo01270a052 |
| 35. | Hamada, Y.; Shibata, M.; Sugiura, T.; Kato, S.; Shioiri, T. J. Org. Chem. 1987, 52, 1252–1255. doi:10.1021/jo00383a014 |
| 36. | Sekine, Y. Atmos. Environ. 2002, 36, 5543–5547. doi:10.1016/S1352-2310(02)00670-2 |
| 9. | Jiménez, O.; de la Rosa, G.; Lavilla, R. Angew. Chem., Int. Ed. 2005, 44, 6521–6525. doi:10.1002/anie.200501548 |
| 12. | Vicente-García, E.; Catti, F.; Ramón, R.; Lavilla, R. Org. Lett. 2010, 12, 860–863. doi:10.1021/ol902913j |
| 12. | Vicente-García, E.; Catti, F.; Ramón, R.; Lavilla, R. Org. Lett. 2010, 12, 860–863. doi:10.1021/ol902913j |
| 9. | Jiménez, O.; de la Rosa, G.; Lavilla, R. Angew. Chem., Int. Ed. 2005, 44, 6521–6525. doi:10.1002/anie.200501548 |
| 12. | Vicente-García, E.; Catti, F.; Ramón, R.; Lavilla, R. Org. Lett. 2010, 12, 860–863. doi:10.1021/ol902913j |
| 35. | Hamada, Y.; Shibata, M.; Sugiura, T.; Kato, S.; Shioiri, T. J. Org. Chem. 1987, 52, 1252–1255. doi:10.1021/jo00383a014 |
| 12. | Vicente-García, E.; Catti, F.; Ramón, R.; Lavilla, R. Org. Lett. 2010, 12, 860–863. doi:10.1021/ol902913j |
| 9. | Jiménez, O.; de la Rosa, G.; Lavilla, R. Angew. Chem., Int. Ed. 2005, 44, 6521–6525. doi:10.1002/anie.200501548 |
| 8. | Smith, C. D.; Gavrilyuk, J. I.; Lough, A. J.; Batey, R. A. J. Org. Chem. 2010, 75, 702–715. doi:10.1021/jo9021106 |
© 2011 Vicente-García et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)