Abstract
By focusing on recent developments on natural and non-natural azasugars (iminocyclitols), this review bolsters the case for the role of olefin metathesis reactions (RCM, CM) as key transformations in the multistep syntheses of pyrrolidine-, piperidine- and azepane-based iminocyclitols, as important therapeutic agents against a range of common diseases and as tools for studying metabolic disorders. Considerable improvements brought about by introduction of one or more metathesis steps are outlined, with emphasis on the exquisite steric control and atom-economical outcome of the overall process. The comparative performance of several established metathesis catalysts is also highlighted.

Graphical Abstract
Review
Introduction
Synthetic and natural polyhydroxylated N-heterocyclic compounds (pyrrolidines, piperidines, piperazines, indolizines, etc., and higher homologues), commonly referred to as azasugars, iminosugars or iminocyclitols, can be considered as carbohydrate mimics in which the endocyclic oxygen atom of sugars has been replaced by an imino group. This vast and highly diversified class has attracted considerable interest due to the remarkable biological profile shown by many of its members which has been detailed in a number of excellent books and reviews [1-12]. Natural iminosugars (i.e., alkaloids mimicking the structures of sugars, widespread in many plants or microorganisms) [12-15], as well as non-natural counterparts, are becoming important leads for drug development in a variety of therapeutic areas, e.g., treatment of cancer [16-20], glycosphingolipid storage disorders [21,22], Gaucher’s disease [23], type-II diabetes [24-26], other metabolic disorders [10], and viral diseases [27,28] such as HIV [29,30] and hepatitis B [31,32] and C [27,33]. Some such products have been already marketed, such as N-hydroxyethyl-1-deoxynojirimycin (Miglitol) and N-butyl-1-deoxynojirimycin (Miglustat) which are active against type-II diabetes and Gaucher’s disease, respectively.
The broad biological activity of iminocyclitols has attracted growing interest in the synthesis of naturally occurring iminocyclitols and in their structural modification. Consequently, efficient and stereoselective synthetic routes have been developed, often starting from an inexpensive chiral-pool of precursors, in particular carbohydrates that share structural features with iminocyclitols. The main hurdles in this approach are the singling out of only one of the hydroxy groups in the open carbohydrate-derived intermediate, converting this hydroxy group into an amino group, and intramolecularly closing this intermediate [8,34-36]. Because of the high density of functional groups, proper protection throughout the overall synthesis scheme is an important feature that must be considered carefully, with full deprotection occurring in the final step.
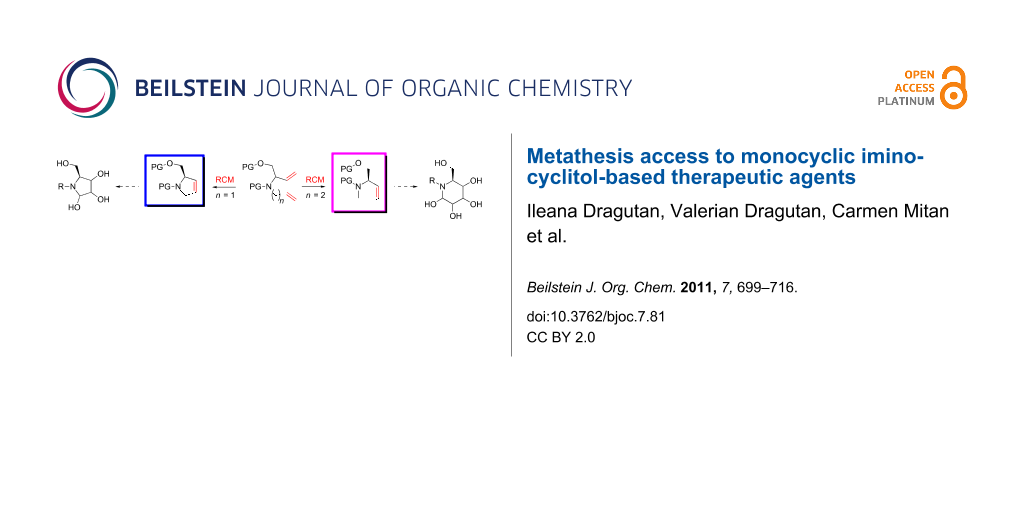
With the advent of well-defined Mo- and Ru-alkylidene metathesis catalysts (e.g., 1–10; Scheme 1) [37-47] the RCM strategy was immediately recognized as central to success in the flexible construction of N-heterocyclic compounds, including azasugars. Moreover, the metathesis approach to azasugars has greatly benefited from the vast synthetic experience acquired in RCM preparation of a host of heterocycles. Any RCM-based protocol to iminocyclitols implies three crucial stages: (i) discovery of a route to obtain stereoselectively, starting from an ordinary substrate, the N-containing prerequisite diene precursor; (ii) RCM cyclization of this diene, with an active catalyst, to access the core cyclic olefin; and (iii) dihydroxylation of the endocyclic double bond in a highly diastereoselective manner to form the target product.
![[1860-5397-7-81-i1]](/bjoc/content/inline/1860-5397-7-81-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Well-defined Mo- and Ru-alkylidene metathesis catalysts.
Scheme 1: Well-defined Mo- and Ru-alkylidene metathesis catalysts.
In comparison to the traditional, lengthier syntheses of iminocyclitols, the metathesis approach has emerged as a highly advantageous method in terms of atom economy. However, before carrying out the RCM reaction, the basic amino group (incompatible with most metathesis catalysts because of chelation to the metal center) [48] must either be protected (as N–Boc, N–Cbz, etc.), masked by incorporation into a cleavable heteroatom-containing cycle (oxazolidine, cyclic ketal, etc.), or deactivated by conversion into amide or carbamate functions. Due to these protective groups even metathesis catalysts sensitive to functionalities can act efficiently under reaction conditions where an adequate balance between activity/stability factors has been met. In addition, the reaction conditions (temperature, solvents) currently employed in olefin metathesis reactions can be productively transferred to the metathesis steps of iminocyclitols synthesis.
By surveying the field of recent azasugar developments, this review focuses on metathesis reactions (mainly RCM, CM) as essential transformations in the multistep synthesis of monocyclic iminocyclitols, while also discussing the successes and failures in effecting the above mentioned three critical stages. New perspectives may open up for practitioners of both glyco- and metathesis chemistry involved in the synthesis and development of iminocyclitols.
Pyrrolidine-based iminocyclitols
Recently, pyrrolidine-based iminocyclitols have assumed increasing importance and some of them have achieved even higher biological significance than the established six-membered piperidine deoxynojirimycin (DNJ) and deoxygalactonojirimycin (DGJ). Five-membered iminocyclitols possessing N-alkyl and C1-alkyl substituents form a class of potent antiviral compounds active, e.g., against hepatitis B virus (HBV), hepatitis C virus (HCV), and human immunodeficiency virus (HIV) [49-52].
Biological activity of this family of iminocyclitols is dictated by the stereochemistry at all carbon atoms of the pyrrolidine ring system which can adopt either a manno or a galacto conformation, therefore inhibiting either α-mannosidases (e.g., 11–13) or α-galactosidases (e.g., 14) (Scheme 2). A characteristic feature in 11–14 is the presence of a 1,2-dihydroxyethyl side chain.
![[1860-5397-7-81-i2]](/bjoc/content/inline/1860-5397-7-81-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Representative pyrrolidine-based iminocyclitols.
Scheme 2: Representative pyrrolidine-based iminocyclitols.
Following the RCM-based strategy (vide supra), an elegant and quite flexible synthesis of five-membered iminocyclitols was achieved by Huwe and Blechert as early as 1997 [53]. Starting from (±)-vinylglycine methyl ester 15 and going successively via amino protection (Cbz) and ester group reduction (LiBH4), a protected racemic diene 16 was obtained; RCM cyclization of the latter using the Grubbs catalyst Cl2(PCy3)2Ru=CH–CH=CPh2 (3) led to the racemic dehydroprolinol derivative 17 in high yield. Subsequent O-protection with trityl chloride and dihydroxylation (with OsO4 or stereoselective epoxidation followed by regioselective epoxide opening) produced the racemic iminocyclitols (18–20) in good overall yields (Scheme 3).
![[1860-5397-7-81-i3]](/bjoc/content/inline/1860-5397-7-81-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of (±)-(2R*,3R*,4S*)-2-hydroxymethylpyrrolidin-3,4-diol (18), (±)-2-hydroxymethylpyrrolidin-3-ol (19) and (±)-(2R*,3R*,4R*)-2-hydroxymethylpyrrolidin-3,4-diol (20).
Scheme 3: Synthesis of (±)-(2R*,3R*,4S*)-2-hydroxymethylpyrrolidin-3,4-diol (18), (±)-2-hydroxymethylpyrrolid...
In addition, Blechert showed that this method was more adaptable as it could also yield enantiopure 18–20, provided that racemization was avoided both during ester group reduction and the subsequent steps. By a similar approach (Scheme 4), these authors also obtained the enantiopure homoiminocyclitol (−)-(2S,3R,4S,5S)-2,5-dihydroxymethylpyrrolidin-3,4-diol (23). Starting from the same racemic vinyl glycine methyl ester and introducing enzymatic resolution in the ester reduction step, enantiopure (+)-21 was obtained. 1st-generation Grubbs catalyst was used for the RCM of (+)-21. It should be noted that the yield of (+)-22 in RCM (10 mol % 2, in benzene) was temperature dependent (88% at room temperature and 98% at 80 °C). Further stereocontrolled dihydroxylation with simultaneous deprotection of (+)-22 gave the final product (−)-23 in good yield.
![[1860-5397-7-81-i4]](/bjoc/content/inline/1860-5397-7-81-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of enantiopure iminocyclitol (−)-(2S,3R,4S,5S)-2,5-dihydroxymethylpyrrolidin-3,4-diol (23).
Scheme 4: Synthesis of enantiopure iminocyclitol (−)-(2S,3R,4S,5S)-2,5-dihydroxymethylpyrrolidin-3,4-diol (23...
