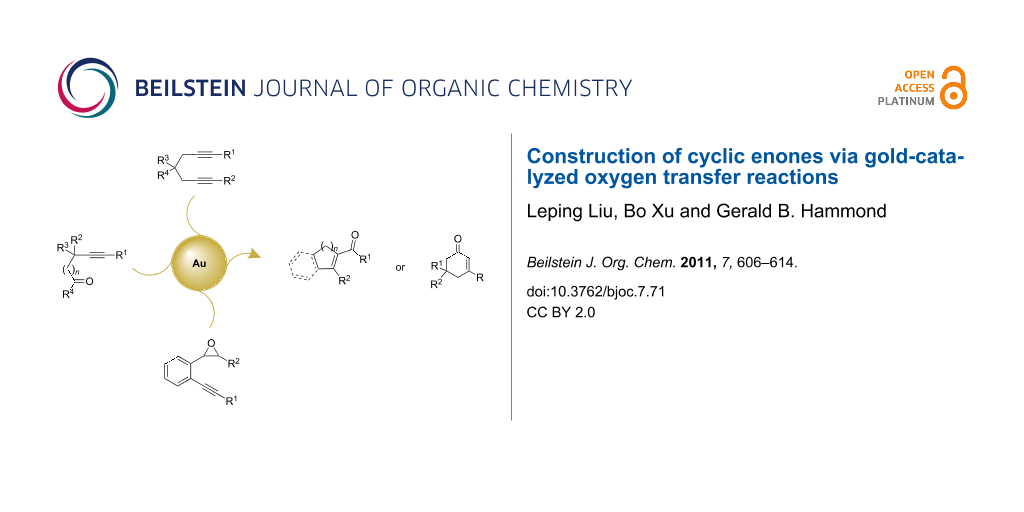
Abstract
During the last decade, gold-catalyzed reactions have become a tour de force in organic synthesis. Recently, the gold-, Brønsted acid- or Lewis acid-catalyzed oxygen transfer from carbonyl to carbon–carbon triple bond, the so-called alkyne–carbonyl metathesis, has attracted much attention because this atom economical transformation generates α,β-unsaturated carbonyl derivatives which are of great interest in synthetic organic chemistry. This mini-review focuses on the most recent achievements on gold-catalyzed oxygen transfer reactions of tethered alkynones, diynes or alkynyl epoxides to cyclic enones. The corresponding mechanisms for the transformations are also discussed.

Graphical Abstract
Review
α,β-Unsaturated carbonyl derivatives are not only important building blocks in synthetic organic chemistry, but are also a significant motif in natural products and biologically active compounds [1-8]. The construction of the conjugated enone substructure has attracted the interest of synthetic chemists for decades. Among numerous methodologies, aldol condensations and Wittig-type reactions have been widely utilized [9-18]. Recently, it was found that conjugated enones could be generated from the oxygen transfer from a carbonyl group to a carbon–carbon triple bond, the so-called alkyne–carbonyl metathesis. This methodology has sparked the attention of the synthetic community, because it could serve as an efficient and atom-economic alternative to the Wittig reaction by the formation of a new carbon–carbon double bond and the simultaneous installation of a carbonyl group. In this regard, several Lewis or Brønsted acid-catalyzed intermolecular or intramolecular alkyne–carbonyl metatheses have been extensively studied (Scheme 1) [19-27].
![[1860-5397-7-71-i1]](/bjoc/content/inline/1860-5397-7-71-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Lewis acid or Brønsted acid-catalyzed alkyne–carbonyl metathesis and a proposed [2 + 2] intermediate.
Scheme 1: Lewis acid or Brønsted acid-catalyzed alkyne–carbonyl metathesis and a proposed [2 + 2] intermediat...
During the early years of this century, organic chemists became aware that gold salts or complexes were highly active catalysts in homogeneous catalysis because of the strong π- and σ-electrophilicity of gold [28-33]. Since then, the number of new gold-catalyzed reactions reported in the literature has increased substantially and gold catalysis has become one of the hottest research fields in synthetic organic chemistry [34-42]. Due to their unique alkynophilicity, gold catalysts are especially suited to the activation of carbon–carbon triple bonds.
Gold-catalyzed formation of cyclic enones from alkynyl ketones
Yamamoto and co-workers were the first to report the gold-catalyzed formation of conjugated cyclic enones under mild conditions using tethered alkynyl ketones as substrates (Scheme 2) [43]. Both, aromatic and aliphatic groups substituted on alkynyl ketones 1 were investigated in this reaction, and the corresponding enone products 2 were isolated in good yields. They employed the alkyne–carbonyl metathesis in the preparation of fused ring systems and obtained two six-membered bicyclic products. However, if the original ring was five- or eight-membered, the reaction produced β,γ-unsaturated bicyclic enones rather than their α,β-unsaturated counterparts.