In an interesting work by Rao and co-workers [54] a Grignard reaction was employed to design the diene with desired stereochemistry for the synthesis of 1,4-dideoxy-1,4-imino-D-allitol (29) and the formal synthesis of (2S,3R,4S)-3,4-dihydroxyproline (30) (Scheme 5). According to their methodology, (R)-2,3-O-isopropylidene-D-glyceraldehyde (24) was treated in a one-pot reaction with benzylamine and then subjected to Grignard addition with vinylmagnesium bromide to provide the alkene 25 as a single diastereomer. The nitrogen atom in 25 was then Boc-protected, debenzylated, and allylated to give the diene 26. RCM of the latter with 1st-generation Grubbs catalyst (10 mol % 2, in dichloromethane, at room temperature) provided the pyrrole scaffold 27. Subsequent stereoselective dihydroxylation (OsO4 and 4-methylmorpholine N-oxide (NMO) in acetone) to yield 28 and final deprotection (MeOH–HCl) gave the imino-D-allitol 29 as the HCl salt. Formal synthesis of (2S,3R,4S)-3,4-dihydroxyproline (30) starting from 24 was carried identically by RCM to afford 27 and subsequent conversion of 28 to 30 was achieved in several steps via the Fleet protocol [55].
![[1860-5397-7-81-i5]](/bjoc/content/inline/1860-5397-7-81-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 1,4-dideoxy-1,4-imino-D-allitol (29) and formal synthesis of (2S,3R,4S)-3,4-dihydroxyproline (30).
Scheme 5: Synthesis of 1,4-dideoxy-1,4-imino-D-allitol (29) and formal synthesis of (2S,3R,4S)-3,4-dihydroxyp...
The tandem RCM/dihydroxylation sequence was also applied by Davis et al. in the synthesis of (−)-2,3-trans-3,4-cis-dihydroxyproline. In this case, an α-amino aldehyde, prepared by addition of a 1,3-dithiane to a chiral N-sulfinyl imine, was used as the chiral starting material [56]. Syntheses of 1,4-dideoxy-1,4-imino derivatives of L-allitol and D-talitol were also accomplished following a similar RCM methodology by Rao and co-workers [57].
1,4-Dideoxy-1,4-imino-D-ribitol (35), known as (+)-DRB, is a potent inhibitor of glucosidases and of eukaryotic DNA polymerases. Its synthesis, as well as that of its dihydroxylated homologue 36, features as the key step five-membered ring formation via RCM induced by the 2nd-generation Grubbs catalyst 5 (Scheme 6) [58].
![[1860-5397-7-81-i6]](/bjoc/content/inline/1860-5397-7-81-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of iminocyclitols 35 and 36.
Scheme 6: Synthesis of iminocyclitols 35 and 36.
A further contribution to new pyrrolidine-based azasugars, characteristically having 1,2-dihydroxyethyl side chains and a quaternary C-atom possessing a hydroxy and a hydroxymethyl group, was made by Vankar et al. [59] (Scheme 7). By ingeniously combining a Baylis–Hillman addition with RCM as the key steps, they obtained, stereoselectively and in high yields, 1,4-dideoxy-1,4-iminohexitols 40 and 44 which showed moderate inhibition of β-galactosidase, and α-galacto- and α-mannosidases, respectively. It should be noted that diene 38 did not cyclize in the presence of 1st-generation Grubbs catalyst, even in refluxing toluene, whereas 2nd-generation Grubbs catalyst afforded (in toluene, at 60 °C) the cyclic products 39 and 43 in 89% and 86% yields, respectively. Interestingly, Upjohn dihydroxylation of 39 or 43 (OsO4, NMO, acetone/H2O/t-BuOH; HCl, MeOH; Ac2O, Et3N, DMAP) gave only one diastereomeric diol, because the bulky acetonide group blocks the β-face of the trisubstituted double bond of the pyrrolidine ring and is thus responsible for the high diastereoselectivity.
![[1860-5397-7-81-i7]](/bjoc/content/inline/1860-5397-7-81-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Total synthesis of iminocyclitols 40 and 44.
Scheme 7: Total synthesis of iminocyclitols 40 and 44.
A metathesis approach has elegantly been used by Trost et al. for the total synthesis of 2,5-dideoxy-2,5-imino-D-mannitol [(+)-DMDP], 49, (−)-bulgecinine, 50, and (+)-broussonetine G, 53 [60,61]. The crucial intermediate, the protected annulated oxazolone 48, resulted from RCM (2nd-generation Grubbs catalyst) of the imino diene 47 (previously produced by a Pd-catalyzed asymmetric transformation). The following three or five steps, respectively, including the enantioselective dihydroxylation of the RCM product 48, occurred smoothly to produce the (+)-DMDP (49) and (−)-bulgecinine (50) (Scheme 8).
![[1860-5397-7-81-i8]](/bjoc/content/inline/1860-5397-7-81-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of 2,5-dideoxy-2,5-imino-D-mannitol [(+)-DMDP] (49) and (−)-bulgecinine (50).
Scheme 8: Synthesis of 2,5-dideoxy-2,5-imino-D-mannitol [(+)-DMDP] (49) and (−)-bulgecinine (50).
The starting point in the synthesis of (+)-broussonetine G, 53, was the same annulated oxazolone 48 which, after conversion into the Weinreb amide 51, was coupled with the alkyl bromide substituted spiro compound 52 (Scheme 9).
![[1860-5397-7-81-i9]](/bjoc/content/inline/1860-5397-7-81-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of (+)-broussonetine G (53).
Scheme 9: Synthesis of (+)-broussonetine G (53).
In fact, the case of broussonetines is much more complicated. This subgroup is currently represented by 30 reported examples, all isolated from plant species and used in folk medicine in China and Japan. Most broussonetines display marked inhibitory activities on various glycosidase types, with selectivities differing from that of other standard iminosugars such as DNJ. In the majority of the broussonetines (54, Scheme 10), a common polyhydroxylated pyrrolidine building block (possibly prepared via protocols including RCM) is bound to a side chain fragment of 13 C-atoms, diversely functionalized. For the introduction of the appropriate side chain, cross-metathesis appeared to be the most versatile method, permitting access to many members of this family, both naturally occurring and analogues. Two types of metathesis processes, RCM and CM, can be thus advantageously intertwined in the synthesis of broussonetines.
![[1860-5397-7-81-i10]](/bjoc/content/inline/1860-5397-7-81-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Structural features of broussonetines 54.
Scheme 10: Structural features of broussonetines 54.
For instance, the syntheses of broussonetines C, D, M, O and P were completed by Falomir, Marco et al. [62,63] in a convergent, stereocontrolled way starting from commercial D-serine (55) as the chiral precursor and by applying the critical step of cross-metathesis (the first-ever synthesis of broussonetines O and P) (Scheme 11).
![[1860-5397-7-81-i11]](/bjoc/content/inline/1860-5397-7-81-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthesis of broussonetines by cross-metathesis.
Scheme 11: Synthesis of broussonetines by cross-metathesis.
The cross-metathesis reaction was promoted by the 2nd-generation Grubbs catalyst (5, in CH2Cl2, by heating under reflux in a N2 atmosphere for 24 h or by heating for 1 h at 100 °C under microwave irradiation). As expected in a cross-metathesis process, a mixture of three products (CM product plus the two homo-metathesis products, all in both stereoisomeric forms) was obtained. Homo-metathesis products from either 56 or the alkene were recycled in the cross-metathesis stage to provide an additional amount of the useful product 57, thus enhancing the overall yield.
Piperidine-based iminocyclitols
During the last decade, polyhydroxylated piperidines have been the target of much cutting-edge synthesis work [8]. Such compounds are of special interest as therapeutic agents and as tools for the study of cellular mechanisms and metabolic diseases. From this class, nojirimycin (NJ, trivial name for 5-amino-5-deoxy-D-glucopyranose) (59), the first alkaloid discovered that mimicks a sugar (originally isolated from Streptomyces filtrate but also found in other bacterial cultures and plant sources), is a potent glycosidase inhibitor. In aqueous solution nojirimycin exists in both the α- and β-forms, each of which is responsible for inhibition of α- or β-glucosidase, respectively. Similar to its other congeners, mannonojirimycin (60; MJ or nojirimycin B) and galactonojirimycin (61; GJ or galactostatin), nojirimycin is unstable because hemiacetal structures can be adopted [8]. 1-Deoxynojirimycin (62; DNJ), the more stable 1-deoxy analogue of nojirimycin, represents the main motif of a large family of iminocyclitols (e.g., 63–66). Although numerous other piperidine iminocyclitols have shown encouraging results against HIV and in cancer therapy, the deoxynojirimycin family is certainly the most interesting. Two deoxynojirimycin derivatives have already found clinical applications: N-butyl-1-deoxynojirimycin (Miglustat, 67), in the treatment of type-II diabetes, and N-hydroxyethyl-1-deoxynojirimycin (Miglitol, 68; FDA approved) in the treatment of Gaucher’s disease (Scheme 12) [8].
![[1860-5397-7-81-i12]](/bjoc/content/inline/1860-5397-7-81-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Representative piperidine-based iminocyclitols.
Scheme 12: Representative piperidine-based iminocyclitols.
Takahata et al. [64] exploited RCM for the contruction of the piperidine ring of 1-deoxynojirimycin (62) and its congeners (1-deoxymannonojirimycin (63), 1-deoxyaltronojirimycin (65), and 1-deoxyallonojirimycin (66), Scheme 13). In their methodology, the D-serine-derived Garner aldehyde 69 provides an attractive starting point since it reacts with organometallic reagents with a high degree of diastereoselectivity and minimal racemization. N-allylation (allyl iodide/NaH; 95% yield) of an intermediate derived from 70 gave the diolefin product 71, which was then subjected to RCM (dichloromethane, 20 °C) in the presence of the 1st-generation Grubbs catalyst [(Cl2(Cy3P)2Ru=CHPh)] (2) to form the chiral tetrahydropyridine building block 72 in 97% yield. The stereochemistry of 72 was unambiguously confirmed by transformation into the known trans-3-hydroxy-2-hydroxymethylpiperidine. The tetrahydropyridine scaffold 72 allowed an efficient synthesis of 1-deoxynojirimycin 62, and its stereoisomers 65 and 66. Thus, acid hydrolysis of the epoxy ring in the anti isomer 73 (H2SO4/dioxane/H2O, 0.2:3:2) gave 1-deoxynojirimycin (62) and 1-deoxyaltronojirimycin (65) in a 1:1 ratio and in 89% yield. Conversely, basic cleavage of the epoxide 73 (KOH/dioxane/H2O) led preferentially to 65 (1:1.5 ratio 62/65; 99% yield). It should be noted that in the case of the syn epoxide 74 both acidic and basic hydrolysis afforded only 1-deoxyaltronojirimycin (65), in 63 and 68% yields, respectively.
![[1860-5397-7-81-i13]](/bjoc/content/inline/1860-5397-7-81-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Total synthesis of 1-deoxynojirimycin (62) and 1-deoxyaltronojirimycin (65).
Scheme 13: Total synthesis of 1-deoxynojirimycin (62) and 1-deoxyaltronojirimycin (65).
Further manipulations based mainly on stereoselective dihydroxylation (K2OsO4·2H2O; NMO as co-oxidant) of the useful building block 72 are at the core of the synthesis of 1-deoxymannonojirimycin (63) and 1-deoxyallonojirimycin (66) (Scheme 14). Although 1-deoxynojirimycin (62) and 1-deoxyaltronojirimycin (65) were obtained in a rather selective manner, a similar route to deoxymannojirimycin (63) and 1-deoxyallonojirimycin (66) did not achieve the same degree of selectivity, presumably due to difficulties in transforming the endocyclic double bond of the RCM product 72 into the targeted trans diols. Clean epoxide opening is frequently troublesome, being governed by a number of factors.
![[1860-5397-7-81-i14]](/bjoc/content/inline/1860-5397-7-81-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Synthesis by RCM of 1-deoxymannonojirimycin (63) and 1-deoxyallonojirimycin (66).
Scheme 14: Synthesis by RCM of 1-deoxymannonojirimycin (63) and 1-deoxyallonojirimycin (66).
An improvement in the selectivity and efficiency of the total synthesis of (+)-1-deoxynojirimycin (62) (24% overall yield) was made by Poisson et al. [65], who developed a one-pot tandem protocol involving enol ether RCM/hydroboration/oxidation, which gave the best results when the Hoveyda–Grubbs catalyst 6 was used in the RCM (Scheme 15).
![[1860-5397-7-81-i15]](/bjoc/content/inline/1860-5397-7-81-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Total synthesis of (+)-1-deoxynojirimycin (62).
Scheme 15: Total synthesis of (+)-1-deoxynojirimycin (62).
Interestingly, in this case the asymmetric synthesis of the protected RCM precursor 78 started from a non-chiral source, the alcohol 75, and proceeded with complete stereocontrol over the 11 steps involved. All attempts to achieve metathesis on another diene precursor having an endocyclic N-atom (the result of N-alkylation of 77 with 3-iodo-2-(methoxymethyloxy)prop-1-ene) led to either recovery of the starting material or olefin isomerization, even in the presence of a number of ruthenium hydride traps. Satisfactory results in RCM were, however, obtained from 78: in the presence of the 2nd-generation Grubbs catalyst 5 and benzoquinone in refluxing toluene, 78 was converted into the cyclized enol ether 79 in 70% yield, while with the Hoveyda–Grubbs catalyst (6, 10 mol %; benzoquinone 10 mol %; in refluxing toluene) 79 was obtained in 85% yield. The three reaction steps leading from 78 to 80, i.e., RCM/hydroboration/oxidation, could be accomplished in one-pot to afford the product as a single isomer (all-trans triol). The prepared (+)-1-deoxynojirimycin (62) displayed spectroscopic data which perfectly matched those of the natural product.
An important precursor for the synthesis of polyhydroxylated piperidines, (3R,4S)-3-hydroxy-4-N-allyl-N-Boc-amino-1-pentene (81), could be obtained as a single diastereomer via the addition of vinyl Grignard to the N-Boc-N-allyl aminoaldehyde, which was derived from the methyl ester of natural, enantiopure L-alanine. Having built the tetrahydropyridine scaffold 82 by RCM of 81 using the 2nd-generation Grubbs catalyst (5; 85% yield), Park et al. [66] were able to effect its stereodivergent dihydroxylation, via a common epoxide intermediate, to yield a range of interesting hydroxylated piperidines. This included ent-1,6-dideoxynojirimycin (ent-1,6-dDNJ, 83) (28% overall yield from N-Boc-L-alanine methyl ester) and 5-amino-1,5,6-trideoxyaltrose (84) (29% overall yield from N-Boc-L-alanine methyl ester), which were produced with excellent diastereoselectivity (>99:1 dr, Scheme 16). It appears that this total synthesis of ent-1,6-dDNJ (83) is the most expeditious to date.
![[1860-5397-7-81-i16]](/bjoc/content/inline/1860-5397-7-81-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Synthesis of ent-1,6-dideoxynojirimycin (83) and 5-amino-1,5,6-trideoxyaltrose (84).
Scheme 16: Synthesis of ent-1,6-dideoxynojirimycin (83) and 5-amino-1,5,6-trideoxyaltrose (84).
It was again Takahata et al. [67] who successfully tackled the synthesis of 1-deoxygalactonojirimycin (64, DGJ) and its congeners, 1-deoxygulonojirimycin (91) and 1-deoxyidonojirimycin (93) (Scheme 17), relying in the first step on the diastereoselective addition of a vinyl organometallic reagent to D-Garner aldehyde. Once more, for construction of the piperidine ring in 86, RCM (1st-generation Grubbs catalyst, 2) was applied to the prerequisite diene 85 bearing a cyclic conformation constraint. The stereochemistry of the chiral building block 86 was confirmed by conversion into the known compound, cis-2-hydroxymethyl-3-hydroxypiperidine (87), (step a). For 1-deoxygulonojirimycin (91) a highly selective dihydroxylation (step f) was performed on 86, under Upjohn conditions. For 1-deoxygalactonojirimycin (64) and 1-deoxyidonojirimycin (93), transformation of 86 proceeded via syn (step c) and anti (step h) epoxidation of the internal double bond in 86, respectively, and subsequent hydrolysis.
![[1860-5397-7-81-i17]](/bjoc/content/inline/1860-5397-7-81-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Synthesis of 1-deoxygalactonojirimycin (64), 1-deoxygulonojirimycin (91) and 1-deoxyidonojirimycin (93) [Step c: m-CPBA, NaH2PO4, CH2Cl2, 0 °C to r.t. Step d: 2,2-dimethoxypropane, PPTS, acetone, r.t. Step e: H2SO4, 1,4-dioxane, H2O, reflux. Step f: K2OsO4·2H2O, NMO, acetone, H2O, r.t. Step g: HCl, MeOH. Step h: Oxone, CF3COCH3, NaHCO3, aqueous Na2·EDTA, CH3CN, 0 °C. Step i: 0.3 M KOH, 1,4-dioxane, H2O, reflux].
Scheme 17: Synthesis of 1-deoxygalactonojirimycin (64), 1-deoxygulonojirimycin (91) and 1-deoxyidonojirimycin (...
Quite recently, an interesting synthesis of three 1-deoxynojirimycin-related iminosugars, L-1-deoxyaltronojirimycin (96), D-1-deoxyallonojirimycin (66), and D-1-deoxygalactonojirimycin (64), was reported by Overkleeft et al. to occur from a single chiral cyanohydrin 94, made available via a chemoenzymatic approach with almond hydroxynitrile lyase (paHNL) [68]. The key steps in the synthetic scheme comprise the cascade Dibal reduction/transimination/NaBH4 reduction of the enantiomerically pure 94, followed by the RCM step (CH2Cl2, 3.5 mol % 1st-generation Grubbs catalyst, reflux under Ar for 48 h) and Upjohn dihydroxylation to afford the target compounds (Scheme 18 for 96).
![[1860-5397-7-81-i18]](/bjoc/content/inline/1860-5397-7-81-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Synthesis of L-1-deoxyaltronojirimycin (96).
Scheme 18: Synthesis of L-1-deoxyaltronojirimycin (96).
RCM promoted by the 1st-generation Grubbs catalyst 2 is starring again in the divergent, flexible methodology disclosed by Singh and Han [69] for the asymmetric synthesis of several deoxyiminocyclitols (1-deoxymannonojirimycin (63), 1-deoxyaltronojirimycin (65), 1-deoxygulonojirimycin (91), 1-deoxyidonojirimycin (93), Scheme 19). Ingeniously selecting as starting material the achiral olefin 97, suitable for electronic and aryl–aryl stacking interactions with the regioselective osmium catalyst, they conducted asymmetric aminohydroxylation (regioselectivity >20:1; enantioselectivity >99% ee) in good yield (70%) to get the RCM precursor diene 98, appropriately protected. Under common RCM conditions (10 mol % 1st-generation Grubbs catalyst 2, toluene, 90 °C, 2 h) 98 was then converted to the key cyclo-olefin intermediate 99 (80% yield). From the latter, the targeted iminocyclitols 63 and 91 have been obtained after artful manipulation of routine protocols (diastereoselective dihydroxylation and protection/deprotection). To access 1-deoxyaltronojirimycin (65) and 1-deoxyidonojirimycin (93), introduction of an additional step involving a cyclic sulfate was necessary.
![[1860-5397-7-81-i19]](/bjoc/content/inline/1860-5397-7-81-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Synthesis of 1-deoxymannonojirimycin (63) and 1-deoxyaltronojirimycin (65).
Scheme 19: Synthesis of 1-deoxymannonojirimycin (63) and 1-deoxyaltronojirimycin (65).