![[1860-5397-7-71-i2]](/bjoc/content/inline/1860-5397-7-71-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Gold-catalyzed cyclization of internal alkynyl ketones.
Scheme 2: Gold-catalyzed cyclization of internal alkynyl ketones.
Yamamoto and co-workers proposed a [2 + 2] mechanism for their gold-catalyzed cyclization of alkynyl ketones (Scheme 3). In their mechanism, the carbonyl group attacks the gold activated triple bond to form an oxonium intermediate, which then generates an oxetenium intermediate. After several electron transfer steps, the cyclic enone product is formed. A similar [2 + 2] pathway has also been invoked for the Brønsted acid- or Lewis acid-mediated intramolecular and intermolecular alkyne–aldehyde metatheses.
![[1860-5397-7-71-i3]](/bjoc/content/inline/1860-5397-7-71-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed [2 + 2] mechanism for the cyclization of alkynyl ketones.
Scheme 3: Proposed [2 + 2] mechanism for the cyclization of alkynyl ketones.
If terminal alkynyl ketone 3 is employed as the substrate, the reaction still furnishes α,β-unsaturated cyclic enone 4, but it necessitates a larger catalyst load (Scheme 4). By carefully monitoring of the reaction, it was found that intermediate 5 was formed together with a mixture of a hydrolyzed derivative of 6 and the final product 4. The isolated intermediate 5 could be transformed into a mixture of 6 and 4 under the reaction conditions, finally yielding 4 via intramolecular aldol condensation.
![[1860-5397-7-71-i4]](/bjoc/content/inline/1860-5397-7-71-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Gold-catalyzed cyclization of terminal alkynyl ketones.
Scheme 4: Gold-catalyzed cyclization of terminal alkynyl ketones.
This gold-catalyzed cyclization of alkynyl ketones to enones was successfully utilized in a cascade reaction by the same authors (Scheme 5) [44]. Using enynones 7 as the substrate, the gold-catalyzed tandem alkyne–carbonyl metathesis/Nazarov reaction generated a number of intriguing fused bicyclic, tricyclic and tetracyclic derivatives of 8 in moderate to good yields and excellent diastereoselectivity. In this case, the gold catalyst exhibited a dual role, namely the activation of alkyne and carbonyl moieties.
![[1860-5397-7-71-i5]](/bjoc/content/inline/1860-5397-7-71-i5.svg?scale=1.9&max-width=1024&background=FFFFFF)
Scheme 5: Gold-catalyzed tandem oxygen transfer/Nazarov cyclizations.
Scheme 5: Gold-catalyzed tandem oxygen transfer/Nazarov cyclizations.
Yamamoto and co-workers attempted to utilize their protocol to build five-membered cyclic enones, however, when they employed alkynyl ketone 9 as the substrate, the gold catalyst did not show good activity, and less than 30% of the desired product 10 was formed [45]. After optimizing the reaction conditions, the authors found that TfOH was the best catalyst for this oxygen transfer reaction in methanol (Scheme 6). This TfOH-mediated cyclization was applied to the synthesis of various fused tricyclic and tetracyclic derivatives of 10.
![[1860-5397-7-71-i6]](/bjoc/content/inline/1860-5397-7-71-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: TfOH-mediated cyclization of alkynyl ketones.
Scheme 6: TfOH-mediated cyclization of alkynyl ketones.
Hammond and co-workers found that the gold-catalyzed oxygen transfer reaction proceeded very smoothly when using alkynyldiketone 11 as the substrate (Scheme 7) [46]. Indeed, this reaction was complete in 5 minutes at room temperature to give the five-membered cyclic enones 12 cleanly and in excellent yields. The large reactivity difference between substrates 9 and 11 prompted the authors to propose an alternative [4 + 2] mechanism for this transformation, rather than the previously proposed and well-accepted [2 + 2] pathway for oxygen transfer reactions.
![[1860-5397-7-71-i7]](/bjoc/content/inline/1860-5397-7-71-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Gold-catalyzed cyclizations of 2-alkynyl-1,5-diketones.
Scheme 7: Gold-catalyzed cyclizations of 2-alkynyl-1,5-diketones.