A similar methodology was used by Han [70] to prepare 5-des(hydroxymethyl)-1-deoxynojirimycin (114) and its mannose analogue 111 (as HCl salts) in a highly stereoselective mode starting from a different common olefin, 107 (Scheme 20). In this case, RCM was promoted by the 2nd-generation Grubbs catalyst 5 which ensured a high yield of the ring closure (89%) under milder conditions (CH2Cl2): all attempts to employ the 1st-generation Grubbs catalyst 2 in RCM failed, supposedly because of an unfavourable steric environment during generation of the Ru–carbene species from 109, as compared to 98 (distinct N-protective groups). Cyclic sulfate chemistry was again invoked for effectively performing the synthesis of 114.
![[1860-5397-7-81-i20]](/bjoc/content/inline/1860-5397-7-81-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of 5-des(hydroxymethyl)-1-deoxymannonojirimycin (111) and 5-des(hydroxymethyl)-1-deoxynojirimycin (114).
Scheme 20: Synthesis of 5-des(hydroxymethyl)-1-deoxymannonojirimycin (111) and 5-des(hydroxymethyl)-1-deoxynoj...
Introducing a genereal strategy for synthesis of deoxyazasugars based on cheap D-glucose, Ghosh et al. also laid groundwork for the preparation of D-1-deoxygulonojirimycin (91) (previously communicated by Takahata [67]; Scheme 17) and L-1-deoxyallonojirimycin (122) (Scheme 21) starting from protected diacetone glucose 115 [71]. Different pathways were devised for 91 and 122 via the epimeric RCM precursors 117 and 120, respectively. High yielding cyclization of these dienes, in the presence of the 1st-generation Grubbs catalyst 2 (10 mol %, in CH2Cl2, under argon, 24 h at 50 °C), led to 118 and 121 with preserved configurations at the stereogenic centre, which therefore allowed the desired stereochemistry in the isomeric final products 91 and 122.
![[1860-5397-7-81-i21]](/bjoc/content/inline/1860-5397-7-81-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Synthesis of D-1-deoxygulonojirimycin (91) and L-1-deoxyallonojirimycin (122).
Scheme 21: Synthesis of D-1-deoxygulonojirimycin (91) and L-1-deoxyallonojirimycin (122).
D-Fagomine (1,2,5-trideoxy-1,5-imino-D-arabinitol or 1,2-dideoxynojirimycin) (129) a natural iminosugar present in buckwheat (widely used in traditional recipes) is an efficient agent for preventing sharp blood glucose peaks after the intake of refined carbohydrates and for positively influencing intestinal microbiota by favouring adhesion of probiotics. It is supposed that fagomine enhances glucose-induced insulin secretion by accelerating the processes which follow glyceraldehyde 3-phosphate formation in the glycolytic pathway.
The total synthesis, involving RCM, of fagomine (129) and its congeners 3-epi-fagomine (126) and 3,4-di-epi-fagomine (130) was achieved by Takahata et al. [72] based again on the Garner aldehyde 69 derived from D-serine. To construct the chiral 1,2,5,6-tetrahydropyridine core 125, the authors resorted to catalytic ring-closing metathesis induced by the 1st-generation Grubbs catalyst 2, with subsequent stereoselective dihydroxylation (under modified Upjohn conditions, Scheme 22). For iminocyclitols containing trans diols at the 3- and 4-positions an epoxy functionality at the double bond in 125 was introduced. While the syn epoxide 128 led to a mixture of fagomine (129) and 3,4-di-epi-fagomine (130), the anti epoxide 127 gave 129 selectively. The 3-epi-fagomine (126) could also be obtained from the RCM product 125 (by conventional dihydroxylation/deprotection; 10 steps from Garner’s aldehyde 69).
![[1860-5397-7-81-i22]](/bjoc/content/inline/1860-5397-7-81-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Total synthesis of fagomine (129), 3-epi-fagomine (126) and 3,4-di-epi-fagomine (130).
Scheme 22: Total synthesis of fagomine (129), 3-epi-fagomine (126) and 3,4-di-epi-fagomine (130).
Adenophorine (α-1-deoxy-1-C-methylhomonojirimycin) is a further important iminocyclitol in whose synthesis RCM proved helpful. (+)-Adenophorine (135), a naturally occurring iminocyclitol with a lipophilic substituent at the anomeric position, is active on α-glucosidase which is a valid proof that α-alkylation at C1 does not supress the glycosidase inhibitory effect. Its lack of activity on β-galactosidase once again indicates that the relative position of hydroxy substituents is critical for selectivity. In the seminal work by Lebreton and coworkers [73], the first asymmetric total synthesis of (+)-adenophorine was achieved in 14 steps (3.5% overall yield, Scheme 23), starting from the Garner’s aldehyde 69. RCM is essential for construction of the 6-membered N-heterocycle in 133. Protection of the amino alcohols trans-132 and cis-132, as the corresponding trans and cis oxazolidinones, afforded a mixture of diastereomers that were not separable on silica gel. After effecting RCM (2nd-generation Grubbs catalyst 5, 5 mol %) on this mixture, separation of the diastereomers by flash chromatography was possible, affording the pure tetrahydropyridine derivative trans-133 in 74% yield. Successive epoxidations on enantiopure trans-133 and then 134, followed each time by regioselective epoxide opening (with a selenium–boron complex and water, respectively), gave finally 135 with good stereoselectivity. This overall synthesis demonstrates rigorous control at every stage of both the steric configuration of the starting materials and the steric effects induced by substituents attached to the piperidine moiety.
![[1860-5397-7-81-i23]](/bjoc/content/inline/1860-5397-7-81-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Total synthesis of (+)-adenophorine (135).
Scheme 23: Total synthesis of (+)-adenophorine (135).
Related studies by Lebreton et al. [74-76] explored the synthesis of a panel of 6-alkyl substituted piperidine iminocyclitols that had been previously isolated by Asano and coworkers [77] from Adenophora spp. These natural products display an unusual structure in that they possess a hydrophobic substituent such as a undecyl, heptyl, butyl or ethyl group at the α position of 1-C. The strategy for (+)-5-deoxyadenophorine (138) and analogues 142–145 began again from D-Garner aldehyde 69 and also used the powerful RCM as the key step (catalyst 5, 5 mol %; CH2Cl2, reflux 1 h) for building the chiral trans-2,6-disubstituted-1,2,5,6-tetrahydropyridine scaffold (72–88% yield, Scheme 24).
![[1860-5397-7-81-i24]](/bjoc/content/inline/1860-5397-7-81-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Total synthesis of (+)-5-deoxyadenophorine (138) and analogues 142–145.
Scheme 24: Total synthesis of (+)-5-deoxyadenophorine (138) and analogues 142–145.
Azepane-based iminocyclitols
Iminocyclitols incorporating the azepane ring system are more flexible than the parent pyrrolidine and piperidine iminosugars, and they adopt quasi-flattened, low-energy conformations which can potentially lead to a more favourable binding with the active site of enzymes. The unusual spatial distribution of the hydroxy groups in these compounds should generate new inhibitory profiles. According to in vitro assays, seven-membered ring iminocyclitols are noted inhibitors of α-mannosidase, an enzyme that plays important roles in glycoprotein biosynthesis. Derivatives of this class bearing hydroxymethyl groups at C-6 have been shown to inhibit powerfully lysosomal α-mannosidase while displaying varying potencies toward α-1,6-mannosidase. On the other hand, N-alkylated polyhydroxylated azepanes with the D-glucose or L-idose configuration proved to be potent β-glucosidase inhibitors that showed only weak activity towards α-glucosidase and α-mannosidase [78-80]. Malto-oligosaccharides and analogues of di- and trisaccharides containing polyhydroxylated azepane moieties are glucosidase or HIV/FIV-protease blockers, or both.
As for the previous classes, in the synthesis of seven-membered iminocyclitols RCM provides a focal point in ring closure being responsible for constructing the azepane framework. For example, 1,6-dideoxy-1,6-iminoheptitols 148 and 149, that can be viewed as higher homologues of fagomine and nojirimycin, respectively, are easily accessed from the protected diene 146. RCM of this diene with 1st-generation Grubbs catalyst (2, CH2Cl2, 45 °C) gives the common N-heterocyclic intermediate 147 (91% yield, Scheme 25). Hydrogenation of the latter gives the iminocyclitol 148 whereas its cis-selective dihydroxylation affords the pentahydroxy derivative 149.
![[1860-5397-7-81-i25]](/bjoc/content/inline/1860-5397-7-81-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Synthesis by RCM of 1,6-dideoxy-1,6-iminoheptitols 148 and 149.
Scheme 25: Synthesis by RCM of 1,6-dideoxy-1,6-iminoheptitols 148 and 149.
Starting from L-serine 150, Lin et al. [81] devised a refined method for the synthesis of structurally diverse stereoisomers of polyhydroxyazepanes. In their complex strategy, RCM (1st-generation Grubbs catalyst, 10 mol %, CH2Cl2, reflux, 12 h) plays a significant role by leading to a panel of oxazolidinyl azacyclic products (e.g., 152 and 154). Remarkably, the authors expertly arranged the positions of the double bonds involved in RCM on the one hand by addition of alkenyl nucleophiles (with different lengths) on aldehyde intermediates, and on the other hand by placing the second double bond at a different distance relative to the nitrogen atom (Scheme 26).
![[1860-5397-7-81-i26]](/bjoc/content/inline/1860-5397-7-81-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Synthesis by RCM of oxazolidinyl azacycles 152 and 154.
Scheme 26: Synthesis by RCM of oxazolidinyl azacycles 152 and 154.
There are two advantageous follow-ups: (i) a desired location of the double bond in the azacyclic RCM product, and therefore of the hydroxyls in the final iminocyclitol products, and (ii) possible extension of the methodology to the construction of other ring sizes (5- to 8-membered). This versatile approach, featuring the basic sequence metathesis/dihydroxylation, led in good yields to a number of stereoisomers of seven-membered iminocyclitols exhibiting glycosidase inhibitory properties (Scheme 27). Of the compounds shown in Scheme 27, compound 161 with L-configuration at C-6 exhibited the highest inhibition.
![[1860-5397-7-81-i27]](/bjoc/content/inline/1860-5397-7-81-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Representative azepane-based iminocyclitols.
Scheme 27: Representative azepane-based iminocyclitols.
As illustrated in Scheme 28, the 2nd-generation Grubbs catalyst 5 found further application in the recent synthesis of seven-membered ring iminocyclitols, e.g., of 7-hydroxymethyl-1-(4-methylphenylsulfonyl)azepane-3,4,5-triol (169). This compound shares a common configuration of the hydroxy groups with its lower cyclic homologue, 1-deoxymannojirimycin (DMJ, 63), a selective inhibitor of α-mannosidase I [82].
![[1860-5397-7-81-i28]](/bjoc/content/inline/1860-5397-7-81-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Synthesis of hydroxymethyl-1-(4-methylphenylsulfonyl)azepane 3,4,5-triol (169).
Scheme 28: Synthesis of hydroxymethyl-1-(4-methylphenylsulfonyl)azepane 3,4,5-triol (169).
Lee et al. [83] also used RCM induced by the 1st-generation Grubbs catalyst 2 or the 2nd-generation Grubbs catalyst 5 (10 mol %, reflux in toluene; 90–91% yield) in an efficient approach to targeted enantiomerically pure, stereochemically defined, six- and seven-membered heterocyclic scaffolds, i.e., the tetrahydropyridin-3-ol 171 and tetrahydroazepin-3-ol 173 (Scheme 29).
![[1860-5397-7-81-i29]](/bjoc/content/inline/1860-5397-7-81-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Synthesis by RCM of tetrahydropyridin-3-ol 171 and tetrahydroazepin-3-ol 173.
Scheme 29: Synthesis by RCM of tetrahydropyridin-3-ol 171 and tetrahydroazepin-3-ol 173.
These diversely substituted N-heterocyclic compounds, endowed with an internal double bond, are versatile precursors suitable for further functionalization. Asymmetric syntheses employing such intermediates could lead to disclosure of further biologically relevant piperidine/azepane alkaloids and iminosugars.
Conclusion
The paper introduces the broad scope of olefin metathesis as a key reaction in synthetic strategies for the preparation of monocyclic iminocyclitols. In comparison with earlier well-established protocols, olefin metathesis (RCM, CM) offers shorter, simpler and atom-economical routes, and preserving at the same time the carefully designed and worked for stereochemistry of the precursors. Whereas RCM is the method of choice for constructing the pyrrolidine, piperidine or azepane cores of monocyclic iminocyclitols, CM rewardingly permits access to a collection of new iminocyclitols simply by using one heterocyclic intermediate endowed with an olefinic side-chain and changing only its olefin partner. The reaction conditions applied in these crucial steps are rather conventional for metathesis processes, with the choice of the temperature and solvent (refluxing CH2Cl2 or toluene) being dictated by steric demands, and hence energetics, for ring-closing or cross-coupling. While the 1st- and 2nd-generation Grubbs catalysts (5–10 mol %) are the catalysts most frequently employed, the 2nd-generation Grubbs and Hoveyda–Grubbs catalysts perform better when harsher conditions are required. Despite the various functionalities existing on the metathesis precursors and products, sensitive metathesis catalysts are quite productive due to inventive protection/deprotection at the O- and N-heteroatoms. Such delicate operations are skillfully conceived so as to either maintain or reverse the geometry at stereogenic centres, as required. In the ensemble of stereocontrolled reactions concentrating on the economical achievement of the targeted number and relative positions of hydroxy, hydroxyalkyl or other substituents, i.e., the overall structure that hinges on the biological activity, metathesis is surely a fine addition which is bound to succeed in creating novel azasugars with a larger therapeutic window. The metathesis approach may ultimately yield benefits for patients suffering from metabolic disorders, cancer and viral diseases.
References
-
Compain, P.; Martin, O. R., Eds. Iminosugars: From Synthesis to Therapeutic Applications; Wiley-VCH: Weinheim, Germany, 2007.
Return to citation in text: [1] -
Stütz, A. E. Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond; Wiley-VCH: Weinheim, Germany, 1999.
Return to citation in text: [1] -
Martin, O. R.; Compain, P., Eds. Iminosugars: Recent Insights into Their Bioactivity Potential as Therapeutic Agents. Curr. Top. Med. Chem. 2003, 3, 471–591.
Return to citation in text: [1] -
Winchester, B.; Fleet, G. W. J. Glycobiology 1992, 2, 199–210. doi:10.1093/glycob/2.3.199
Return to citation in text: [1] -
Stocker, B. L.; Dangerfield, E. M.; Win-Mason, A. L.; Haslett, G. W.; Timmer, M. S. M. Eur. J. Org. Chem. 2010, 1615–1637. doi:10.1002/ejoc.200901320
Return to citation in text: [1] -
Davis, B. G. Tetrahedron: Asymmetry 2009, 20, 652–671. doi:10.1016/j.tetasy.2009.03.013
Return to citation in text: [1] -
Compain, P.; Chagnault, V.; Martin, O. R. Tetrahedron: Asymmetry 2009, 20, 672–711. doi:10.1016/j.tetasy.2009.03.031
Return to citation in text: [1] -
Pearson, M. S. M.; Mathé-Allainmat, M.; Fargeas, V.; Lebreton, J. Eur. J. Org. Chem. 2005, 2159–2191. doi:10.1002/ejoc.200400823
Return to citation in text: [1] [2] [3] [4] [5] -
Compain, P.; Martin, O. R. Curr. Top. Med. Chem. 2003, 3, 541–560. doi:10.2174/1568026033452474
Return to citation in text: [1] -
Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102, 515–554. doi:10.1021/cr000433k
Return to citation in text: [1] [2] -
Moreno-Clavijo, E.; Carmona, A. T.; Moreno-Vargas, A. J.; Molina, L.; Robina, I. Curr. Org. Synth. 2011, 8, 102–133.
Return to citation in text: [1] -
Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/S0957-4166(00)00113-0
Return to citation in text: [1] [2] -
Asano, N. Naturally Occurring Iminosugars and Related Alkaloids: Structure, Activity and Applications. In Iminosugars: From Synthesis to Therapeutic Applications; Compain, P.; Martin, O. R., Eds.; Wiley-VCH: Weinheim, Germany , 2007; pp 7–24. doi:10.1002/9780470517437.ch2
Return to citation in text: [1] -
Asano, N. Curr. Top. Med. Chem. 2003, 3, 471–484. doi:10.2174/1568026033452438
Return to citation in text: [1] -
Watson, A. A.; Fleet, G. W. J.; Asano, N.; Molyneux, R. J.; Nash, R. J. Phytochemistry 2001, 56, 265–295. doi:10.1016/S0031-9422(00)00451-9
Return to citation in text: [1] -
Goss, P. E.; Baptiste, J.; Fernandes, B.; Baker, M. A.; Dennis, J. W. Cancer Res. 1994, 54, 1450–1457.
Return to citation in text: [1] -
Goss, P. E.; Baker, M. A.; Carver, J. P.; Dennis, J. W. Clin. Cancer Res. 1995, 1, 935–944.
Return to citation in text: [1] -
Asano, N. Glycobiology 2003, 13, 93R–104R. doi:10.1093/glycob/cwg090
Return to citation in text: [1] -
Nishimura, Y. Curr. Top. Med. Chem. 2003, 3, 575–591. doi:10.2174/1568026033452492
Return to citation in text: [1] -
Nishimura, Y. Iminosugar-based antitumoural agents. In Iminosugars: From Synthesis to Therapeutic Applications; Compain, P.; Martin, O. R., Eds.; Wiley-VCH: Weinheim, Germany, 2007; pp 269–294. doi:10.1002/9780470517437.ch12
Return to citation in text: [1] -
Butters, T. D.; Dwek, R. A.; Platt, F. M. Chem. Rev. 2000, 100, 4683–4696. doi:10.1021/cr990292q
Return to citation in text: [1] -
Fan, J. Q. Trends Pharmacol. Sci. 2003, 24, 355–360. doi:10.1016/S0165-6147(03)00158-5
Return to citation in text: [1] -
Yu, Z.; Sawkar, A. R.; Whalen, L. J.; Wong, C.; Kelly, J. W. J. Med. Chem. 2007, 50, 94–100. doi:10.1021/jm060677i
Return to citation in text: [1] -
Mitrakou, A.; Tountas, N.; Raptis, A. E.; Bauer, R. J.; Schulz, H.; Raptis, S. A. Diabetic Med. 1998, 15, 657–660. doi:10.1002/(SICI)1096-9136(199808)15:8<657::AID-DIA652>3.0.CO;2-7
Return to citation in text: [1] -
Andersen, B.; Rassov, A.; Westergaard, N.; Lundgren, K. Biochem. J. 1999, 342, 545–550.
Return to citation in text: [1] -
Somsak, L.; Nagy, V.; Hadazy, Z.; Docsa, T.; Gergely, P. Curr. Pharm. Des. 2003, 9, 1177–1189.
Return to citation in text: [1] -
Durantel, D.; Branza-Nichita, N.; Carrouee-Durantel, S.; Butters, T. D.; Dwek, R. A.; Zitzmann, N. J. Virol. 2001, 75, 8987–8998. doi:10.1128/JVI.75.19.8987-8998.2001
Return to citation in text: [1] [2] -
Greimel, P.; Spreitz, J.; Stütz, A. E.; Wrodnigg, T. M. Curr. Top. Med. Chem. 2003, 3, 513–523. doi:10.2174/1568026033452456
Return to citation in text: [1] -
Gruters, R. A.; Neefjes, J. J.; Tersmette, M.; de Goede, R. E.; Tulp, A.; Huisman, H. G.; Miedema, F.; Ploegh, H. L. Nature 1987, 330, 74–77. doi:10.1038/330074a0
Return to citation in text: [1] -
Walker, B. D.; Kowalski, M.; Goh, W. C.; Kozarsky, K.; Krieger, M.; Rosen, C.; Rohrschneider, L.; Haseltine, W. A.; Sodroski, J. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 8120–8124.