An isotopic labeling experiment was designed to elucidate the pathway responsible for the gold-catalyzed intramolecular oxygen transfer of 2-alkynyl-1,5-diketones (Scheme 8). By introducing an 18O atom into one of the carbonyls of the substrate, and using the 13C NMR spectra of the substrate and product to locate the 18O atom, the authors hoped to elucidate the more favorable mechanistic pathway. The alkynyldiketone [18O]-11 was chosen as a model substrate. If the reaction follows a [2 + 2] route then 18O would end up on the left carbonyl group in [18O]-12a (Scheme 8, top), whereas it would be incorporated on the benzoyl group in [18O]-12b if the reaction follows a [4 + 2] pathway (Scheme 8, bottom).
![[1860-5397-7-71-i8]](/bjoc/content/inline/1860-5397-7-71-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Designed isotopic labeling experiment for mechanistic studies.
Scheme 8: Designed isotopic labeling experiment for mechanistic studies.
The result of this isotopic experiment is outlined in Scheme 9. Substrate [18O]-11 was synthesized from the 18O exchange of compound 11 with H218O under acidic conditions, and its 13C NMR spectrum showed that the 18O exchange happened only at the methyl carbonyl group (carbon 1). This substrate was subjected to the gold-catalyzed oxygen transfer reaction conditions and the product [18O]-12 was obtained in quantitative yield without any 18O loss. It was later found that the 18O was only incorporated into the benzoyl group (carbon 4) in product [18O]-12, as determined from its 13C NMR spectrum. The absence of any detectable 18O incorporation at carbon 3 demonstrates that the [2 + 2] pathway is disfavored, and instead it is the [4 + 2] pathway that is the favored mechanism in the gold-catalyzed intramolecular oxygen transfer of 2-alkynyl-1,5-diketones.
![[1860-5397-7-71-i9]](/bjoc/content/inline/1860-5397-7-71-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
The discovery of a [4 + 2] cycloaddition of a furanium intermediate to a carbonyl group was further verified by quantum chemical calculations. The competing [2 + 2] and [4 + 2] reaction coordinates were computed for the simplified substrate 11a, shown in Scheme 10. In accordance with the experimental findings, the [4 + 2] pathway is found to be the more favorable. The rate-limiting step in each pathway is the intramolecular nucleophilic addition to the Au-coordinated alkyne – the barrier for this step is computed to be 6.8 kcal/mol lower for the formation of the five-membered ring oxonium intermediate C than for the seven-membered ring oxonium A. This energetic preference is also observed in the stabilities of the oxoniums themselves, with C considerably more stable by 16.1 kcal/mol. The subsequent transformations are all computed to be feasible, with the barrier to [4 + 2] cyclization lying only 4.4 kcal/mol above the starting complex. Further calculations on the barrier for transition states were also consistent with the rapid conversion that was observed in the experiments. Overall, the large energetic preference of the intermediates and transition states for the [4 + 2] pathway over the [2 + 2] pathway supports the postulate that the [4 + 2] pathway is dominant in the gold-catalyzed oxygen transfer of 2-alkynyl-1,5-diketones, which is exactly in accordance with the 18O isotopic experiments.
![[1860-5397-7-71-i10]](/bjoc/content/inline/1860-5397-7-71-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: B2PLYP/6-311+G(d,p)//B2PLYP/6-31G(d) computed reaction profile, relative energies in kcal/mol.
Scheme 10: B2PLYP/6-311+G(d,p)//B2PLYP/6-31G(d) computed reaction profile, relative energies in kcal/mol.
Chan and co-workers developed a gold-catalyzed tandem intramolecular rearrangement of alkynyl arylaldehydes 13 to benzoxepinones 14 with good regioselectivity (Scheme 11) [47]. This transformation was effectively promoted by the addition of benzyl alcohol and the sequential addition of p-toluenesulfonic acid. However, in the absence of p-toluenesulfonic acid, benzyl ether 15 was isolated as the major product. The latter was considered to be an intermediate in the reaction and moreover, the isolated compound 15 could be transformed into the final product 14 under the mediation of p-toluenesulfonic acid.
![[1860-5397-7-71-i11]](/bjoc/content/inline/1860-5397-7-71-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Gold-catalyzed cyclization of tethered alkynyl arylaldehydes.
Scheme 11: Gold-catalyzed cyclization of tethered alkynyl arylaldehydes.
Gold-catalyzed formation of cyclic enones from diynes
Zhang and co-workers reported gold-catalyzed cyclizations to cyclohexenones 17, employing terminal 1,6-diynes 16 as substrates in the presence of a Brønsted acid and 1 equiv of water (Scheme 12) [48]. None of the desired products were obtained in the absence of the gold catalyst, the Brønsted acid or water. Interestingly, when the diacid 1,6-diyne (R1 = R2 = COOH) was employed in the reaction, only the esterified product (R1 = R2 = COOMe) was isolated, albeit in low yield. The authors also carried out this gold-catalyzed transformation in an ionic liquid [49]. This modification enabled the separation of the gold catalyst from the organic mixture and the recovered gold catalyst in the ionic liquid was re-used as many as five times without loss of activity.