Return to citation in text: [1] -
Block, T. M.; Lu, X.; Platt, F. M.; Foster, G. R.; Gerlich, W. H.; Blumberg, B. S.; Dwek, R. A. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 2235–2239.
Return to citation in text: [1] -
Mehta, A.; Carrouee, S.; Conyers, B.; Jordan, R.; Butters, T. D.; Dwek, R. A.; Block, T. M. Hepatology (Hoboken, NJ, U. S.) 2001, 33, 1488–1495. doi:10.1053/jhep.2001.25103
Return to citation in text: [1] -
Durantel, D.; Carrouée-Durantel, S.; Branza-Nichita, N.; Dwek, R. A.; Zitzmann, N. Antimicrob. Agents Chemother. 2004, 48, 497–504. doi:10.1128/AAC.48.2.497-504.2004
Return to citation in text: [1] -
Ye, X.-S.; Sun, F.; Liu, M.; Li, Q.; Wang, Y.; Zhang, G.; Zhang, L.-H.; Zhang, X. L. J. Med. Chem. 2005, 48, 3688–3691. doi:10.1021/jm050169t
Return to citation in text: [1] -
Désiré, J.; Dransfield, P. J.; Gore, P. M.; Shipman, M. Synlett 2001, 1329–1331. doi:10.1055/s-2001-16039
Return to citation in text: [1] -
Kumar, V.; Ramesh, N. G. Tetrahedron 2006, 62, 1877–1885. doi:10.1016/j.tet.2005.11.037
Return to citation in text: [1] -
Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592–4633. doi:10.1002/anie.200300576
Return to citation in text: [1] -
Grubbs, R. H., Ed. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003; Vol. 1.
Return to citation in text: [1] -
Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424
Return to citation in text: [1] -
Dunne, A. M.; Mix, S.; Blechert, S. Tetrahedron Lett. 2003, 44, 2733–2736. doi:10.1016/S0040-4039(03)00346-0
Return to citation in text: [1] -
Bieniek, M.; Michrowska, A.; Usanov, D. L.; Grela, K. Chem.–Eur. J. 2008, 14, 806–818. doi:10.1002/chem.200701340
Return to citation in text: [1] -
Sauvage, X.; Borguet, Y.; Zaragoza, G.; Demonceau, A.; Delaude, L. Adv. Synth. Catal. 2009, 351, 441–455. doi:10.1002/adsc.200800664
Return to citation in text: [1] -
Nolan, S. P.; Clavier, H. Chem. Soc. Rev. 2010, 39, 3305–3316. doi:10.1039/b912410c
Return to citation in text: [1] -
Ding, F.; Sun, Y. G.; Monsaert, S.; Dragutan, I.; Dragutan, V.; Verpoort, F. Curr. Org. Synth. 2008, 5, 291–304.
Return to citation in text: [1] -
Krehl, S.; Geissler, D.; Hauke, S.; Kunz, O.; Staude, L.; Schmidt, B. Beilstein J. Org. Chem. 2010, 6, 1188–1198. doi:10.3762/bjoc.6.136
Return to citation in text: [1] -
Dragutan, V.; Dragutan, I.; Balaban, A. T. Platinum Met. Rev. 2000, 44, 112–118.
Return to citation in text: [1] -
Dragutan, I.; Dragutan, V.; Delaude, L.; Demonceau, A.; Noels, A. F. Rev. Roum. Chim. 2007, 52, 1013–1025.
Return to citation in text: [1] -
Felpin, F.-X.; Lebreton, J. Eur. J. Org. Chem. 2003, 3693–3712. doi:10.1002/ejoc.200300193
Return to citation in text: [1] -
Saotome, C.; Wong, C.-H.; Kanie, O. Chem. Biol. 2001, 8, 1061–1070. doi:10.1016/S1074-5521(01)00074-6
Return to citation in text: [1] -
Behling, J. R.; Campbell, A. L.; Babiak, K. A.; Ng, J. S.; Medic, J.; Farid, P.; Fleet, G. W. J. Tetrahedron 1993, 49, 3359–3366. doi:10.1016/S0040-4020(01)90163-2
Return to citation in text: [1] -
Moriarty, R. M.; Mitan, C. I.; Branza-Nichita, N.; Phares, K. R.; Parrish, D. Org. Lett. 2006, 8, 3465–3467. doi:10.1021/ol061071r
Return to citation in text: [1] -
Cren, S.; Wilson, C.; Thomas, N. R. Org. Lett. 2005, 7, 3521–3523. doi:10.1021/ol051232b
Return to citation in text: [1] -
Huwe, C. M.; Blechert, S. Synthesis 1997, 61–67. doi:10.1055/s-1997-1497
Return to citation in text: [1] -
Madhan, A.; Rao, B. V. Tetrahedron Lett. 2003, 44, 5641–5643. doi:10.1016/S0040-4039(03)01366-2
Return to citation in text: [1] -
Fleet, G. W. J.; Son, J. C. Tetrahedron 1988, 44, 2637–2647. doi:10.1016/S0040-4020(01)81716-6
Return to citation in text: [1] -
Davis, F. A.; Ramachandar, T.; Chai, J.; Skucas, E. Tetrahedron Lett. 2006, 47, 2743–2746. doi:10.1016/j.tetlet.2006.02.092
Return to citation in text: [1] -
Chandrasekhar, B.; Madhan, A.; Rao, B. V. Tetrahedron 2007, 63, 8746–8751. doi:10.1016/j.tet.2007.06.044
Return to citation in text: [1] -
Murruzzu, C.; Riera, A. Tetrahedron: Asymmetry 2007, 18, 149–154. doi:10.1016/j.tetasy.2006.12.023
Return to citation in text: [1] -
Doddi, V. R.; Vankar, Y. D. Eur. J. Org. Chem. 2007, 5583–5589. doi:10.1002/ejoc.200700719
Return to citation in text: [1] -
Trost, B. M.; Horne, D. B.; Woltering, M. J. Chem.–Eur. J. 2006, 12, 6607–6620. doi:10.1002/chem.200600202
Return to citation in text: [1] -
Trost, B. M.; Horne, D. B.; Woltering, M. J. Angew. Chem., Int. Ed. 2003, 42, 5987–5990. doi:10.1002/anie.200352857
Return to citation in text: [1] -
Ribes, C.; Falomir, E.; Murga, J.; Carda, M.; Marco, J. A. Org. Biomol. Chem. 2009, 7, 1355–1360. doi:10.1039/B821431J
Return to citation in text: [1] -
Ribes, C.; Falomir, E.; Murga, J.; Carda, M.; Marco, J. A. Tetrahedron 2009, 65, 10612–10616. doi:10.1016/j.tet.2009.10.066
Return to citation in text: [1] -
Takahata, H.; Banba, Y.; Sasatani, M.; Nemoto, H.; Kato, A.; Adachi, I. Tetrahedron 2004, 60, 8199–8205. doi:10.1016/j.tet.2004.06.112
Return to citation in text: [1] -
Danoun, G.; Ceccon, J.; Greene, A. E.; Poisson, J.-F. Eur. J. Org. Chem. 2009, 4221–4224. doi:10.1002/ejoc.200900595
Return to citation in text: [1] -
Rengasamy, R.; Curtis-Long, M. J.; Seo, W. D.; Jeong, S. H.; Jeong, I.-Y.; Park, K. H. J. Org. Chem. 2008, 73, 2898–2901. doi:10.1021/jo702480y
Return to citation in text: [1] -
Takahata, H.; Banba, Y.; Ouchi, H.; Nemoto, H. Org. Lett. 2003, 5, 2527–2529. doi:10.1021/ol034886y
Return to citation in text: [1] [2] -
van den Nieuwendijk, A. M. C. H.; Ruben, M.; Engelsma, S. E.; Risseeuw, M. D. P.; van den Berg, R. J. B. H. N.; Boot, R. G.; Aerts, J. M.; Brussee, J.; van der Marel, G. A.; Overkleeft, H. S. Org. Lett. 2010, 12, 3957–3959. doi:10.1021/ol101556k
Return to citation in text: [1] -
Singh, O. V.; Han, H. Tetrahedron Lett. 2003, 44, 2387–2391. doi:10.1016/S0040-4039(03)00218-1
Return to citation in text: [1] -
Han, H. Tetrahedron Lett. 2003, 44, 1567–1569. doi:10.1016/S0040-4039(03)00014-5
Return to citation in text: [1] -
Ghosh, S.; Shashidhar, J.; Dutta, S. K. Tetrahedron Lett. 2006, 47, 6041–6044. doi:10.1016/j.tetlet.2006.06.110
Return to citation in text: [1] -
Banba, Y.; Abe, C.; Nemoto, H.; Kato, A.; Adachi, I.; Takahata, H. Tetrahedron: Asymmetry 2001, 12, 817–819. doi:10.1016/S0957-4166(01)00136-7
Return to citation in text: [1] -
Pearson, M. S. M.; Evain, M.; Mathé-Allainmat, M.; Lebreton, J. Eur. J. Org. Chem. 2007, 4888–4894. doi:10.1002/ejoc.200700459
Return to citation in text: [1] -
Felpin, F.-X.; Boubekeur, K.; Lebreton, J. J. Org. Chem. 2004, 69, 1497–1503. doi:10.1021/jo035522m
Return to citation in text: [1] -
Felpin, F.-X.; Lebreton, J. Tetrahedron Lett. 2003, 44, 527–530. doi:10.1016/S0040-4039(02)02586-8