![[1860-5397-7-71-i12]](/bjoc/content/inline/1860-5397-7-71-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Gold-catalyzed cyclization of terminal diynes.
Scheme 12: Gold-catalyzed cyclization of terminal diynes.
A hydrolysis/cyclization mechanism was proposed for the transformation (Scheme 13). Although this mechanism is plausible, another option for the cyclization step might exist. One of the key intermediates in the catalytic cycle is the hydrolyzed product – the alkynyl ketone from hydrolysis of one triple bond – which is the same as the substrate that was employed by Yamamoto and co-workers. Thus, a similar diketone intermediate 6' could also have been formed before being transformed into the final product via intramolecular aldol condensation.
![[1860-5397-7-71-i13]](/bjoc/content/inline/1860-5397-7-71-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Proposed hydrolysis/cyclization mechanism.
Scheme 13: Proposed hydrolysis/cyclization mechanism.
Fiksdahl and co-workers investigated a similar gold-catalyzed transformation of internal 1,6-diynes 18 in methanol at room temperature (Scheme 14) [50,51]. Interestingly, a non-conjugated five-membered cyclic enone 19 was isolated as the product, instead of the conjugated cyclohexenone that was obtained from terminal 1,6-diynes. However, the scope of this transformation was limited to just a few substituent variations on the alkynes. When both R1 and R2 were ethyl groups, this cyclization was dramatically retarded and only traces of the desired product were obtained. Under the mediation of aluminium oxide, this non-conjugated cyclopentylidene ketone product isomerized to the conjugated cyclopentenyl ketone 20.
![[1860-5397-7-71-i14]](/bjoc/content/inline/1860-5397-7-71-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Gold-catalyzed cyclization of internal diynes.
Scheme 14: Gold-catalyzed cyclization of internal diynes.
The authors proposed a solvolysis/cyclization mechanism for this gold-catalyzed cyclization, which was supported by a deuterium isotopic experiment (Scheme 15). Two molecules of methanol were involved in the transformation and a dimethoxyketal intermediate was formed: The final product was derived from the hydrolysis of this ketal intermediate. When d4-methanol was used as the solvent, a highly deuterated product was isolated, which provided strong support for the proposed mechanism.
![[1860-5397-7-71-i15]](/bjoc/content/inline/1860-5397-7-71-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Proposed solvolysis/cyclization mechanism.
Scheme 15: Proposed solvolysis/cyclization mechanism.
Gold-catalyzed formation of cyclic enones from alkynyl epoxides
Hashmi and co-workers synthesized a number of 2-alkynyl aryl epoxides 21 intended to be used as substrates for a gold-catalyzed rearrangement to naphthols. Surprisingly, acylindene 22 turned out to be the product of this reaction, rather than the expected naphthol (Scheme 16) [52]. However, when a bulky group was substituted on the triple bond, this gold-catalyzed transformation was completely suppressed. Moreover, none of the desired product could be obtained when a terminal alkyne, a TMS-substituted alkyne, or even an ester-substituted epoxide was used as the starting material.
![[1860-5397-7-71-i16]](/bjoc/content/inline/1860-5397-7-71-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Gold-catalyzed cyclization of alkynyl epoxides and the 18O isotopic labeling experiment.
Scheme 16: Gold-catalyzed cyclization of alkynyl epoxides and the 18O isotopic labeling experiment.
An 18O isotopic experiment helped the authors to propose an intramolecular oxygen transfer mechanism for the above transformation (Scheme 17). When employing the 18O incorporated substrate in the reaction, the authors found that the isolated product still contained the isotopic atom which excludes the involvement of external water in the reaction. A cross-over experiment with a mixture of two substrates (one with 18O, the other without) was also conducted, and no 18O scramble was found in the products, which clearly supported the intramolecular nature of the oxygen transfer.
![[1860-5397-7-71-i17]](/bjoc/content/inline/1860-5397-7-71-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Proposed oxygen transfer mechanism.
Scheme 17: Proposed oxygen transfer mechanism.
Liu and co-workers independently reported a very similar gold-catalyzed cyclization of 2-alkynyl aryl epoxide 21 to acylindene 22 (Scheme 18) [53]. A deuterium isotopic experiment was conducted to support the intramolecular oxygen transfer mechanism. However, when a trisubstituted epoxide 23 was employed in the reaction, the gold catalyst did not promote the transformation. By contrast, when AgSbF6 was used as the catalyst, the 1,2-alkyl shifted product 24 was obtained.
![[1860-5397-7-71-i18]](/bjoc/content/inline/1860-5397-7-71-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Gold or silver-catalyzed cyclization of alkynyl epoxides and the corresponding deuterium labeling experiment.
Scheme 18: Gold or silver-catalyzed cyclization of alkynyl epoxides and the corresponding deuterium labeling e...
Conclusion
This short review compiles recently reported gold-catalyzed oxygen transfer reactions used to build cyclic enones from tethered alkynyl ketones, 1,6-diynes or 2-alkynyl aryl epoxides. Most of these reactions take place under mild conditions and the corresponding products were isolated in good yields. The mechanisms for these transformations were also comparatively discussed. Similar Brønsted acid or other metal mediated transformations and their applications to cascade cyclizations were additionally described. Given gold’s strong π-electrophilicity, it is expected that novel applications of gold catalysts in reactions of alkynes, allenes, and even alkenes, will continue to attract the attention of synthetic chemists.
References
-
Perlmutter, P. Conjugate Addition Reactions in Organic Synthesis; Pergamon: Oxford, 1992.
Return to citation in text: [1] -
Sibi, M. P.; Manyem, S. Tetrahedron 2000, 56, 8033. doi:10.1016/S0040-4020(00)00618-9
Return to citation in text: [1] -
Feringa, B. L. Acc. Chem. Res. 2000, 33, 346. doi:10.1021/ar990084k
Return to citation in text: [1] -
Woodward, S. Chem. Soc. Rev. 2000, 29, 393. doi:10.1039/b002690p
Return to citation in text: [1] -
Alexakis, A.; Benhaim, C. Eur. J. Org. Chem. 2002, 3221. doi:10.1002/1099-0690(200210)2002:19<3221::AID-EJOC3221>3.0.CO;2-U
Return to citation in text: [1] -
López, F.; Minnaard, A. J.; Feringa, B. L. Acc. Chem. Res. 2007, 40, 179. doi:10.1021/ar0501976
Return to citation in text: [1] -
Sulzer-Mossé, S.; Alexakis, A. Chem. Commun. 2007, 3123. doi:10.1039/b701216k
Return to citation in text: [1] -
Touré, B. B.; Hall, D. G. Chem. Rev. 2009, 109, 4439. doi:10.1021/cr800296p
Return to citation in text: [1] -
Mahrwald, R., Ed. Modern Aldol Reactions; Wiley-VCH: Weinheim, 2004.
Return to citation in text: [1] -
Trost, B. M.; Brindle, C. S. Chem. Soc. Rev. 2010, 39, 1600. doi:10.1039/b923537j
Return to citation in text: [1] -
Brodmann, T.; Lorenz, M.; Schaeckel, R.; Simsek, S.; Kalesse, M. Synlett 2009, 174. doi:10.1055/s-0028-1087520
Return to citation in text: [1] -
Mlynarski, J.; Paradowska, J. Chem. Soc. Rev. 2008, 37, 1502. doi:10.1039/b710577k
Return to citation in text: [1] -
Ramazani, A.; Kazemizadeh, A. R.; Ahmadi, E.; Noshiranzadeh, N.; Souldozi, A. Curr. Org. Chem. 2008, 12, 59. doi:10.2174/138527208783330055
Return to citation in text: [1] -
Pietrusiewicz, K. M.; Zablocka, M. Chem. Rev. 1994, 94, 1375. doi:10.1021/cr00029a009
Return to citation in text: [1] -
Cobridge, D. E. C. Phosphorus: An Outline of Chemistry, Biochemistry and Uses, 5th ed.; Elsevier: Amsterdam, 1995.
Return to citation in text: [1] -
Engel, R. Synthesis of Carbon-Phosphorus Bonds; CRC: Boca Raton, FL, 1988.
Return to citation in text: [1] -
Cadogan, J. I. G. Organophosphorus Reagents in Organic Synthesis; Academic Press: New York, 1979.