Return to citation in text: [1] -
Felpin, F.-X.; Boubekeur, K.; Lebreton, J. Eur. J. Org. Chem. 2003, 4518–4527. doi:10.1002/ejoc.200300423
Return to citation in text: [1] -
Ikeda, K.; Takahashi, M.; Nishida, M.; Miyauchi, M.; Kizu, H.; Kameda, Y.; Arisawa, M.; Watson, A. A.; Nash, R. J.; Fleet, G. W. J.; Asano, N. Carbohydr. Res. 1999, 323, 73–80. doi:10.1016/S0008-6215(99)00246-3
Return to citation in text: [1] -
Butters, T. D.; Alonzi, D. S.; Kukushkin, N. V.; Ren, Y.; Blériot, Y. Glycoconjugate J. 2009, 26, 1109–1116. doi:10.1007/s10719-009-9231-3
Return to citation in text: [1] -
Li, H.; Liu, T.; Zhang, Y.; Favre, S.; Bello, C.; Vogel, P.; Butters, T. D.; Oikonomakos, N. G.; Marrot, J.; Blériot, Y. ChemBioChem 2008, 9, 253–260. doi:10.1002/cbic.200700496
Return to citation in text: [1] -
Li, H.; Blériot, Y.; Chantereau, C.; Mallet, J.-M.; Sollogoub, M.; Zhang, Y.; Rodriquez-García, E.; Vogel, P.; Jiménez-Barbero, J.; Sinaÿ, P. Org. Biomol. Chem. 2004, 2, 1492–1499. doi:10.1039/b402542c
Return to citation in text: [1] -
Lin, C.-C.; Pan, Y.-s.; Patkar, L. N.; Lin, H.-M.; Tzou, D.-L. M.; Subramanian, T.; Lin, C.-C. Bioorg. Med. Chem. 2004, 12, 3259–3267. doi:10.1016/j.bmc.2004.03.064
Return to citation in text: [1] -
Chang, M.-Y.; Kung, Y.-H.; Ma, C.-C.; Chen, S.-T. Tetrahedron 2007, 63, 1339–1344. doi:10.1016/j.tet.2006.12.002
Return to citation in text: [1] -
Lee, H. K.; Im, J. H.; Jung, S. H. Tetrahedron 2007, 63, 3321–3327. doi:10.1016/j.tet.2007.02.045
Return to citation in text: [1]
| 64. | Takahata, H.; Banba, Y.; Sasatani, M.; Nemoto, H.; Kato, A.; Adachi, I. Tetrahedron 2004, 60, 8199–8205. doi:10.1016/j.tet.2004.06.112 |
| 65. | Danoun, G.; Ceccon, J.; Greene, A. E.; Poisson, J.-F. Eur. J. Org. Chem. 2009, 4221–4224. doi:10.1002/ejoc.200900595 |
| 66. | Rengasamy, R.; Curtis-Long, M. J.; Seo, W. D.; Jeong, S. H.; Jeong, I.-Y.; Park, K. H. J. Org. Chem. 2008, 73, 2898–2901. doi:10.1021/jo702480y |
| 1. | Compain, P.; Martin, O. R., Eds. Iminosugars: From Synthesis to Therapeutic Applications; Wiley-VCH: Weinheim, Germany, 2007. |
| 2. | Stütz, A. E. Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond; Wiley-VCH: Weinheim, Germany, 1999. |
| 3. | Martin, O. R.; Compain, P., Eds. Iminosugars: Recent Insights into Their Bioactivity Potential as Therapeutic Agents. Curr. Top. Med. Chem. 2003, 3, 471–591. |
| 4. | Winchester, B.; Fleet, G. W. J. Glycobiology 1992, 2, 199–210. doi:10.1093/glycob/2.3.199 |
| 5. | Stocker, B. L.; Dangerfield, E. M.; Win-Mason, A. L.; Haslett, G. W.; Timmer, M. S. M. Eur. J. Org. Chem. 2010, 1615–1637. doi:10.1002/ejoc.200901320 |
| 6. | Davis, B. G. Tetrahedron: Asymmetry 2009, 20, 652–671. doi:10.1016/j.tetasy.2009.03.013 |
| 7. | Compain, P.; Chagnault, V.; Martin, O. R. Tetrahedron: Asymmetry 2009, 20, 672–711. doi:10.1016/j.tetasy.2009.03.031 |
| 8. | Pearson, M. S. M.; Mathé-Allainmat, M.; Fargeas, V.; Lebreton, J. Eur. J. Org. Chem. 2005, 2159–2191. doi:10.1002/ejoc.200400823 |
| 9. | Compain, P.; Martin, O. R. Curr. Top. Med. Chem. 2003, 3, 541–560. doi:10.2174/1568026033452474 |
| 10. | Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102, 515–554. doi:10.1021/cr000433k |
| 11. | Moreno-Clavijo, E.; Carmona, A. T.; Moreno-Vargas, A. J.; Molina, L.; Robina, I. Curr. Org. Synth. 2011, 8, 102–133. |
| 12. | Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/S0957-4166(00)00113-0 |
| 23. | Yu, Z.; Sawkar, A. R.; Whalen, L. J.; Wong, C.; Kelly, J. W. J. Med. Chem. 2007, 50, 94–100. doi:10.1021/jm060677i |
| 49. | Saotome, C.; Wong, C.-H.; Kanie, O. Chem. Biol. 2001, 8, 1061–1070. doi:10.1016/S1074-5521(01)00074-6 |
| 50. | Behling, J. R.; Campbell, A. L.; Babiak, K. A.; Ng, J. S.; Medic, J.; Farid, P.; Fleet, G. W. J. Tetrahedron 1993, 49, 3359–3366. doi:10.1016/S0040-4020(01)90163-2 |
| 51. | Moriarty, R. M.; Mitan, C. I.; Branza-Nichita, N.; Phares, K. R.; Parrish, D. Org. Lett. 2006, 8, 3465–3467. doi:10.1021/ol061071r |
| 52. | Cren, S.; Wilson, C.; Thomas, N. R. Org. Lett. 2005, 7, 3521–3523. doi:10.1021/ol051232b |
| 72. | Banba, Y.; Abe, C.; Nemoto, H.; Kato, A.; Adachi, I.; Takahata, H. Tetrahedron: Asymmetry 2001, 12, 817–819. doi:10.1016/S0957-4166(01)00136-7 |
| 21. | Butters, T. D.; Dwek, R. A.; Platt, F. M. Chem. Rev. 2000, 100, 4683–4696. doi:10.1021/cr990292q |
| 22. | Fan, J. Q. Trends Pharmacol. Sci. 2003, 24, 355–360. doi:10.1016/S0165-6147(03)00158-5 |
| 73. | Pearson, M. S. M.; Evain, M.; Mathé-Allainmat, M.; Lebreton, J. Eur. J. Org. Chem. 2007, 4888–4894. doi:10.1002/ejoc.200700459 |
| 16. | Goss, P. E.; Baptiste, J.; Fernandes, B.; Baker, M. A.; Dennis, J. W. Cancer Res. 1994, 54, 1450–1457. |
| 17. | Goss, P. E.; Baker, M. A.; Carver, J. P.; Dennis, J. W. Clin. Cancer Res. 1995, 1, 935–944. |
| 18. | Asano, N. Glycobiology 2003, 13, 93R–104R. doi:10.1093/glycob/cwg090 |
| 19. | Nishimura, Y. Curr. Top. Med. Chem. 2003, 3, 575–591. doi:10.2174/1568026033452492 |
| 20. | Nishimura, Y. Iminosugar-based antitumoural agents. In Iminosugars: From Synthesis to Therapeutic Applications; Compain, P.; Martin, O. R., Eds.; Wiley-VCH: Weinheim, Germany, 2007; pp 269–294. doi:10.1002/9780470517437.ch12 |
| 37. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592–4633. doi:10.1002/anie.200300576 |
| 38. | Grubbs, R. H., Ed. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003; Vol. 1. |
| 39. | Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424 |
| 40. | Dunne, A. M.; Mix, S.; Blechert, S. Tetrahedron Lett. 2003, 44, 2733–2736. doi:10.1016/S0040-4039(03)00346-0 |
| 41. | Bieniek, M.; Michrowska, A.; Usanov, D. L.; Grela, K. Chem.–Eur. J. 2008, 14, 806–818. doi:10.1002/chem.200701340 |
| 42. | Sauvage, X.; Borguet, Y.; Zaragoza, G.; Demonceau, A.; Delaude, L. Adv. Synth. Catal. 2009, 351, 441–455. doi:10.1002/adsc.200800664 |
| 43. | Nolan, S. P.; Clavier, H. Chem. Soc. Rev. 2010, 39, 3305–3316. doi:10.1039/b912410c |
| 44. | Ding, F.; Sun, Y. G.; Monsaert, S.; Dragutan, I.; Dragutan, V.; Verpoort, F. Curr. Org. Synth. 2008, 5, 291–304. |
| 45. | Krehl, S.; Geissler, D.; Hauke, S.; Kunz, O.; Staude, L.; Schmidt, B. Beilstein J. Org. Chem. 2010, 6, 1188–1198. doi:10.3762/bjoc.6.136 |
| 46. | Dragutan, V.; Dragutan, I.; Balaban, A. T. Platinum Met. Rev. 2000, 44, 112–118. |
| 47. | Dragutan, I.; Dragutan, V.; Delaude, L.; Demonceau, A.; Noels, A. F. Rev. Roum. Chim. 2007, 52, 1013–1025. |
| 67. | Takahata, H.; Banba, Y.; Ouchi, H.; Nemoto, H. Org. Lett. 2003, 5, 2527–2529. doi:10.1021/ol034886y |
| 12. | Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/S0957-4166(00)00113-0 |
| 13. | Asano, N. Naturally Occurring Iminosugars and Related Alkaloids: Structure, Activity and Applications. In Iminosugars: From Synthesis to Therapeutic Applications; Compain, P.; Martin, O. R., Eds.; Wiley-VCH: Weinheim, Germany , 2007; pp 7–24. doi:10.1002/9780470517437.ch2 |