Return to citation in text: [1] -
Maryanoff, B. E.; Reitz, A. B. Chem. Rev. 1989, 89, 863. doi:10.1021/cr00094a007
Return to citation in text: [1] -
Viswanathan, G. S.; Li, C.-J. Tetrahedron Lett. 2002, 43, 1613. doi:10.1016/S0040-4039(02)00082-5
Return to citation in text: [1] -
Curini, M.; Epifano, F.; Maltese, F.; Rosati, O. Synlett 2003, 552. doi:10.1055/s-2003-37515
Return to citation in text: [1] -
Rhee, J. U.; Krische, M. J. Org. Lett. 2005, 7, 2493. doi:10.1021/ol050838x
Return to citation in text: [1] -
Saito, A.; Umakoshi, M.; Yagyu, N.; Hanazawa, Y. Org. Lett. 2008, 10, 1783. doi:10.1021/ol800539a
Return to citation in text: [1] -
González-Rodríguez, C.; Escalante, L.; Varela, J. A.; Castedo, L.; Saá, C. Org. Lett. 2009, 11, 1531. doi:10.1021/ol900142r
Return to citation in text: [1] -
Kurtz, K. C. M.; Hsung, R. P.; Zhang, Y. Org. Lett. 2006, 8, 231. doi:10.1021/ol052487s
Return to citation in text: [1] -
Hsung, R. P.; Zificsak, C. A.; Wei, L.-L.; Douglas, C. J.; Xiong, H.; Mulder, J. A. Org. Lett. 1999, 1, 1237. doi:10.1021/ol990211c
Return to citation in text: [1] -
Miura, K.; Yamamoto, K.; Yamanobe, A.; Ito, K.; Kinoshita, H.; Ichikawa, J.; Hosomi, A. Chem. Lett. 2010, 39, 766. doi:10.1246/cl.2010.766
Return to citation in text: [1] -
Saito, A.; Kasai, J.; Odaira, Y.; Fukaya, H.; Hanzawa, Y. J. Org. Chem. 2009, 74, 5644. doi:10.1021/jo900857c
Return to citation in text: [1] -
Ito, Y.; Sawamura, M.; Hayashi, T. J. Am. Chem. Soc. 1986, 108, 6405. doi:10.1021/ja00280a056
Return to citation in text: [1] -
Fukuda, Y.; Utimoto, K. Synthesis 1991, 975. doi:10.1055/s-1991-26621
Return to citation in text: [1] -
Hashmi, A. S. K.; Schwarz, L.; Choi, J.-H.; Frost, T. M. Angew. Chem., Int. Ed. 2000, 39, 2285. doi:10.1002/1521-3773(20000703)39:13<2285::AID-ANIE2285>3.0.CO;2-F
Return to citation in text: [1] -
Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553. doi:10.1021/ja005570d
Return to citation in text: [1] -
Hashmi, A. S. K. Gold Bull. 2003, 36, 3. doi:10.1007/BF03214859
Return to citation in text: [1] -
Hashmi, A. S. K. Gold Bull. 2004, 37, 51. doi:10.1007/BF03215517
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180. doi:10.1021/cr000436x
Return to citation in text: [1] -
Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766. doi:10.1039/b615629k
Return to citation in text: [1] -
Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239. doi:10.1021/cr068434l
Return to citation in text: [1] -
Arcadi, A. Chem. Rev. 2008, 108, 3266. doi:10.1021/cr068435d
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326. doi:10.1021/cr0684319
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351. doi:10.1021/cr068430g
Return to citation in text: [1] -
Sohel, S. M. A.; Liu, R.-S. Chem. Soc. Rev. 2009, 38, 2269. doi:10.1039/b807499m
Return to citation in text: [1] -
Hashmi, A. S. K. Angew. Chem., Int. Ed. 2010, 49, 5232. doi:10.1002/anie.200907078
Return to citation in text: [1] -
Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657. doi:10.1021/cr100414u
Return to citation in text: [1] -
Jin, T.; Yamamoto, Y. Org. Lett. 2007, 9, 5259. doi:10.1021/ol702455v
Return to citation in text: [1] -
Jin, T.; Yamamoto, Y. Org. Lett. 2008, 10, 3137. doi:10.1021/ol801265s
Return to citation in text: [1] -
Jin, T.; Yang, F.; Liu, C.; Yamamoto, Y. Chem. Commun. 2009, 3533. doi:10.1039/b905954g
Return to citation in text: [1] -
Liu, L.-P.; Malhotra, D.; Paton, R. S.; Houk, K. N.; Hammond, G. B. Angew. Chem., Int. Ed. 2010, 49, 9132. doi:10.1002/anie.201005514
Return to citation in text: [1] -
Sze, E. M. L.; Rao, W.; Koh, M. J.; Chan, P. W. H. Chem.–Eur. J. 2011, 17, 1437. doi:10.1002/chem.201003096
Return to citation in text: [1] -
Zhang, C.; Cui, D.-M.; Yao, L.-Y.; Wang, B.-S.; Hu, Y.-Z.; Hayashi, T. J. Org. Chem. 2008, 73, 7811. doi:10.1021/jo800974x
Return to citation in text: [1] -
Cui, D.-M.; Ke, Y.-N.; Zhuang, D.-W.; Wang, Q.; Zhang, C. Tetrahedron Lett. 2010, 51, 980. doi:10.1016/j.tetlet.2009.12.055
Return to citation in text: [1] -
Sperger, C.; Fiksdahl, A. Org. Lett. 2009, 11, 2449. doi:10.1021/ol900681b
Return to citation in text: [1] -
Sperger, C.; Strand, L. H. S.; Fiksdahl, A. Tetrahedron 2010, 66, 7749. doi:10.1016/j.tet.2010.07.071
Return to citation in text: [1] -
Hashmi, A. S. K.; Bührle, M.; Salathé, R.; Bats, J. W. Adv. Synth. Catal. 2008, 350, 2059. doi:10.1002/adsc.200800385
Return to citation in text: [1] -
Lin, G.-Y.; Li, C.-W.; Hung, S.-H.; Liu, R.-S. Org. Lett. 2008, 10, 5059. doi:10.1021/ol802047g
Return to citation in text: [1]
| 1. | Perlmutter, P. Conjugate Addition Reactions in Organic Synthesis; Pergamon: Oxford, 1992. |
| 2. | Sibi, M. P.; Manyem, S. Tetrahedron 2000, 56, 8033. doi:10.1016/S0040-4020(00)00618-9 |
| 3. | Feringa, B. L. Acc. Chem. Res. 2000, 33, 346. doi:10.1021/ar990084k |
| 4. | Woodward, S. Chem. Soc. Rev. 2000, 29, 393. doi:10.1039/b002690p |
| 5. | Alexakis, A.; Benhaim, C. Eur. J. Org. Chem. 2002, 3221. doi:10.1002/1099-0690(200210)2002:19<3221::AID-EJOC3221>3.0.CO;2-U |
| 6. | López, F.; Minnaard, A. J.; Feringa, B. L. Acc. Chem. Res. 2007, 40, 179. doi:10.1021/ar0501976 |
| 7. | Sulzer-Mossé, S.; Alexakis, A. Chem. Commun. 2007, 3123. doi:10.1039/b701216k |
| 8. | Touré, B. B.; Hall, D. G. Chem. Rev. 2009, 109, 4439. doi:10.1021/cr800296p |
| 34. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180. doi:10.1021/cr000436x |
| 35. | Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766. doi:10.1039/b615629k |
| 36. | Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239. doi:10.1021/cr068434l |
| 37. | Arcadi, A. Chem. Rev. 2008, 108, 3266. doi:10.1021/cr068435d |
| 38. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326. doi:10.1021/cr0684319 |
| 39. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351. doi:10.1021/cr068430g |
| 40. | Sohel, S. M. A.; Liu, R.-S. Chem. Soc. Rev. 2009, 38, 2269. doi:10.1039/b807499m |
| 41. | Hashmi, A. S. K. Angew. Chem., Int. Ed. 2010, 49, 5232. doi:10.1002/anie.200907078 |
| 42. | Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657. doi:10.1021/cr100414u |
| 53. | Lin, G.-Y.; Li, C.-W.; Hung, S.-H.; Liu, R.-S. Org. Lett. 2008, 10, 5059. doi:10.1021/ol802047g |
| 28. | Ito, Y.; Sawamura, M.; Hayashi, T. J. Am. Chem. Soc. 1986, 108, 6405. doi:10.1021/ja00280a056 |
| 29. | Fukuda, Y.; Utimoto, K. Synthesis 1991, 975. doi:10.1055/s-1991-26621 |
| 30. | Hashmi, A. S. K.; Schwarz, L.; Choi, J.-H.; Frost, T. M. Angew. Chem., Int. Ed. 2000, 39, 2285. doi:10.1002/1521-3773(20000703)39:13<2285::AID-ANIE2285>3.0.CO;2-F |
| 31. | Hashmi, A. S. K.; Frost, T. M.; Bats, J. W. J. Am. Chem. Soc. 2000, 122, 11553. doi:10.1021/ja005570d |