| 14. | Asano, N. Curr. Top. Med. Chem. 2003, 3, 471–484. doi:10.2174/1568026033452438 |
| 15. | Watson, A. A.; Fleet, G. W. J.; Asano, N.; Molyneux, R. J.; Nash, R. J. Phytochemistry 2001, 56, 265–295. doi:10.1016/S0031-9422(00)00451-9 |
| 48. | Felpin, F.-X.; Lebreton, J. Eur. J. Org. Chem. 2003, 3693–3712. doi:10.1002/ejoc.200300193 |
| 71. | Ghosh, S.; Shashidhar, J.; Dutta, S. K. Tetrahedron Lett. 2006, 47, 6041–6044. doi:10.1016/j.tetlet.2006.06.110 |
| 29. | Gruters, R. A.; Neefjes, J. J.; Tersmette, M.; de Goede, R. E.; Tulp, A.; Huisman, H. G.; Miedema, F.; Ploegh, H. L. Nature 1987, 330, 74–77. doi:10.1038/330074a0 |
| 30. | Walker, B. D.; Kowalski, M.; Goh, W. C.; Kozarsky, K.; Krieger, M.; Rosen, C.; Rohrschneider, L.; Haseltine, W. A.; Sodroski, J. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 8120–8124. |
| 27. | Durantel, D.; Branza-Nichita, N.; Carrouee-Durantel, S.; Butters, T. D.; Dwek, R. A.; Zitzmann, N. J. Virol. 2001, 75, 8987–8998. doi:10.1128/JVI.75.19.8987-8998.2001 |
| 33. | Durantel, D.; Carrouée-Durantel, S.; Branza-Nichita, N.; Dwek, R. A.; Zitzmann, N. Antimicrob. Agents Chemother. 2004, 48, 497–504. doi:10.1128/AAC.48.2.497-504.2004 |
| 69. | Singh, O. V.; Han, H. Tetrahedron Lett. 2003, 44, 2387–2391. doi:10.1016/S0040-4039(03)00218-1 |
| 27. | Durantel, D.; Branza-Nichita, N.; Carrouee-Durantel, S.; Butters, T. D.; Dwek, R. A.; Zitzmann, N. J. Virol. 2001, 75, 8987–8998. doi:10.1128/JVI.75.19.8987-8998.2001 |
| 28. | Greimel, P.; Spreitz, J.; Stütz, A. E.; Wrodnigg, T. M. Curr. Top. Med. Chem. 2003, 3, 513–523. doi:10.2174/1568026033452456 |
| 8. | Pearson, M. S. M.; Mathé-Allainmat, M.; Fargeas, V.; Lebreton, J. Eur. J. Org. Chem. 2005, 2159–2191. doi:10.1002/ejoc.200400823 |
| 34. | Ye, X.-S.; Sun, F.; Liu, M.; Li, Q.; Wang, Y.; Zhang, G.; Zhang, L.-H.; Zhang, X. L. J. Med. Chem. 2005, 48, 3688–3691. doi:10.1021/jm050169t |
| 35. | Désiré, J.; Dransfield, P. J.; Gore, P. M.; Shipman, M. Synlett 2001, 1329–1331. doi:10.1055/s-2001-16039 |
| 36. | Kumar, V.; Ramesh, N. G. Tetrahedron 2006, 62, 1877–1885. doi:10.1016/j.tet.2005.11.037 |
| 70. | Han, H. Tetrahedron Lett. 2003, 44, 1567–1569. doi:10.1016/S0040-4039(03)00014-5 |
| 10. | Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102, 515–554. doi:10.1021/cr000433k |
| 67. | Takahata, H.; Banba, Y.; Ouchi, H.; Nemoto, H. Org. Lett. 2003, 5, 2527–2529. doi:10.1021/ol034886y |
| 24. | Mitrakou, A.; Tountas, N.; Raptis, A. E.; Bauer, R. J.; Schulz, H.; Raptis, S. A. Diabetic Med. 1998, 15, 657–660. doi:10.1002/(SICI)1096-9136(199808)15:8<657::AID-DIA652>3.0.CO;2-7 |
| 25. | Andersen, B.; Rassov, A.; Westergaard, N.; Lundgren, K. Biochem. J. 1999, 342, 545–550. |
| 26. | Somsak, L.; Nagy, V.; Hadazy, Z.; Docsa, T.; Gergely, P. Curr. Pharm. Des. 2003, 9, 1177–1189. |
| 31. | Block, T. M.; Lu, X.; Platt, F. M.; Foster, G. R.; Gerlich, W. H.; Blumberg, B. S.; Dwek, R. A. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 2235–2239. |
| 32. | Mehta, A.; Carrouee, S.; Conyers, B.; Jordan, R.; Butters, T. D.; Dwek, R. A.; Block, T. M. Hepatology (Hoboken, NJ, U. S.) 2001, 33, 1488–1495. doi:10.1053/jhep.2001.25103 |
| 68. | van den Nieuwendijk, A. M. C. H.; Ruben, M.; Engelsma, S. E.; Risseeuw, M. D. P.; van den Berg, R. J. B. H. N.; Boot, R. G.; Aerts, J. M.; Brussee, J.; van der Marel, G. A.; Overkleeft, H. S. Org. Lett. 2010, 12, 3957–3959. doi:10.1021/ol101556k |
| 56. | Davis, F. A.; Ramachandar, T.; Chai, J.; Skucas, E. Tetrahedron Lett. 2006, 47, 2743–2746. doi:10.1016/j.tetlet.2006.02.092 |
| 54. | Madhan, A.; Rao, B. V. Tetrahedron Lett. 2003, 44, 5641–5643. doi:10.1016/S0040-4039(03)01366-2 |
| 74. | Felpin, F.-X.; Boubekeur, K.; Lebreton, J. J. Org. Chem. 2004, 69, 1497–1503. doi:10.1021/jo035522m |
| 75. | Felpin, F.-X.; Lebreton, J. Tetrahedron Lett. 2003, 44, 527–530. doi:10.1016/S0040-4039(02)02586-8 |
| 76. | Felpin, F.-X.; Boubekeur, K.; Lebreton, J. Eur. J. Org. Chem. 2003, 4518–4527. doi:10.1002/ejoc.200300423 |
| 55. | Fleet, G. W. J.; Son, J. C. Tetrahedron 1988, 44, 2637–2647. doi:10.1016/S0040-4020(01)81716-6 |
| 77. | Ikeda, K.; Takahashi, M.; Nishida, M.; Miyauchi, M.; Kizu, H.; Kameda, Y.; Arisawa, M.; Watson, A. A.; Nash, R. J.; Fleet, G. W. J.; Asano, N. Carbohydr. Res. 1999, 323, 73–80. doi:10.1016/S0008-6215(99)00246-3 |
| 78. | Butters, T. D.; Alonzi, D. S.; Kukushkin, N. V.; Ren, Y.; Blériot, Y. Glycoconjugate J. 2009, 26, 1109–1116. doi:10.1007/s10719-009-9231-3 |
| 79. | Li, H.; Liu, T.; Zhang, Y.; Favre, S.; Bello, C.; Vogel, P.; Butters, T. D.; Oikonomakos, N. G.; Marrot, J.; Blériot, Y. ChemBioChem 2008, 9, 253–260. doi:10.1002/cbic.200700496 |
| 80. | Li, H.; Blériot, Y.; Chantereau, C.; Mallet, J.-M.; Sollogoub, M.; Zhang, Y.; Rodriquez-García, E.; Vogel, P.; Jiménez-Barbero, J.; Sinaÿ, P. Org. Biomol. Chem. 2004, 2, 1492–1499. doi:10.1039/b402542c |
| 8. | Pearson, M. S. M.; Mathé-Allainmat, M.; Fargeas, V.; Lebreton, J. Eur. J. Org. Chem. 2005, 2159–2191. doi:10.1002/ejoc.200400823 |
| 8. | Pearson, M. S. M.; Mathé-Allainmat, M.; Fargeas, V.; Lebreton, J. Eur. J. Org. Chem. 2005, 2159–2191. doi:10.1002/ejoc.200400823 |
| 62. | Ribes, C.; Falomir, E.; Murga, J.; Carda, M.; Marco, J. A. Org. Biomol. Chem. 2009, 7, 1355–1360. doi:10.1039/B821431J |
| 63. | Ribes, C.; Falomir, E.; Murga, J.; Carda, M.; Marco, J. A. Tetrahedron 2009, 65, 10612–10616. doi:10.1016/j.tet.2009.10.066 |
| 8. | Pearson, M. S. M.; Mathé-Allainmat, M.; Fargeas, V.; Lebreton, J. Eur. J. Org. Chem. 2005, 2159–2191. doi:10.1002/ejoc.200400823 |
| 59. | Doddi, V. R.; Vankar, Y. D. Eur. J. Org. Chem. 2007, 5583–5589. doi:10.1002/ejoc.200700719 |
| 83. | Lee, H. K.; Im, J. H.; Jung, S. H. Tetrahedron 2007, 63, 3321–3327. doi:10.1016/j.tet.2007.02.045 |
| 60. | Trost, B. M.; Horne, D. B.; Woltering, M. J. Chem.–Eur. J. 2006, 12, 6607–6620. doi:10.1002/chem.200600202 |
| 61. | Trost, B. M.; Horne, D. B.; Woltering, M. J. Angew. Chem., Int. Ed. 2003, 42, 5987–5990. doi:10.1002/anie.200352857 |
| 57. | Chandrasekhar, B.; Madhan, A.; Rao, B. V. Tetrahedron 2007, 63, 8746–8751. doi:10.1016/j.tet.2007.06.044 |
| 81. | Lin, C.-C.; Pan, Y.-s.; Patkar, L. N.; Lin, H.-M.; Tzou, D.-L. M.; Subramanian, T.; Lin, C.-C. Bioorg. Med. Chem. 2004, 12, 3259–3267. doi:10.1016/j.bmc.2004.03.064 |
| 58. | Murruzzu, C.; Riera, A. Tetrahedron: Asymmetry 2007, 18, 149–154. doi:10.1016/j.tetasy.2006.12.023 |
| 82. | Chang, M.-Y.; Kung, Y.-H.; Ma, C.-C.; Chen, S.-T. Tetrahedron 2007, 63, 1339–1344. doi:10.1016/j.tet.2006.12.002 |
© 2011 Dragutan et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)