| 32. | Hashmi, A. S. K. Gold Bull. 2003, 36, 3. doi:10.1007/BF03214859 |
| 33. | Hashmi, A. S. K. Gold Bull. 2004, 37, 51. doi:10.1007/BF03215517 |
| 19. | Viswanathan, G. S.; Li, C.-J. Tetrahedron Lett. 2002, 43, 1613. doi:10.1016/S0040-4039(02)00082-5 |
| 20. | Curini, M.; Epifano, F.; Maltese, F.; Rosati, O. Synlett 2003, 552. doi:10.1055/s-2003-37515 |
| 21. | Rhee, J. U.; Krische, M. J. Org. Lett. 2005, 7, 2493. doi:10.1021/ol050838x |
| 22. | Saito, A.; Umakoshi, M.; Yagyu, N.; Hanazawa, Y. Org. Lett. 2008, 10, 1783. doi:10.1021/ol800539a |
| 23. | González-Rodríguez, C.; Escalante, L.; Varela, J. A.; Castedo, L.; Saá, C. Org. Lett. 2009, 11, 1531. doi:10.1021/ol900142r |
| 24. | Kurtz, K. C. M.; Hsung, R. P.; Zhang, Y. Org. Lett. 2006, 8, 231. doi:10.1021/ol052487s |
| 25. | Hsung, R. P.; Zificsak, C. A.; Wei, L.-L.; Douglas, C. J.; Xiong, H.; Mulder, J. A. Org. Lett. 1999, 1, 1237. doi:10.1021/ol990211c |
| 26. | Miura, K.; Yamamoto, K.; Yamanobe, A.; Ito, K.; Kinoshita, H.; Ichikawa, J.; Hosomi, A. Chem. Lett. 2010, 39, 766. doi:10.1246/cl.2010.766 |
| 27. | Saito, A.; Kasai, J.; Odaira, Y.; Fukaya, H.; Hanzawa, Y. J. Org. Chem. 2009, 74, 5644. doi:10.1021/jo900857c |
| 50. | Sperger, C.; Fiksdahl, A. Org. Lett. 2009, 11, 2449. doi:10.1021/ol900681b |
| 51. | Sperger, C.; Strand, L. H. S.; Fiksdahl, A. Tetrahedron 2010, 66, 7749. doi:10.1016/j.tet.2010.07.071 |
| 9. | Mahrwald, R., Ed. Modern Aldol Reactions; Wiley-VCH: Weinheim, 2004. |
| 10. | Trost, B. M.; Brindle, C. S. Chem. Soc. Rev. 2010, 39, 1600. doi:10.1039/b923537j |
| 11. | Brodmann, T.; Lorenz, M.; Schaeckel, R.; Simsek, S.; Kalesse, M. Synlett 2009, 174. doi:10.1055/s-0028-1087520 |
| 12. | Mlynarski, J.; Paradowska, J. Chem. Soc. Rev. 2008, 37, 1502. doi:10.1039/b710577k |
| 13. | Ramazani, A.; Kazemizadeh, A. R.; Ahmadi, E.; Noshiranzadeh, N.; Souldozi, A. Curr. Org. Chem. 2008, 12, 59. doi:10.2174/138527208783330055 |
| 14. | Pietrusiewicz, K. M.; Zablocka, M. Chem. Rev. 1994, 94, 1375. doi:10.1021/cr00029a009 |
| 15. | Cobridge, D. E. C. Phosphorus: An Outline of Chemistry, Biochemistry and Uses, 5th ed.; Elsevier: Amsterdam, 1995. |
| 16. | Engel, R. Synthesis of Carbon-Phosphorus Bonds; CRC: Boca Raton, FL, 1988. |
| 17. | Cadogan, J. I. G. Organophosphorus Reagents in Organic Synthesis; Academic Press: New York, 1979. |
| 18. | Maryanoff, B. E.; Reitz, A. B. Chem. Rev. 1989, 89, 863. doi:10.1021/cr00094a007 |
| 52. | Hashmi, A. S. K.; Bührle, M.; Salathé, R.; Bats, J. W. Adv. Synth. Catal. 2008, 350, 2059. doi:10.1002/adsc.200800385 |
| 46. | Liu, L.-P.; Malhotra, D.; Paton, R. S.; Houk, K. N.; Hammond, G. B. Angew. Chem., Int. Ed. 2010, 49, 9132. doi:10.1002/anie.201005514 |
| 48. | Zhang, C.; Cui, D.-M.; Yao, L.-Y.; Wang, B.-S.; Hu, Y.-Z.; Hayashi, T. J. Org. Chem. 2008, 73, 7811. doi:10.1021/jo800974x |
| 45. | Jin, T.; Yang, F.; Liu, C.; Yamamoto, Y. Chem. Commun. 2009, 3533. doi:10.1039/b905954g |
| 49. | Cui, D.-M.; Ke, Y.-N.; Zhuang, D.-W.; Wang, Q.; Zhang, C. Tetrahedron Lett. 2010, 51, 980. doi:10.1016/j.tetlet.2009.12.055 |
| 47. | Sze, E. M. L.; Rao, W.; Koh, M. J.; Chan, P. W. H. Chem.–Eur. J. 2011, 17, 1437. doi:10.1002/chem.201003096 |
© 2011 Liu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)